
Abstract
Background
Amino acids are the basic components of protein and an important index to evaluate meat quality. With the rapid development of genomics, candidate regions and genes affecting amino acid content in livestock and poultry have been gradually revealed. Hence, genome-wide association study (GWAS) can be used to screen candidate loci associated with amino acid content in duck meat.
Result
In the current study, the content of 16 amino acids was detected in 358 duck breast muscles. The proportion of Glu to the total amino acid content was relatively high, and the proportion was 0.14. However, the proportion of Met content was relatively low, at just 0.03. By comparative analysis, significant differences were found between males and females in 3 amino acids, including Ser, Met, and Phe. In addition, 12 SNPs were significantly correlated with Pro content by GWAS analysis, and these SNPs were annotated by 7 protein-coding genes; 8 significant SNPs were associated with Tyr content, and these SNPs were annotated by 6 protein-coding genes. At the same time, linkage disequilibrium (LD) analysis was performed on these regions with significant signals. The results showed that three SNPs in the 55–56 Mbp region of chromosome 3 were highly correlated with the leader SNP (chr3:55526954) that affected Pro content (r2 > 0.6). Similarly, LD analysis showed that there were three SNPs in the 21.2–21.6 Mbp region of chromosome 13, which were highly correlated with leader SNP (chr13:21421661) (r2 > 0.6). Moreover, Through functional enrichment analysis of all candidate genes. The results of GO enrichment analysis showed that several significant GO items were associated with amino acid transport function, including amino acid transmembrane transport and glutamine transport. The results further indicate that these candidate genes are closely associated with amino acid transport. Among them, key candidate genes include SLC38A1. For KEGG enrichment analysis, CACNA2D3 and CACNA1D genes were covered by significant pathways.
Conclusion
In this study, GWAS analysis found a total of 28 significant SNPs affecting amino acid content. Through gene annotation, a total of 20 candidate genes were screened. In addition, Through LD analysis and enrichment analysis, we considered that SERAC1, CACNA2D3 and SLC38A1 genes are important candidate genes affecting amino acid content in duck breast muscle.
Similar content being viewed by others

Background
With the improvement in living standards, consumers pay more and more attention to the meat quality of livestock and poultry [1]. The nutritional value of protein in meat directly reflects its quality [2]. Amino acids are important components of muscle protein, and the content and composition of amino acids can be used as important indicators for evaluating the nutritional value of meat [3, 4]. The content of essential amino acids in beef is higher than that of other meat products, making beef have high nutritional value [5]. In addition, in pork and mutton studies, different amino acid contents and ratios affect meat quality differently [6, 7]. Asparagine (Asp) and glutamate (Glu) produce umami, and alanine (Ala) and proline (Pro) produce sweetness, which makes meat more delicious. Also, the amino acid content has an extremely important effect on livestock and poultry meat quality.
The amino acid content and proportion in meat are affected by genetics, nutrition, environment, and other aspects [8]. Genetic factors are an important influencing factor [9,10,11]. Meanwhile, At the beginning of this century, scholars have gradually investigated the underlying genetic mechanism of some chemical compositions in meat. Some studies have revealed quantitative trait loci (QTL) and candidate genes affecting fatty acid content in livestock based on whole-genome scanning and gene linkage analysis with microsatellite markers [12,13,14]. In recent years, with the rapid development of nucleotide sequencing technology, genome-wide association analysis (GWAS) has been widely used to reveal the genetic foundations of amino acids in meat [15]. Sasago et al. used 22 free amino acids in 574 Japanese black beef as phenotypes for GWAS and The result shows that, the SLC6A6 gene may affect taurine content, and STT3B and GADL1 genes may affect β-alanine content [16]. In addition, in some bivalves, Meng et al. used the 18 free amino acid contents of 426 oysters as phenotypes for GWAS analysis and identified a total of 787 significant SNPs (P < 10− 6) [17]. These studies indicate that the GWAS method can be used to study the genetic basis of amino acid content differences in meat.
Duck meat is a high-quality poultry meat widely consumed in a variety of livestock and poultry products, and its rich nutritional value is vital to consumers [18]. Moreover, China is the country with the largest production of duck meat in the world, and the breeding quantity is increasing at an annual rate of 10-15% [19]. Meanwhile, scientists are becoming more concerned about improving the quality of duck meat nowadays. Hence, this study used the GWAS method to locate some QTL regions and candidate genes that can affect amino acid content in duck breast muscle. These findings have important significance for optimizing duck meat production and quality improvement and provide useful references for genetic improvement of livestock and poultry breeding.
Results
Descriptive statistics of phenotypic traits
This study used a fully automated amino acid analyzer to determine meat’s amino acid contents, with an amino acid standard of 17 amino acids. Sixteen amino acids were finally detected, including 11 essential amino acids (EAA) and 5 non-essential amino acids (NEAA) (Table 1). However, high concentrations of hydrochloric acid destroyed (Cys)2 during pre-treatment, so it was not detected. In addition, among the 16 amino acids detected, Glu accounted for the highest proportion of total amino acids, and Met accounted for the lowest (Figure S1a). Moreover, among the 11 essential amino acids, the content of Lys and Arg accounted for a relatively high proportion of the total essential amino acids. (Figure S1b) For non-essential amino acids, Glu and Tyr accounted for a relatively high proportion of total non-essential amino acids (Figure S1c).
Moreover, by comparative analysis, the contents of 3 amino acids showed an apparent difference between the genders (P < 0.05), including Ser, Met, and Phe (Table S1). Furthermore, the normal distribution test shows that the 16 amino acid content values were normally distributed (Fig. 1). In addition, we performed principal component analysis (PCA) on the content of 16 amino acids in all individuals. The results showed that amino acid content did not show significant population stratification (Figure S2).In addition, through correlation analysis, except for Arg and Pro, which had low correlation with other amino acids, the correlation among the other 14 amino acids was relatively high, with Pearson correlation coefficients ranging from 0.33 to 0.96 (Fig. 2). Among them, Val, Gly, Ala, Glu, and Tyr were highly correlated, and their Pearson correlation coefficients ranged from 0.50 to 0.89.

Distribution of frequency of 16 amino acids in duck breast muscle. Asp, Asparagine; Thr, Threonine; Ser, Serine; Glu, Glutamate; Gly, Glycine; Ala, Alanine; Val, Valine; Met, Methionine; Ile, Isoleucine; Leu, Leucine; Tyr, Tyrosine; Phe, Phenylalanine; Lys, Lysine; His, Histidine; Arg, Arginine; Pro, Proline. The same is below

The correlation coefficient matrix heat map of 16 amino acids. Pearson’s correlation coefficients and p-values were calculated using the cor. Test function in the R stats package. *p < 0.05 was considered statistically significant; **p < 0.01 was considered very significant; and ***p < 0.001 was considered highly significant
Genome-wide association analysis
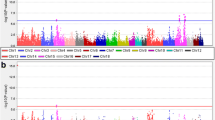
In this study, GWAS analysis was conducted using the absolute and relative contents of 16 amino acids in the breast muscles of 120-day-old ducks as phenotypes (Correction threshold = 8.59). The absolute content represents the actual value of the amino acid we measured, and the relative content is equal to the absolute content divided by the total amino acid content. Their meanings are different. At the same time, relative content can be used as a phenotype to verify the GWAS results of absolute content. Significant SNP signals were observed for Pro, Tyr, Val, and Glu in the Manhattan plot when using absolute content as the phenotype. Additionally, using the relative content as the phenotype, the GWAS results showed significant SNP signals for Tyr, Pro, Ala, Val, and Gly. Furthermore, the corresponding Q-Q plots demonstrated that the analysis model used was reasonable, with most p-values consistent with expected values and significant SNPs found, demonstrating the reliability of the above association results. Manhattan plots and Q-Q plots for other traits that showed no significant signals can be found in Figure S3 and S4, respectively.
GWAS based on absolute content
The absolute content of amino acids was used as a phenotype for GWAS analysis, and several characters with significant signals were obtained. Among them, the Manhattan map of Pro has significant SNP signals on chromosomes 3 and 11 (Fig. 3a). A total of 9 SNPs reached the significant threshold level, including three on chromosome 3 and 6 on chromosome 11. Through functional gene annotation of SNPs, SERAC1, NPTN, ZNF592, and SH3GL3 were screened (Table 2). GWAS results from Val showed only two SNPs that reached the significant threshold level (Fig. 3b), and these SNP-annotated genes include TTC7A and LOC110351546 (Table 2). The Manhattan result of Tyr showed a significant SNP signal on chromosome 2 (Fig. 3c), which was distributed on CDH18 (Table 2). The results of Glu showed a significant SNP signal on chromosome 1 (Fig. 3d), within the region of the LOC110351546 gene (Table 2).

Manhattan plots of genome-wide association analysis results. The absolute contents of amino acids as phenotypes were analyzed by GWAS to obtain the Manhattan map. Pro = Proline; Val = Valine; Tyr = Tyrosine; Glu = Glutamate. The x-axis shows the physical positions of each marker along the chromosomes, and the y-axis shows the − Log10P values for the association tests. The dashed line represents the threshold line (Correction threshold = 8.59)
GWAS based on relative content
Similarly, GWAS analysis using amino acid relative content as phenotype also obtained some characters with significant signals. The Manhattan result of Tyr showed 7 significant SNPs (Fig. 4a), of which four were located on chromosome 1 and three on chromosome 13. The functional gene annotations of SNPS are shown in Table 3, including SYN3, BICD1, MYO16, and CACNA1D and CACNA2D3. The Manhattan result of Ala showed significant SNP signals on chromosome 5 (Fig. 4b). The SYT16 and TNNI2 genes were obtained by SNP annotation (Table 3). The Manhattan map of Pro showed only three significant SNP signals (Fig. 4c), and their SNP-annotated genes include SLC38A1 (Table 3). In addition, Val’s GWAS results showed significant SNP signals on chromosome 3 (Fig. 4d), and PAK5 and HSF2 were screened out through SNP annotation (Table 3). The Manhattan map of Gly showed a significant SNP signal on chromosome 3 (Fig. 4e), which was annotated to acquire the INTS9 gene (Table 3).

Manhattan plots of genome-wide association analysis results. The relative content of amino acids was used as the phenotype for GWAS analysis to obtain the Manhattan map. Tyr = Tyrosine; Ala = Alanine; Pro = Proline; Val = Valine; Gly = Glycine. The x-axis shows the physical positions of each marker along the chromosomes, and the y-axis shows the − Log10P values for the association tests. The dashed line represents the threshold line (Correction threshold = 8.59)
LD analysis of significant candidate regions
According to the GWAS results, Pro had significant candidate regions on chromosomes 3 and 11(Fig. 3a). In this region (55.35-55.54Mbp), the correlation between leader SNP (chr3:55526954) and the surrounding SNPs was obtained through LD analysis, and 3 SNPs were highly correlated (r2 > 0.6; Fig. 5a). Finally, we screened the SERAC1 gene (Table S2). Similarly, Tyr has significant SNPs on chromosome 13 (Fig. 4a). Through LD analysis, the correlation between leader SNP (chr13:21421661) and other SNPs was calculated in this region (21.2-21.6Mbp), of which 3 SNPs were highly correlated (r2 > 0.6; Fig. 5b). Finally, the CACNA2D3 gene was screened by gene annotation (Table S3).

LD analysis in the region with significant SNPs. (A) represents the LD analysis and related genes in the c significant SNPs region (the Manhattan map showed a significant signal after GWAS phenotypic with the absolute content of Pro). (B) represents LD analysis and related genes in the significant SNPs region of Tyr (Manhattan map showed significant signal after GWAS with the relative content of Tyr as the phenotype)
Functional enrichment analysis of candidate genes located in SNPs
To further investigate the functional and regulatory relationships of these significant SNP markers and their various candidate genes, we used GO and KEGG databases for functional enrichment analysis of 134 genes, including all the annotated genes of significant SNPs and the significant locus near the high-LD region (r2 > 0.2) of annotated genes (Table S4). GO enrichment results showed amino acid transmembrane transport as a significant GO term in the biological process. Similarly, glutamine transport was also significant. There are 3 GO subcategories in the molecular function, two associated with amino acid transport and transport function, including amino acid transmembrane transporter activity and L-glutamine transmembrane transporter activity. Among them, the important candidate genes associated with amino acid transport function include SLC38A1 and SLC38A2 genes (Fig. 6, Table S5). For KEGG enrichment analysis, only one significant pathway (Cardiac muscle contraction pathway) was enriched (Table S6). Genes enriched in this pathway include CACNA2D3, CACNA1D and CACNA2D4.

GO enrichment analysis for 134 candidate genes. The x-axis indicates the number of genes for each GO term; the y-axis corresponds to the GO terms. The color of the bar represents the P value. The red boxes represent the GO terms associated with amino acids
Discussion
Amino acids are the raw materials of protein in poultry meat. At the same time, the amino acid content is an important index of meat quality [20]. This study detected the content of 16 amino acids in the breast muscles of 358 ducks. The content of Glu (5.61 g/100 g) and Asp (4.00 g/100 g) is relatively high, and the content of Met (1.23 g/100 g) is relatively low. Studies have shown that Glu and Asp have relatively high amino acid content in duck meat [21, 22], which is consistent with our results. However, the content values of some amino acids obtained in this study differ greatly from the results of other studies. Amino acids such as glutamate and asparagine acid content values were 2 times higher than those of other studies [23]. This may be related to many factors, among which we think it is closely associated with the age factor, because our slaughter age is 120 days old, and other studies slaughter time and our slaughter time gap is larger. At the same time, Studies have shown that within a certain range, the older the poultry animals are, the higher the amino acid content will be [24]. Of course, it is closely associated with the breed, sex, and nutritional factors. In addition, compared with other livestock and poultry meat, the contents of various amino acids are also different [25]. Zagorska et al. showed that the content of Glu, Lys, Asp, and Leu in pork was relatively high, and its content was between 6% and 13% of total amino acids (dry weight) [26]. This is quite different from our results. This may be due to the differences in the amino acid content of duck meat and other poultry meat due to genetic background, nutrition, and feeding methods [27].
Moreover, we also compared and analyzed the differences in these amino acids between males and females and found that only 3 amino acids had significant differences between males and females (P < 0.05), including Ser, Met, and Phe. In previous studies, Yin et al. showed that the 11 amino acids of Guangyuan grey chickens showed significant differences between genders [28]. Ji et al. also compared the amino acid content in different parts of Duroc pigs and found significant differences in various amino acids between genders [29]. Although the finding is inconsistent with our results, it is also a normal phenomenon due to the difference in breed, nutrition, and other factors.
Phenotypic correlation analysis showed that most amino acids had a high correlation, such as Lys and Leu, Tyr and Phe. In the study of amino acid metabolism, the results show that the metabolism of most amino acids is interrelated [30]. For example, Tyr can be synthesized from Phe, and the synthesis of Pro requires the participation of Glu [31,32,33]. However, the specific influencing mechanism needs further systematic study.
Based on GWAS analysis of different phenotypic values, significant SNPs were found in Pro, Tyr, and other traits. We found candidate genes affecting Pro content on chromosomes 3 and 11 through gene annotation, including SLC38A1, SERAC1, ZNF592, SH3GL3, and NPTN. According to reports, the SLC38A1 belongs to the SLC38A family. It is involved in the transport of Ala and Gln in mammals and participates in the TCA cycle [34, 35]. On the other hand, studies have reported that Gln is involved in Pro metabolism [36]. Therefore, we speculate that the SLC38A1 gene is closely associated with the metabolic process of Pro. In addition, studies have shown that Pro’s metabolic process occurs in mitochondria [37]. Notably, SERAC1 plays a key role in phosphatidylglycerol remodeling, which is essential for both mitochondrial function and intracellular cholesterol transport [38]. However, whether this is relevant for pro-synthesis in mitochondria needs further study. Similarly, we screened candidate genes that affect Tyr content, including CACNA2D3, CDH18, SYN3, BICD1, MYO16, and CACNA1D. Interestingly, Yang et al. presented candidate genes affecting Australian Boer goat muscle development through GWAS analysis, including the CACNA2D3 gene [39]. As an important amino acid constituting muscle protein [40], it is speculated that this may be indirectly associated with the metabolic process of tyrosine. However, whether there is a link between the two needs to be further studied.
Through GO and KEGG analyses, Significant GO items of candidate gene enrichment were associated with amino acid transmembrane transport activity. Interestingly, both SLC38A1 and SLC38A2 genes are included in the GO terms associated with amino acid transport. Therefore, we speculate that these candidate genes may be important in material transport during amino acid production. For KEGG analysis, only one significant pathway was enriched in this study, which included CACNA2D3 and CACNA1D. However, whether this pathway is involved in amino acid regulation needs further study.
Amino acids have an important effect on meat quality. However, the genetic mapping of amino acid-related genes in poultry has not been reported. Therefore, the candidate genes affecting amino acid content in duck breast muscle were identified by GWAS for the first time in this study. Due to the extremely complex amino acid gene regulation and metabolism process, small sample size, and differences in detection methods, the substantive evidence we finally obtained is still limited. However, these candidate genes and candidate regions can provide a useful reference value for breeding breeds with better meat quality by genetic means.
Materials and methods
Animals and sampling
A total of 358 ducks (Tianfu Nonghua duck) were provided by the Waterfowl Breeding Farm of Sichuan Agricultural University. All ducks were raised under the same conditions and had free access to food and water. At 60 days old, 5 mL of whole blood samples were collected using venipuncture and stored at -20 ℃. At 120 days old, breast muscles were collected during slaughter and stored in a -4℃ refrigerator. The raising of animals, and sampling processes were achieved through the guidelines of the Sichuan Agricultural University institution and met the feeding standards of the Animal Welfare Committee.
Determination of amino acid content
Amino acid content was detected in the breast muscles of 358 ducks at 120 days of age. The detection process was divided into pre-processing and on-machine operation.
The duck breast muscle was pre-treated by acid hydrolysis. First, an accurate weight of 0.5 g duck breast muscle sample collected from the same location was carefully added into the hydrolytic tube. Addition of 6 mol/L HCL10 mL containing phenol was made, sealed, and baked in an oven at 110℃ for 24 h. Secondly, the volume was set to 100 mL after the sample filtration with ultra-pure water. Third, ultrafiltration was carried out using a C18 extractor. Fourthly, after ultrafiltration, the sample solution was absorbed into a 1.5mL EP tube and deacidified in a vacuum deacidifier (about 2–3 h). Finally, 2mL of sample buffer was added to the deacidified sample and mixed evenly. Then, a small amount of liquid was absorbed by a needle tube and filtered through a 0.25 μm filter into a small brown bottle ready for machine analysis.
In addition, 17 amino acid standards were used (Sigma, AAS18). After pretreatment, amino acids were determined by an automatic amino acid analyzer (A300, membraPure GmbH, Germany). For specific operation methods and processes refer to the manual (Version 1.3 of the A300). The peak of amino acid content is shown in Figure S5.
DNA extraction and whole genome re-sequencing
DNA was extracted from the duck’s blood (n = 358) using the phenol-chloroform protocol. DNA quality was detected by Nano Drop-2000 and agarose gel electrophoresis. The evaluated samples were constructed by a paired-end library. A small fragment library with a fragment length of 150 base pairs (bp) (PE150) was constructed, and a successful library was constructed. All libraries were sequenced on the Illumina®Hiseq X-Ten platform of Bio-company (Biomarker Technologies, Beijing, China). Each sample’s mean sequence coverage of the raw reads sequence was 5×.
Quality control analysis of raw reads was performed using Trimmomatic (v0.36) after removing reads containing joint sequences. The clean reads obtained were then compared with the duck reference genome (ZJU1.0, GCA_015476344.1) using Burrows-Wheeler Aligner (BWA aln) [41]. Meanwhile, a total of 12,996,047,606 clean reads were generated after quality filtering. HaplotypeCaller in GATK was used to identify and analyze SNPs and InDels, and VCFtools was used to filter SNP data further [42, 43]. Finally, a total of 19,628,941 SNPs were obtained by VCFtools for GWAS analysis. The specific methods and statistical results have been sorted out by our research group in the early stage [44].
Genome-wide association analysis (GWAS)
GWAS uses the mixed linear model program Emmax to identify SNPS [45]. To correct for population stratification, the model uses fixed effects that include the first three principal component values (PCA eigenvectors) from the genome-wide SNP genotypes [46]. Meanwhile, random effects are present in the kinship estimation matrix of all individual genome-wide SNP genotypes. In addition, the effect of sex was further analyzed as a fixed effect in GWAS. The R package (v3.5.1) was used to draw Manhattan and QQ plots. Finally, the Correction threshold (− Log10P ≥ 8.59) was used to identify significant SNPs, and the calculation formula is P = 0.05/The total number of SNPs. Meanwhile, the QQ plots are used to detect if SNP are false positives due to population stratification.
LD analysis
After GWAS analysis, we screened some important candidate regions and SNPs. To explore the relationship between the most significant SNPs (the leader SNPs) and other SNPs in the significant candidate region, this study used Plink (version 1.90) software for LD analysis, and the Locuszoom graph was generated by R (version 3.5.1).
Gene annotation and enrichment analysis
Based on the duck reference genome (ZJU1.0, GCA_015476345.1), SnpEff software annotated significant SNPs to obtain candidate genes for different traits [47]. David website (https://david.ncifcrf.gov/) was used for Gene Ontology (GO) function enrichment, and KOBAS 3.0 (http://kobas.cbi.pku.edu.cn/) was used for the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. In the enrichment analysis, a significant level of P < 0.05 was the standard for significant enrichment of GO terms and KEGG pathways.
Statistical analysis
Microsoft Excel 2021 software was used to analyze each trait’s mean value, standard deviation, and coefficient of variation. SPSS software (version 22.0, Windows, SPSS Inc., Chicago, IL) was used for the normal distribution test. The data that did not conform to the normal distribution was processed by logarithm, and Rdrew the correlation analysis and correlation heat map between various traits. The graphs were drawn with GraphPad Prism (version 8.0.2) and R Studio (version 4.1.1).
Data availability
The genome re-sequencing raw data was available in NCBI’s SRA database (https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=studies&f=study&term= &go = Go; Accession number: PRJNA907492 and PRJNA907501).
Abbreviations
- GWAS:
-
Genome-wide association studies
- QTLs:
-
Quantitative trait loci
- LD:
-
Linkage disequilibrium
- SNP:
-
Single nucleotide polymorphism
- PCA:
-
Principal component analysis
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GO:
-
Gene ontology
- Asp:
-
Asparagine
- Thr:
-
Threonine
- Ser:
-
Serine
- Glu:
-
Glutamate
- Gly:
-
Glycine
- Ala:
-
Alanine
- Val:
-
Valine
- Met:
-
Methionine
- Ile:
-
Isoleucine
- Leu:
-
Leucine
- Tyr:
-
Tyrosine
- Phe:
-
Phenylalanine
- Lys:
-
Lysine
- His:
-
Histidine
- Arg:
-
Arginine
- Pro:
-
Proline
- EAAs:
-
essential amino acids
- NEAAs:
-
non-essential amino acids
- TAAs:
-
total amino acids
References
Jing Z, Wang X, Cheng Y, Wei C, Hou D, Li T, Li W, Han R, Li H, Sun G, et al. Detection of CNV in the SH3RF2 gene and its effects on growth and carcass traits in chickens. BMC Genet. 2020;21(1):22.
Long Y, Han Y, Zhao Y, Chen D, Wang D, Yang Y, Su C, Shen X. Effect of Mulberry Leaf TMR on Growth Performance, Meat Quality and Expression of Meat Quality Master Genes (ADSL, H-FABP) in Crossbred Black Goats. Foods (Basel, Switzerland) 2022, 11(24).
Deng S, Xing T, Li C, Xu X, Zhou G. The Effect of Breed and Age on the growth performance, carcass traits and metabolic Profile in breast muscle of Chinese indigenous chickens. Foods (Basel Switzerland) 2022, 11(3).
Zhang J, Zhang Y, Wang J, Jin H, Qian S, Chen P, Wang M, Chen N, Ding L. Comparison of antioxidant capacity and muscle amino acid and fatty acid composition of nervous and calm Hu Sheep. Antioxid (Basel Switzerland) 2023, 12(2).
Algammal AM, Elsayed ME, Hashem HR, Ramadan H, Sheraba NS, El-Diasty EM, Abbas SM, Hetta HF. Molecular and HPLC-based approaches for detection of aflatoxin B(1) and ochratoxin a released from toxigenic aspergillus species in processed meat. BMC Microbiol. 2021;21(1):82.
Farmer LJ, Mottram DS, Whitfield, FBJJotSoF. Agriculture: volatile compounds produced in Maillard reactions involving cysteine. Ribose Phospholipid. 1989;49(3):347–68.
Yim DG, Choi YS, Nam KC. Sea tangle (Laminaria Japonica) supplementation on meat quality of Korean native black goat. J Anim Sci Technol. 2019;61(6):352–8.
Muroya S, Ueda S, Komatsu T, Miyakawa T, Ertbjerg P. MEATabolomics: muscle and meat metabolomics in domestic animals. Metabolites 2020, 10(5).
Ou Z, Shi Y, Li Q, Wu Y, Chen F. Effects of Sex on the Muscle Development and Meat Composition in Wuliangshan Black-Bone Chickens. Animals: an open access journal from MDPI 2022, 12(19).
Goto T, Ohya K, Takaya M. Genotype affects free amino acids of egg yolk and albumen in Japanese indigenous breeds and commercial Brown layer chickens. Poult Sci. 2022;101(2):101582.
Zhang R, Pavan E, Ross AB, Deb-Choudhury S, Dixit Y, Mungure TE, Realini CE, Cao M, Farouk MM. Molecular insights into quality and authentication of sheep meat from proteomics and metabolomics. J Proteom. 2023;276:104836.
Pérez-Enciso M, Clop A, Noguera J, Ovilo C, Coll A, Folch J, Babot D, Estany J, Oliver M. Diaz IJJoAS: a QTL on pig chromosome 4 affects fatty acid metabolism: evidence from an Iberian by Landrace Intercross. 2000, 78(10):2525–31.
Tshipuliso N, Alexander L, Geary T, Snelling W, Rule D, Koltes J, Mote B. MacNeil MJSAJoAS: mapping QTL for fatty acid composition that segregates between the Japanese Black and Limousin cattle breeds. 2008, 38(2):126–30.
Gutierrez-Gil B, Wiener P, Richardson R, Wood J, Williams, JJMs. Identification of QTL with effects on fatty acid composition of meat in a Charolais× holstein cross population. 2010, 85(4):721–9.
van Son M, Enger EG, Grove H, Ros-Freixedes R, Kent MP, Lien S. Grindflek EJBg: genome-wide association study confirm major QTL for backfat fatty acid composition on SSC14 in Duroc pigs. 2017, 18(1):1–13.
Sasago N, Takeda M, Ohtake T, Abe T, Sakuma H, Kojima T, Sasaki S, Uemoto Y. Genome-wide association studies identified variants for taurine concentration in Japanese black beef. Anim Sci J. 2018;89(8):1051–9.
Meng J, Song K, Li C, Liu S, Shi R, Li B, Wang T, Li A, Que H, Li L, et al. Genome-wide association analysis of nutrient traits in the oyster Crassostrea gigas: genetic effect and interaction network. BMC Genomics. 2019;20(1):625.
Bai H, Bao Q, Zhang Y, Song Q, Liu B, Zhong L, Zhang X, Wang Z, Jiang Y, Xu Q, et al. Research note: effects of the rearing method and stocking density on carcass traits and proximate composition of meat in small-sized meat ducks. Poult Sci. 2020;99(4):2011–6.
Xiu L, Liu Y, Wu W, Chen S, Zhong Z, Wang H. Prevalence and multilocus sequence typing of Clostridium perfringens isolated from 4 duck farms in Shandong province, China. Poult Sci. 2020;99(10):5105–17.
Luo Q, Li J, Li H, Zhou D, Wang X, Tian Y, Qin J, Tian X, Lu Q. The Effects of Purple Corn Pigment on Growth Performance, Blood Biochemical Indices, Meat Quality, Muscle Amino Acids, and Fatty Acids of Growing Chickens. Foods (Basel, Switzerland) 2022, 11(13).
Cao Z, Gao W, Zhang Y, Huo W, Weng K, Zhang Y, Li B, Chen G, Xu Q. Effect of marketable age on proximate composition and nutritional profile of breast meat from Cherry Valley broiler ducks. Poult Sci. 2021;100(11):101425.
Gumułka M, Połtowicz K. Comparison of carcass traits and meat quality of intensively reared geese from a Polish genetic resource flock to those of commercial hybrids. Poult Sci. 2020;99(2):839–47.
Zhang X, Deng Y, Hu S, Hu X, Ma J, Hu J, Hu B, He H, Li L, Liu HJPS. Comparative analysis of amino acid content and protein synthesis-related genes expression levels in breast muscle among different duck breeds/strains. 2023, 102(1):102277.
Beski SSM, Swick RA, Iji PA. Specialized protein products in broiler chicken nutrition: a review. Anim Nutr (Zhongguo Xu Mu Shou Yi Xue Hui). 2015;1(2):47–53.
Maikanov BS, Ismagulova GT, Auteleyeva LT, Kemeshov ZO, Zhanabayeva DK. Assessment of quality and safety of meats from various animal species in the Shuchinsk-Burabay resort zone, Kazakhstan. Veterinary World. 2021;14(6):1615–21.
Zagorska J, Degola L, Strazdins I, Gramatina I, Kince T, Galoburda R. Effects of Lactobionic Acid on Pig Growth Performance and Chemical composition of pork. Animals: Open Access J MDPI 2022, 12(9).
Juárez M, Lam S, Bohrer BM, Dugan MER, Vahmani P, Aalhus J, Juárez A, López-Campos O, Prieto N, Segura J. Enhancing the Nutritional Value of Red Meat through genetic and feeding strategies. Foods (Basel Switzerland) 2021, 10(4).
Yin L, Xu M, Huang Q, Zhang D, Lin Z, Wang Y, Liu Y. Nutrition and Flavor Evaluation of Amino Acids in Guangyuan Grey Chicken of Different Ages, Genders and Meat Cuts. Animals: an open access journal from MDPI 2023, 13(7).
Xia JQ, Liu DY, Liu J, Jiang XP, Wang L, Yang S, Liu D. Sex effects on carcass characteristics, meat quality traits and meat amino acid and fatty acid compositions in a novel Duroc line pig. J Anim Physiol Anim Nutr. 2023;107(1):129–35.
Cui C, Zhu L, Wang Q, Liu R, Xie D, Guo Y, Yu D, Wang C, Chen D, Jiang P. A GC-MS-based untargeted metabolomics approach for comprehensive metabolic profiling of Vancomycin-induced toxicity in mice. Heliyon. 2022;8(7):e09869.
Wurihan A, Bilige, Lili, Sirguleng, Aduqinfu, Bai M. Metabonomics study of liver and kidney subacute toxicity induced by garidi-5 in rats. Chin Herb Med. 2022;14(3):422–31.
Yang HC, Yu H, Liu YC, Chen TL, Stern A, Lo SJ, Chiu DT. IDH-1 deficiency induces growth defects and metabolic alterations in GSPD-1-deficient Caenorhabditis elegans. J Mol Med. 2019;97(3):385–96.
Lu M, Sanderson SM, Zessin A, Ashcraft KA, Jones LW, Dewhirst MW, Locasale JW, Hsu DS. Exercise inhibits tumor growth and central carbon metabolism in patient-derived xenograft models of colorectal cancer. Cancer Metabolism. 2018;6:14.
Kandasamy P, Gyimesi G, Kanai Y, Hediger MAJT. Amino acid transporters revisited: New views in health and disease. 2018, 43(10):752–789.
Niknafs S, Fortes MR, Cho S, Black JL, Roura, EJBg. Alanine-specific appetite in slow growing chickens is associated with impaired glucose transport and TCA cycle. 2022, 23(1):393.
Nagasawa T, Ebisu K, Inoue Y, Miyano K, Tamura M. A new role of Pro-73 of p47phox in the activation of neutrophil NADPH oxidase. Arch Biochem Biophys. 2003;416(1):92–100.
Wu G, Bazer FW, Burghardt RC, Johnson GA, Kim SW, Knabe DA, Li P, Li X, McKnight JR, Satterfield MC, et al. Proline and hydroxyproline metabolism: implications for animal and human nutrition. Amino Acids. 2011;40(4):1053–63.
Wortmann SB, Vaz FM, Gardeitchik T, Vissers LE, Renkema GH, Schuurs-Hoeijmakers JH, Kulik W, Lammens M, Christin C. Kluijtmans LAJNg: mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. 2012, 44(7):797–802.
Yang BG, Yuan Y, Zhou DK, Ma YH, Mahrous KF, Wang SZ, He YM, Duan XH, Zhang WY. Genome-wide selection signal analysis of Australian Boer goat reveals artificial selection imprinting on candidate genes related to muscle development. Anim Genet. 2021;52(4):550–5.
Ouattara DA, Remolue L, Becker J, Perret M, Bunescu A, Hennig K, Biliaut E, Badin A, Giacomini C, Reynier F, et al. An integrated transcriptomics and metabolomics study of the immune response of newly hatched chicks to the cytosine-phosphate-guanine oligonucleotide stimulation. Poult Sci. 2020;99(9):4360–72.
Li H, Durbin RJ. Fast and accurate short read alignment with Burrows–Wheeler transform. 2009, 25(14):1754–60.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Del Angel G, Rivas MA. Hanna MJNg: a framework for variation discovery and genotyping using next-generation DNA sequencing data. 2011, 43(5):491–8.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry STJB. The variant call format and VCFtools. 2011, 27(15):2156–8.
Ma S, Li P, Liu H, Xi Y, Xu Q, Qi J, Wang J, Li L, Wang J, Hu J, et al. Genome-wide association analysis of the primary feather growth traits of duck: identification of potential loci for growth regulation. Poult Sci. 2023;102(1):102243.
Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences 1998, 95(25):14863–14868.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA. Reich DJNg: principal components analysis corrects for stratification in genome-wide association studies. 2006, 38(8):904–9.
Pablo C, Adrian P, Lily WL, Melissa C, Tung N, Luan W, Xiangyi JLS, L, M RD.: A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. %J Fly. 2012, 6(2).
Du Sert NP, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U. Emerson MJPb: reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020;18(7):e3000411.
Acknowledgements
Thanks to all the authors for their contributions to the study.
Funding
This work was supported by grants from the National Key R&D Program of China (2022YFF1000000), the Key Technology Support Program of Sichuan Province (2021JDJQ0008).
Author information
Authors and Affiliations
Contributions
RW and HL were responsible for the experiment design, experimental performance, and manuscript drafting. In addition, YL, YX, JQ, ZS, GT performed the data analysis of the whole genome sequencing. JH, and LB participated in the sample collection and preparation work. JW, LL and HL participated in the writing instruction and revision of the manuscript. All listed authors have made substantial contributions to the research and publication. Thank you to all the authors for their contributions to the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All methods were carried out following relevant guidelines and regulations. The Institutional Animal Care and Use Committee (IACUC) guidelines of Sichuan Agricultural University approved all the experiments and protocols. All methods were carried out in accordance with relevant guidelines and regulations. The protocols for all animal experiments were approved by the Animal Welfare Committee of Sichuan Agricultural University, and all methods strictly obeyed the Guide for the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines 2.0 [48].
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, R., Lu, Y., Qi, J. et al. Genome-wide association analysis explores the genetic loci of amino acid content in duck’s breast muscle. BMC Genomics 25, 486 (2024). https://doi.org/10.1186/s12864-024-10287-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10287-1