
Abstract
Long-read sequencing is revolutionizing de-novo genome assemblies, with continued advancements making it more readily available for previously understudied, non-model organisms. Stony corals are one such example, with long-read de-novo genome assemblies now starting to be publicly available, opening the door for a wide array of ‘omics-based research. Here we present a new de-novo genome assembly for the endangered Caribbean star coral, Orbicella faveolata, using PacBio circular consensus reads. Our genome assembly improved the contiguity (51 versus 1,933 contigs) and complete and single copy BUSCO orthologs (93.6% versus 85.3%, database metazoa_odb10), compared to the currently available reference genome generated using short-read methodologies. Our new de-novo assembled genome also showed comparable quality metrics to other coral long-read genomes. Telomeric repeat analysis identified putative chromosomes in our scaffolded assembly, with these repeats at either one, or both ends, of scaffolded contigs. We identified 32,172 protein coding genes in our assembly through use of long-read RNA sequencing (ISO-seq) of additional O. faveolata fragments exposed to a range of abiotic and biotic treatments, and publicly available short-read RNA-seq data. With anthropogenic influences heavily affecting O. faveolata, as well as its increasing incorporation into reef restoration activities, this updated genome resource can be used for population genomics and other ‘omics analyses to aid in the conservation of this species.
Similar content being viewed by others

Background
Advances in sequencing technologies are providing new opportunities in genome assembly and research, specifically, long-read sequencing methodologies such as PacBio and Oxford Nanopore. Longer stretches of DNA can reduce the number of contigs and improve the classification of highly repetitive regions such as telomeric and centromeric repeats [1,2,3,4] which is commonly a problem and more difficult with short-read sequencing. In addition, reductions in the cost have made long-read sequencing methodologies highly accessible and attainable for use in non-model organisms, facilitating new research into inter/intra-population variation, as well as investigations into areas such as gene function and gene coding sequences.
Stony corals (order Scleractinia) are keystone organisms, providing the framework for subtropical and tropical coral reef ecosystems. At present, the 10 long-read coral genomes that are publicly available all represent those of Pacific coral species [5,6,7,8,9,10], with no long-read genomic resources available for Caribbean corals. Orbicella faveolata (Fig. 1A) is an important reef-building coral in the family Merulinidae in the Caribbean. While historically inhabiting back and fore reefs at a range of depths throughout the Caribbean [11, 12], it is now listed as “threatened” under the US Endangered Species Act [13] and “endangered” on the IUCN red list [14]. Despite ongoing protection efforts, populations of O. faveolata have continued to decrease in the Caribbean due to bleaching [15, 16] and disease events, specifically stony coral tissue loss disease [17, 18]. As a result, Caribbean reef restoration activities are currently incorporating O. faveolata [19,20,21]. A highly contiguous and complete reference genome would be an invaluable resource to support restoration efforts through use in ‘omics analysis such as population genetic studies [16, 22] and transcriptomics [23,24,25]. With local and global anthropogenic influences having drastic effects on coral assemblages and populations [26,27,28,29], a well annotated and contiguous genome can also lay the foundations for studies aiming to identify resistance biomarkers within and between populations [30] to variables such as heat stress, disease, and ocean acidification.

Orbicella faveolata on a reefscape and gamete bundle collection methodology. A. Picture of an adult Orbicella faveolata colony at Horseshoe reef during the day. B. Gamete bundle collection methodology apparatus. Picture shows the top of the net which is placed over the adult coral colony, with an attached 50 ml conical centrifuge tube allowing collection of gamete bundles
Previously, an O. faveolata genome was assembled using short-read technology (NCBI accession GCA_002042975.1) [31]. Here, we used long-read PacBio circular consensus sequencing (CCS) of high molecular weight (HMW) DNA extracted from O. faveolata sperm collected in the field (Fig. 1B) to assemble a more contiguous and complete de-novo genome assembly of O. faveolata. We achieved a highly contiguous and complete de-novo genome assembly, with long-read RNA-seq (ISO-seq) resulting in better gene prediction. We then discuss how to further improve our resource using approaches such as optical mapping or Hi-C sequencing, and provide applications for the implementation of this genetic resource into ongoing conservation and restoration initiatives.
Results
De-novo genome assembly
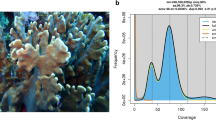
High molecular weight (HMW) DNA extracted from Orbicella faveolata sperm yielded 2,604,886 HiFi reads (average length = 12,688 base paris (bp), total length = 32,999,915,949 bp). A BLASTn [32] search of the raw HiFi reads identified 54,828 reads which were considered as contaminants (prokaryotic, viral, and UniVec databases) with a bit score > 1000 (2.1% of the raw HiFi reads). After removal of these sequences, 2,550,058 contaminant free (CF) HiFi reads (average length = 12,671 bp, total length = 32,313,799,715 bp) remained and were used for de-novo genome assembly. The CF HiFi reads had an estimated sequencing coverage of 99x, a predicted genome length of 469,984,355 bp, ploidy of two, homozygosity of 98.7%, heterozygosity of 1.26%, and duplication of 0.552 (Fig. 2A).

GenomeScope2 and Merqury analyses of the cleaned raw HiFi reads, primary, and alternative assemblies using HiFiasm. A. GenomeScope2 linear k-mer distributions for O. faveolata generated from Merqury output. Black line shows a theoretical diploid model for this species, with computed homozygosity (aa) and heterozygosity (ab) of 98.7% and 1.2% respectively. B. Merqury plot of the primary and alternative assemblies using HiFiASM in --primary mode. Default parameters showed duplicates were purged adequately (red and blue lines) resulting in no additional filtering before scaffolding, downstream gene prediction, and annotation steps
CF HiFi reads were assembled into primary and alternate pseudo-haplotype assemblies with HiFiasm [33]. Kmer profile analysis of primary and alternate assemblies with Merqury [34] confirmed successful duplicate purging (Fig. 2B). The primary assembly consisted of 62 contigs, with a largest contig of 40,246,328 bp, N50 of 33,295,526 bp, L50 of 7, and GC content of 39.49% as identified by Quast [35, 36]. BUSCO [37] analysis of the primary assembly identified 94.86% of metazoan single copy orthologs to be complete (single copy: 93.61%, duplicated: 1.26%), fragmented to be 2.31%, and missing 2.83%. For Quast [35, 36] and BUSCO [37] results of the alternate pseudo-haplotype, please see Supplementary File 1. Scaffolding of the primary assembly with ntLink [38] yielded 51 scaffolds and unchanged quality metrics from the primary assembly (Fig. 3A). BUSCO [37] results were also unchanged as a result of scaffolding (Fig. 3B). Hard and soft masking of scaffolds resulted in 50.20% (247,928,041 bp) of bases masked (Fig. 4). For a full breakdown of masking results, please see Supplementary File 2.

Quast and BUSCO analysis results of long-read stony coral genomes. A. Results from Quast analysis of our de-novo assembly, previous short read Orbicella faveolata assembly, and all publicly available long-read stony coral genomes. B. Results of BUSCO analysis using our O. faveolata de-novo assembly, the previous short read O. faveolata assembly, and all publicly available long-read stony coral genomes with the metazoa_odb10 database. Completeness is split into single copy (light blue) and duplicated (dark blue). Fragmented = yellow, missing = red. Percentages for each metric are present in each bar: Csc = complete and single copy, Cd = complete and duplicated, Fr = fragmented, M = missing. For both (A) and (B) “Orbicella faveolata (short-read)” is the previously assembled short-read genome, and “Orbicella faveolata (long-read)” is the de-novo assembly using PacBio HiFi reads

Visualization of scaffolded genome assembly of Orbicella faveolata. A. GC content calculated with a sliding window of 50,000 base pairs (bp). Y-axis shows the percentage calculated for GC content over each 50,000 bp sliding window. B. Repeat content plotted using a sliding window of 50,000 base pairs and the gff output file from RepeatMasker. Y-axis shows counts of repetitive regions for each sliding window of 50,000 base pairs. C. Telomeric repeats generated with a sliding window of 50,000 base pairs and the repeat pattern of “TTAGGG”. Y-axis shows the counts of the telomeric repeat for each sliding window of 50,000 base pairs. Telomeric repeats can be identified by peaks at either the start or end of each scaffold. D. Gene density calculated with a sliding window of 50,000 base pairs and the “gene” identifiers from the gff file generated from funannotate::annotate. Y-axis shows the counts of genes for each sliding window of 50,000 base pairs
Telomere analysis, using tidk (https://github.com/tolkit/telomeric-identifier), identified 19 scaffolded contigs with telomeric repeats (TeloScafs) at either one (telocentric, 12 of the 19 scaffolds), or both (metacentric, 7 of the 19 scaffolds), ends (Fig. 4). BUSCO [37] analysis of the 19 TeloScafs identified 90.2% metazoan single ortholog copies to be complete (single copy: 89.4%,duplicated: 0.8%), fragmented of 2.4%, and missing of 7.4%.
Genome annotation
There were 10 RNA-seq samples (Table 1) that were successfully pooled in equal concentration and used for ISO-seq library prep. ISO-seq (https://github.com/PacificBiosciences/IsoSeq) processing of the CCS HiFi reads resulted in 310,755 high quality (hq) transcripts with an average transcript count of 14. Prior to PASA [39] an initial cleaning, using seqClean [40], of the hq transcripts resulted in validation of 310,572 transcripts (1,926 trimmed) and the removal of 183 transcripts (by dust: 10, by short: 173). PASA [39] analysis resulted in 57,279 gene model assemblies. From this, TransDecoder (https://github.com/TransDecoder/TransDecoder) identified 56,835 coding sequences (CDs), with 53,673 open reading frames (ORFs) which could be propagated to the genome. These ORFs were used as input to Funannotate::predict [41] to train ab-initio gene predictors and generate consensus gene models.
Funannotate::predict [41] identified an initial 32,280 gene models from Evidence Modeler (EVM), which was reduced to 28,663 mRNA gene models after filtering (too short: 5, transposable elements: 3,612). tRNAscan-SE [42] identified 6,033 valid non-overlapping models, resulting in a total of 34,496 (EVM + tRNA-scan) gene models (Supplementary File 3). Refinement of gene models and untranscribed regions (UTRs), using funannotate::update [41], resulted in 1,881 new gene models with this comprising 32,172 protein coding genes, 5,762 tRNAs, and an average gene length of 5977.66 bp (Supplementary File 3). UTRs were also successfully updated for 14,722 gene models. BUSCO [37] analysis of the protein coding genes (database metazoa_odb10) identified complete orthologs of 95.1% (single copy 88.7%, duplicated 6.4%), fragmented as 2.5%, and missing as 2.4% (Supplementary Fig. 1A). For comparison of our protein coding genes to other long-read coral genomes, an Orthofinder [43] analysis was undertaken using the protein fasta files from other publicly available long-read coral genomes [5,6,7,8,9,10]. Ortholog analysis of the protein coding genes, using Orthofinder [43], identified 29,917 (93%) of genes present in orthogroups, 2,255 (7%) genes not assigned orthogroups, 18,199 (55.7%) genes shared between the coral species s, and 1,903 (5.9%) of genes only present in a single species (Supplementary Fig. 1B, and Supplementary File 4).
Comparison to previous O. faveolata reference genome and other long-read coral genomes
Comparison of BUSCO [37] and Quast [35, 36] metrics demonstrated improvement of our de-novo assembly in comparison to the current O. faveolata reference assembly (NCBI accession GCA_002042975.1) generated using short-read technology [31]. Scaffold number decreased from 1933 to 51, N50 increased from 4,771,691 bp to 40,246,328 bp, and L50 decreased from 124 to 7 (Fig. 3A). There was also an 8.3% increase of BUSCO [37] completeness (single and duplicated) from 85.3 to 93.6% (Fig. 3B). Alignment of the previous reference genome to our de-novo assembly resulted in 99.48% of contigs mapping to our new de-novo assembly.
When comparing coding genes, the short-read assembly housed 32,587, while the de-novo assembly housed 32,172 (a decrease of 415). Using the protein coding genes, there was also an increase of complete (single copy and duplicated) BUSCOs of 7.9% between the short-read assembly (87.2%) and our de-novo genome assembly (95.1%) (Supplementary Fig. 1A).
Comparison of our de-novo assembled genome to other coral long-read genomes [5,6,7,8,9,10] identified comparable BUSCO completeness (single and duplicated) of > 90% (Fig. 3B). Interestingly, Quast [35, 36] identified that our assembly showed lower scaffold count than other long-read assemblies, with the next most contiguous assembly comprising 212 scaffolds compared with the present studies 51 scaffolds (Fig. 3A). When comparing the longest contig and N50 with other publicly available genomes, our de-novo assembly was second to the Montipora capitata V3 [9] genome resource (Fig. 3A). Comparison of protein coding genes identified comparable BUSCO [37] completion of > 90% between our de-novo assembly and other long-read coral genomes (Supplementary Fig. 1A). Once taking into account the total starting number of protein coding genes (Supplementary Fig. 1B), ortholog analysis between the other available long-read coral genomes identified comparable statistics, with > 92% of genes present in all the coral species, and < 7.5% remaining unassigned (Supplementary Fig. 1C). The percentage of orthogroups between species ranged between 50 and 60%, with the percentage of genes in species-specific orthogroups ranging from 2 to 12% (Supplementary Fig. 1C).
Mitochondrial genome
MitoHiFi [44] identified a complete mitochondrial genome sequence present in the raw HiFi reads with a length of 17,083 bp, successful circulation, and the following genes: two transfer RNAs (tRNAs), 13 protein coding genes, and two ribosomal RNAs (rRNAs). Phylogenetic comparison against all available coral mito genomes on the NCBI identified our O. faveolata mitochondrial genome to be placed ‘sister’ to the previous O. faveolata mitochondrial genome [45] and other Orbicella species (Supplementary Fig. 2). This is most likely due to inherent differences between long and short read sequencing technologies [46, 47].
Discussion
Long-read sequencing provides a highly contiguous reference genome resource for Orbicella faveolata
In the present study we have demonstrated that long-read PacBio CCS sequencing dramatically improves the genome resource of Orbicella faveolata. The previous reference assembly for O. faveolata [31] utilized short-read sequencing methodologies on HiSeq 2500 and MiSeq machines, which pose computational challenges for the construction of a highly contiguous assembly [1,2,3,4]. Long-read technology, such as PacBio Sequel sequencing, can span repetitive regions of the genome, resulting in fewer contigs. This advantage is clearly demonstrated in our final assembly which consisted of 51 scaffolds, nearly 40 times fewer than the 1,932 scaffolds in the previous short-read O. faveolata reference assembly [31]. This increase in contiguity is further reflected in improved N50 (40,246,328 versus 4,771,691), L50 (7 versus 124), and BUSCO completeness (single copy and duplicated, 85.3% versus 93.6%, Fig. 3B) metrics. Despite these improvements, our new de-novo assembly identified similarities for GC content (de-novo: 39.49%, short-read: 38.5%, Fig. 3A), overall genome length (de-novo: 494,730,336 bp, short-read: 485,548,939 bp, Fig. 3A) with the short-read O. faveolata genome resource [31], as well as a ploidy of two (Fig. 2A). Comparison between protein coding genes also identified an improvement between our de-novo assembly and the previous O. faveolata reference, with this identified through an increase of BUSCO completeness (single copy and duplicated) from 87.2 to 95.1% (Supplementary Fig. 1B). These results highlight how long-read methodologies can improve upon older genomic resources that used short-read methodologies.
We also compared our assembly to other publicly available long-read stony coral genome assemblies [5,6,7,8,9,10]. Despite only using HiFi reads for our assembly and scaffolding, our assembly attains approximately equal completeness and contiguity as measured by Quast [35, 36] (Fig. 3A) and BUSCO [37] (Fig. 3B) when compared to assemblies that incorporated auxiliary scaffolding techniques [7, 9]. With continued improvement and cost reduction of long-read sequencing methodologies, the results of our study show that the generation of a high quality reference genome for stony corals can be achieved without additional methods such as Hi-C, optical mapping, or supplemental short-read sequencing. Using these additional methods are still advantageous, allowing additional decreases in contig number, as well as generation of chromosomal level assemblies. BUSCO completion (single copy and duplicated) of the protein coding genes were also comparable between our de-novo assembly and the other coral long-read genomes, indicating it is of comparable quality despite only using HiFi reads (Supplementary Fig. 1A). Orthofinder [43] analysis identified 93% of protein coding genes from our O. faveolata de-novo assembly to be assigned to orthogroups when analyzed with other long-read coral genome resources [5,6,7,8,9,10], with 5.9% of these genes being species specific to O. faveolata (Supplementary 4). This suggests our gene prediction and annotation pipeline is of comparable quality to other coral long-read genome assemblies. As more long-read coral genomes become available, an in depth analysis of orthologs and paralogs should be undertaken to identify core coral gene function, and potential processes which could be species specific.
Potential chromosomes are recovered from HiFi reads without additional sequencing information
Previous work has suggested that the potential karyotype of O. faveolata is 16 chromosomes [48]. In our study, telomeric repeat analysis identified regions at either one (telocentric, 12 scaffolds) or both (metacentric, seven scaffolds) ends of 19 of the 51 scaffolds (TeloScafs) (Fig. 4). Telomeric repeats are indicative of chromosome ends, suggesting several scaffolds in our assembly represent complete, telomere-to-telomere sequences, and thus that we may have captured some full chromosomes in our assembly. BUSCO [37] analysis also identified 90.2% of metazoan universal single copy orthologs as complete in telomere containing scaffolds, as compared to 94.86% in the entire scaffold set (Fig. 3A). With the high percentage BUSCO completion of the telomere containing scaffolds, this further suggests that several scaffolds likely represent complete chromosomes. The number of identified TeloScafs is however larger than the potential karyotype of 16 for O. faveolata [48] suggesting that we missed some repetitive sections of the genome such as centromeric repeats. This may also be due to only utilizing HiFi reads for our assembly. Future work should re-assemble our HiFi reads using additional methodologies such as optical mapping [49, 50] or Hi-C sequencing [51, 52] to achieve a true telomere-to-telomere chromosome scale assembly and resolve any discrepancies in the number of telomere-containing scaffolds. Additionally, the karyotype of O. faveolata, as well as other coral species, should be defined experimentally rather than relying on bioinformatic methods to infer karyotype. Historically, working with coral gametes has been difficult due to them only spawning once a year. With developments in ex-situ spawning, there is now higher availability of coral gametes throughout the year [53,54,55] making gamete based research easier and more accessible for coral species. This, paired with new karyotyping methodologies for non-model invertebrate organisms [56], will allow experimental identification of coral species karyotype to occur, paving the way for improved genome assemblies due to known chromosome number.
Future directions and conclusions
In this study we provide an updated genome resource for the endangered coral species O. faveolata at near-chromosome scale using only PacBio HiFi long reads. Despite improvements in completeness and contiguity over the current O. faveolata reference assembly [31], our assembly may yet be improved to a bonafide chromosomal level with additional sequencing (specifically Hi-C). Use of this updated resource will also assist efforts to functionally characterize genes, an area of research that is just starting to occur within coral species [57]. Additionally, we hope that this resource will facilitate more in-depth ‘omic analyses utilizing O. faveolata as the focal species. As this species continues to be integrated into reef restoration activities [19,20,21], a thorough understanding of its population structure and response to anthropogenic stressors will be key to its preservation.
Methods
Tissue collection, nucleic acid extractions, library preparation, and sequencing
To generate high molecular weight (HMW) DNA for de-novo genome assembly, gamete bundles (sperm and eggs) were collected from one spawning colony of Orbicella faveolata (Fig. 1A) on the 18th August 2022 at roughly 00:15 local time at Horseshoe Reef (Key Largo, FL, USA; 25.1388°N, 80.2950°W). Gamete bundles were collected in a conical mesh net with a 50 ml conical centrifuge tube at the apex (Fig. 1B), then capped and brought to the boat. Onboard the vessel, as gamete bundles started to break apart, they were diluted with filtered seawater to reach a sperm concentration of ~ 108 cells/ml [58]. After transport to the University of Miami Rosenstiel School, eggs were separated from sperm using a Corning 70 μm sterile cell strainer. Eggs caught on the filter were discarded, and filtrate was inspected under a microscope to remove any residual eggs. Six 1.5 ml tubes with 1 ml of the sperm filtrate, in seawater, were then centrifuged at 3,000 g for five minutes. The supernatant was removed, and 1 ml of additional sperm filtrate was added to each tube and repeated 8x. Each tube had a total of 8 ml of filtrate processed. Pelleted sperm was then resuspended in 1x PBS (pH 7.2) using a wide pipette tip, and centrifuged at 3,000 g for five minutes. The supernatant was removed without disturbing the pellet, and each tube was then flash frozen in liquid nitrogen and stored at -80ºC. Frozen sperm was then shipped to the University of California (UC) Davis Genome Center for HMW DNA extractions, library preparation, and sequencing on one flow cell of a PacBio Sequel II. For detailed methods, please see Supplementary File 5.
To generate a high quality and complete annotated transcriptome, the largest transcriptional snapshot of mRNA was desired to capture all transcripts that are present within the O. faveolata genome. As such, we exposed fragments (~ 5 cm2) from one genet of O. faveolata to different biotic and abiotic stimuli to maximize the range of mRNA expression (Table 1). This O. faveolata genet was a rescue coral that had been housed in the Experimental Reef Lab (Miami, FL) for three months prior to use in the biotic and abiotic exposures for RNA expression profiles. Following stimuli exposure, coral fragments were sampled using a hammer and chisel and placed in a 2 ml bead beating tube filled with 0.1 and 0.5 mm beads, and 1.2 ml of DNA/RNA shield (Zymo, Irvine). Bead beating tubes were then bead beat for 30 min on a VortexGenie at max speed before being centrifuged at 16,000 rpm for 1 min. A total of 400 µl of supernatant was transferred to a new tube and total RNA was extracted with the Quick RNA Miniprep kit (Zymo, Irvine) including the fifteen minute DNase I digestion step. Total RNA was eluted with 80 µL of pre-heated (60 o C) RNase-free water, with a three minute incubation on the spin column matrix. Eluted total RNA was cleaned and concentrated with the Clean and Concentrate − 5 RNA kit (Zymo, Irvine), with an elution volume of 25 µl of pre-heated (60 oC) RNase-free water. The purity and concentration of the RNA was assessed using a Nanodrop and a Qubit V4 (Invitrogen), respectively. RNA was then sent to UC Davis DNA Technology Core (Davis CA) for additional QC (TapeStation), library prep, and sequencing on one flow cell of a PacBio Sequel II. For detailed methods, please see Supplementary File 5.
Mitochondrial genome assembly
The mitochondrial genome was assembled from the HiFi reads, prior to contaminant removal, using MitoHiFi [44, 59] with key parameters -o 5 (invertebrate parameter). Due to unsuccessful circulation using the publicly available O. faveolata mitochondrial genome [45], MitoHiFi was run using closely related stony coral species mitochondrial genomes available on the NCBI (Supplementary File 5). For our final mitochondrial genome assembly, we used Platygrya carnosa (Nucleotide accession = NC_020049.1) [60] as the reference in MitoHiFi which allowed successful circulation. Phylogenetic analysis was undertaken with our O. faveolata mitochondrial genome and all available Scleractinia coral mitochondrial genomes on the NCBI (Supplementary File 6). Briefly, all reference genomes were concatenated into one fasta and run through trimal [61] with the following parameters: -gt 0.3, -st 0.001, -cons 30. Circulator [62] was used to orient all mitochondrial genomes in the same order, before multi-sequence alignment with mafft [63]. RAxML [64] was then used to generate the phylogenetic tree (-x 10, -p 10, -#100, -m GTRCAT) with the default value of 100 bootstraps. The phylogenetic tree was visualized with figtree (http://tree.bio.ed.ac.uk/software/figtree/), ggplot [65] and ggtree [66] in R [67] and RStudio.
De-novo genome assembly
A schematic of the bioinformatic pipeline used for de-novo genome assembly can be found in Supplementary Fig. 3. Raw HiFi reads first underwent a contamination screening, following the methodology in [68], using BLASTn [32, 68] against the assembled mitochondrial O. faveolata genome and the following databases: common eukaryote contaminant sequences (ftp.ncbi.nlm.nih.gov/pub/kitts/contam_in_euks.fa.gz), NCBI viral (ref_viruses_rep_genomes) and prokaryote (ref_prok_rep_genomes) representative genome sets downloaded with blast::update_blastdb.pl. All raw HiFi reads with a bit score > 1000 were removed. Prior to assembly, the kmer profile of cleaned raw HiFi reads was generated with Meryl [34], and used for genome profiling with GenomeScope2 [69] to estimate genome size, repetitiveness, heterozygosity, and ploidy. The cleaned raw HiFi reads were then assembled with HiFiasm [33] (key parameters:–primary, -s 0.55,–purge-max 150) into a primary and alternative assembly. Assembly statistics were obtained using Quast [35, 36], BUSCO [37] (organism metazoa_odb10), and Merqury [34]. A subsequent BLASTn [32] was run to identify additional contaminants using the previously mentioned databases. Scaffolding of the primary assembly was done using the clean raw HiFi reads and nt-links (key parameters: g 100, rounds 5) [38, 70] resulting in the scaffolded assembly. Final assembly statistics were generated with BUSCO [37] and Quast [36].
The scaffolded assembly was then analyzed with RepeatModeler2 [71] to generate a de-novo library of repetitive elements. RepeatModeler2 [71] results were uploaded to the Dfam database (https://www.dfam.org/home) as requested in the user documentation. Output from RepeatModeler2 [71] was then used in RepeatMasker (https://github.com/rmhubley/RepeatMasker) to generate hard masked (default parameters) and soft masked (-xsmall) versions of the scaffolded assembly with accompanying gff files.
Identification of telomeric repeats in the scaffolded contigs
To identify potential telomeres in our scaffolded contigs (TeloScafs), the Telomere Identification Toolkit (tidk; https://github.com/tolkit/telomeric-identifier) with the coral telomeric repeat “TTAGGG” [72, 73] was used with following parameters: search,–window 50,000. Scaffolded contigs with telomeric repeats at either one (telocentric) or both (metacentric) ends were then used in a BUSCO [37] (database = metazoa_odb10) analysis as to allow comparison of BUSCO completeness between the set of TeloScafs, and the scaffolded de-novo assembly.
Annotation of de-novo genome and transcriptome assemblies
A combination of PASA [39] and funannotate [41] were used to annotate the de-novo assembled genome. PASA [39] was used to model gene structures using the scaffolded genome assembly and the high quality (hq) transcripts. The hq transcripts were cleaned using seqClean [40] before being used for transcript alignment and alignment assembly with the scaffolded genome assembly (pasa::Launch_PASA_pipeline.py, key parameters: -R, -T,–ALIGNERS blat,minimap2,–TRANSDECODER,–ALT_SPLICE). A high quality dataset for downstream ab initio gene prediction containing gene models with coordinates based on the genome sequences was generated from PASA transcript assemblies with pasa::pasa_asmbls_to_training_set.dbi. The soft masked scaffolded genome and transcript based gene models from PASA [39] were then input into funannotate::predict [41] (key parameters:--organism other,–repeats2evm,–keep_evm,–optimize_augustus) to train the ab-initio gene predictors (Augustus [74], GeneMark-ES/ET [75], snap [76], glimmerhmm [77]), before running Evidence Modeler [78, 79] to generate consensus gene models. Transfer RNA’s (tRNAs) were identified using tRNAscan-SE [42]. Gene model predictions and untranscribed regions (UTRs) were then refined using funannotate::update [41] using the hq transcripts and a de-novo assembled transcriptome of O.faveolata (from [80]) using Trinity [81] with key parameter:–trimmomatic. InterproScan [82] was then run on the updated gene models to classify proteins into families, and predict domains. Interproscan [82] results were then incorporated with the results from funannotate::update [41] and used in funannotate::annotate [41] to assign functional annotation to the protein-coding genes, with the optional addition of eggNOG-mapper [83,84,85].
Comparisons to other coral genome resources
To compare our final de-novo assembly to the previous Orbicella faveolata reference genome [31] BUSCO [37] (database = metazoa_odb10) and QUAST [35, 36] were used. Percentage mapping of reads between the two genomes was done using Minimap2 [86, 87] (key parameters: -ax asm5) and samtools [88] (key parameter: flagstat). Comparison of coding genes was done using the protein fasta files in BUSCO [37] using the proteins flag (-m) and database metazo_odb10.
Our final de-novo assembly was compared against all other publicly available long-read coral genomes [5,6,7,8,9,10] using QUAST [35, 36] and BUSCO [37] (database = metazoa_odb10). An additional analysis of coding genes was run using the protein fasta files from the long-read assemblies in BUSCO [37] with key parameters -m protein and database metazo_odb10. Finally, Orthofinder [43] was used to identify ortholog groups between all the long-read coral genomes with results visualized using ggplot2 [65].
Summary circos plot generation
Circos [89] was used to generate a circular summary figure of the de-novo assembled genome. For visualization, all contigs less than 1 mb were combined. Additional quality metrics were calculated as follows, with outputs formatted for Circos using tidyverse [90] and SeqinR [91] in Rstudio. GC content and skew were identified using GCcalc (https://github.com/WenchaoLin/GCcalc) with key parameters: -w 50,000, and -s 250,000. For repeat content, the GFF from repeatmasker (https://github.com/rmhubley/RepeatMasker) was first converted to a bed file using Bedops [92] before being used in deepStats::dsComputerBEDdensity [93] with a sliding window of 50,000 (-w 50,000). For gene content, the GFF file from funannotate::update [41] was processed in the same manner as repeat content above. The output from the telomere analysis, using tidk (https://github.com/tolkit/telomeric-identifier), was also incorporated in the final Circos [89] summary figure.
Data availability
The O. faveolata de-novo genome assembly presented here is publicly available at PRJNA970355 on the NCBI, as well as at https://doi.org/10.5281/zenodo.10151798. Full analysis scripts, pipeline, and tool versions are avaliable at https://github.com/benyoung93/orbicella_faveolata_pacbio_genome_transcriptome. All program versions are available in Supplementary File 7 and in the github repository. The mitochondrial genome is available at GenBank accession OR906199, as well as at https://doi.org/10.5281/zenodo.10151798.
References
Nath S, Shaw DE, White MA. Improved contiguity of the threespine stickleback genome using long-read sequencing. G3 GenesGenomesGenetics.2021;11(2):jkab007.
van Rengs WMJ, Schmidt MHW, Effgen S, Le DB, Wang Y, Zaidan MWAM. A chromosome scale tomato genome built from complementary PacBio and Nanopore sequences alone reveals extensive linkage drag during breeding. Plant J. 2022;110(2):572–88.
Yekefenhazi D, He Q, Wang X, Han W, Song C, Li W. Chromosome-level genome assembly of Nibea coibor using PacBio HiFi reads and Hi-C technologies. Sci Data. 2022;9(1):670.
Tham CY, Poon L, Yan T, Koh JYP, Ramlee MK, Teoh VSI. High-throughput telomere length measurement at nucleotide resolution using the PacBio high fidelity sequencing platform. Nat Commun. 2023;14(1):281.
Shumaker A, Putnam HM, Qiu H, Price DC, Zelzion E, Harel A. Genome analysis of the rice coral Montipora capitata. Sci Rep. 2019;9(1):2571.
Cooke I, Ying H, Forêt S, Bongaerts P, Strugnell JM, Simakov O. Genomic signatures in the coral holobiont reveal host adaptations driven by Holocene climate change and reef specific symbionts. Sci Adv. 2020;6(48):eabc6318.
Fuller ZL, Mocellin VJL, Morris LA, Cantin N, Shepherd J, Sarre L. Population genetics of the coral Acropora millepora: toward genomic prediction of bleaching. Science. 2020;369(6501):eaba4674.
Shinzato C, Khalturin K, Inoue J, Zayasu Y, Kanda M, Kawamitsu M. Eighteen coral genomes reveal the Evolutionary Origin of Acropora Strategies to accommodate environmental changes. Mol Biol Evol. 2021;38(1):16–30.
Stephens TG, Lee J, Jeong Y, Yoon HS, Putnam HM, Majerová E. High-quality genome assemblies from key hawaiian coral species. GigaScience. 2022;11:giac098.
Noel B, Denoeud F, Rouan A, Buitrago-López C, Capasso L, Poulain J. Pervasive tandem duplications and convergent evolution shape coral genomes. Genome Biol. 2023;24(1):123.
Goreau TF, Wells JW. The shallow-water Scleractinia of Jamaica: revised list of species and their Vertical distribution range. Bull Mar Sci. 1967;17(2):442–53.
Holstein DM, Smith TB, Gyory J, Paris CB. Fertile fathoms: deep reproductive refugia for threatened shallow corals. Sci Rep. 2015;5(1):12407.
United States.TheEndangeredSpeciesActasamendedbyPublicLaw97–304(theEndangeredSpeciesActamendmentsof1982)[Internet].Washington:U.S.G.P.O., 1983.;1983.Availablefrom:https://search.library.wisc.edu/catalog/999606103702121.
.IUCN, Orbicella faveolata R, Hoeksema B, Rivera-Sosa A, Villamizar E, Vermeij M, Croquer A, Banaszak A. TheIUCNRedListofThreatenedSpecies2022:e.T133373A165855828[Internet].2021[cited2023Sep11].Availablefrom:https://www.iucnredlist.org/species/133373/165855828.
Grottoli AG, Warner ME, Levas SJ, Aschaffenburg MD, Schoepf V, McGinley M. The cumulative impact of annual coral bleaching can turn some coral species winners into losers. Glob Change Biol. 2014;20(12):3823–33.
Dziedzic KE, Elder H, Tavalire H, Meyer E. Heritable variation in bleaching responses and its functional genomic basis in reef-building corals (Orbicella faveolata). Mol Ecol. 2019;28(9):2238–53.
.FDEP, Case Defintion. StonyCoralTissueLossDisease(SCTLD)[Internet].2018.Availablefrom:https://floridadep.gov/sites/default/files/Copy%20of%20StonyCoralTissueLossDisease_CaseDefinition%20final%2010022018.pdf.
Cróquer A, Weil E, Rogers CS, Similarities. andDifferencesBetweenTwoDeadlyCaribbeanCoralDiseases:WhitePlagueandStonyCoralTissueLossDisease.FrontMarSci[Internet].2021[cited2023Sep7];8.Availablefrom:https://www.frontiersin.org/articles/https://doi.org/10.3389/fmars.2021.709544.
Page CA, Muller EM, Vaughan DE. Microfragmenting for the successful restoration of slow growing massive corals. Ecol Eng. 2018;123:86–94.
Rivas N, Hesley D, Kaufman M, Unsworth J, D’Alessandro M, Lirman D. Developing best practices for the restoration of massive corals and the mitigation of predation impacts: influences of physical protection, colony size, and genotype on outplant mortality. Coral Reefs. 2021;40(4):1227–41.
Raker C, Olmeda-Saldaña M, Williams SM, Weil E, Prada C. UseofpredatorexclusioncagestoenhanceOrbicellafaveolatamicro-fragmentsurvivorshipandgrowthduringrestoration.FrontMarSci[Internet].2023[cited2023Aug3];10.Availablefrom:https://www.frontiersin.org/articles/https://doi.org/10.3389/fmars.2023.1122369.
Alegría-Ortega A, Sanín-Pérez MJ, Quan-Young LI, Londoño-Mesa MH. Genetic structure of Orbicella faveolata population reveals high connectivity among a marine protected area and Varadero Reef in the Colombian caribbean. Aquat Conserv Mar Freshw Ecosyst. 2021;31(4):764–76.
Traylor-Knowles N, Connelly MT, Young BD, Eaton K, Muller EM, Paul VJ. etal.GeneExpressionResponsetoStonyCoralTissueLossDiseaseTransmissioninM.cavernosaandO.faveolataFromFlorida.FrontMarSci[Internet].2021[cited2023Aug2];8.Availablefrom:https://www.frontiersin.org/articles/https://doi.org/10.3389/fmars.2021.681563.
Beavers KM, Van Buren EW, Rossin AM, Emery MA, Veglia AJ, Karrick CE. Stony coral tissue loss disease induces transcriptional signatures of in situ degradation of dysfunctional Symbiodiniaceae. Nat Commun. 2023;14(1):2915.
Studivan MS, Eckert RJ, Shilling E, Soderberg N, Enochs IC, Voss JD. Stonycoraltissuelossdiseaseinterventionwithamoxicillinleadstoareversalofdisease-modulatedgeneexpressionpathways.MolEcol.2023;n/a(n/a):1–20.
Hughes TP, Baird AH, Bellwood DR, Card M, Connolly SR, Folke C. Climate Change, Human impacts, and the resilience of Coral Reefs. Science. 2003;301(5635):929–33.
Hoegh-Guldberg O. Coral reef ecosystems and anthropogenic climate change. Reg Environ Change. 2011;11(1):215–27.
Cramer KL, Jackson JBC, Angioletti CV, Leonard-Pingel J, Guilderson TP. Anthropogenic mortality on coral reefs in Caribbean Panama predates coral disease and bleaching. Ecol Lett. 2012;15(6):561–7.
Camp EF, Schoepf V, Mumby PJ, Hardtke LA, Rodolfo-Metalpa R, Smith DJ. The future of Coral Reefs subject to Rapid Climate Change: lessons from Natural Extreme environments. Front Mar Sci. 2018;5:4.
Traylor-Knowles N, Baker AC, Beavers KM, Garg N, Guyon JR, Hawthorn A. Advancesincoralimmunity‘omicsinresponsetodiseaseoutbreaks.FrontMarSci[Internet].2022[cited2023Sep7];9.Availablefrom:https://www.frontiersin.org/articles/https://doi.org/10.3389/fmars.2022.952199.
Prada C, Hanna B, Budd AF, Woodley CM, Schmutz J, Grimwood J. Empty niches after extinctions increase Population sizes of Modern corals. Curr Biol. 2016;26(23):3190–4.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421.
Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18(2):170–5.
Rhie A, Walenz BP, Koren S, Phillippy AM. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020;21(1):245.
Mikheenko A, Valin G, Prjibelski A, Saveliev V, Gurevich A. Icarus: visualizer for de novo assembly evaluation. Bioinformatics. 2016;32(21):3321–3.
Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics. 2018;34(13):i142–50.
Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO Update: Novel and Streamlined Workflows along with broader and deeper phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and viral genomes. Mol Biol Evol. 2021;38(10):4647–54.
Coombe L, Warren RL, Wong J, Nikolic V, Birol I. ntLink: a Toolkit for De Novo Genome Assembly Scaffolding and Mapping using long reads. Curr Protoc. 2023;3(4):e733.
Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Hannick LI. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31(19):5654–66.
Lee Y, Tsai J, Sunkara S, Karamycheva S, Pertea G, Sultana R. The TIGR Gene indices: clustering and assembling EST and known genes and integration with eukaryotic genomes. Nucleic Acids Res. 2005;33(Database Issue):D71–4.
Palmer JM, Stajich J. Funannotatev1.8.1:Eukaryoticgenomeannotation[Internet].Zenodo;2020[cited2023May5].Availablefrom:https://zenodo.org/record/4054262.
Chan PP, Lin BY, Mak AJ, Lowe TM. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021;49(16):9077–96.
Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20(1):238.
Uliano-Silva M, Ferreira JGRN, Krasheninnikova K, Consortium DTof, Formenti L. L,etal.MitoHiFi:apythonpipelineformitochondrialgenomeassemblyfromPacBioHighFidelityreads[Internet].bioRxiv;2023[cited2023May5].p.2022.12.23.521667.Availablefrom:https://www.biorxiv.org/content/https://doi.org/10.1101/2022.12.23.521667v2.
Fukami H, Knowlton N. Analysis of complete mitochondrial DNA sequences of three members of the Montastraea annularis coral species complex (Cnidaria, Anthozoa, Scleractinia). Coral Reefs. 2005;24(3):410–7.
Minhas BF, Beck EA, Cheng CHC, Catchen J. Novel mitochondrial genome rearrangements including duplications and extensive heteroplasmy could underlie temperature adaptations in Antarctic notothenioid fishes. Sci Rep. 2023;13(1):6939.
Morgan B, Wang TY, Chen YZ, Moctezuma V, Burgos O, Le MH. Long-read sequencing data reveals dynamic evolution of mitochondrial genome size and the phylogenetic utility of mitochondrial DNA in Hercules Beetles (Dynastes; Scarabaeidae). Genome Biol Evol. 2022;14(10):evac147.
Snelling J, Dziedzic K, Guermond S, Meyer E. DevelopmentofanintegratedgenomicmapforathreatenedCaribbeancoral(Orbicellafaveolata)[Internet].bioRxiv;2017[cited2023Aug8].p.183467.Availablefrom:https://www.biorxiv.org/content/https://doi.org/10.1101/183467v2.
Leinonen M, Salmela L. Optical map guided genome assembly. BMC Bioinformatics. 2020;21(1):285.
Yuan Y, Chung CYL, Chan TF. Advances in optical mapping for genomic research. Comput Struct Biotechnol J. 2020;18:2051–62.
Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. Hi–C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58(3):268–76.
Yamaguchi K, Kadota M, Nishimura O, Ohishi Y, Naito Y, Kuraku S. Technical considerations in Hi-C scaffolding and evaluation of chromosome-scale genome assemblies. Mol Ecol. 2021;30(23):5923–34.
Craggs J, Guest JR, Davis M, Simmons J, Dashti E, Sweet M. Inducing broadcast coral spawning ex situ: closed system mesocosm design and husbandry protocol. Ecol Evol. 2017;7(24):11066–78.
O’Neil KL, Serafin RM, Patterson JT, Craggs JRK. RepeatedexsituSpawninginTwoHighlyDiseaseSusceptibleCoralsintheFamilyMeandrinidae.FrontMarSci[Internet].2021[cited2023Jul21];8.Availablefrom:https://www.frontiersin.org/articles/https://doi.org/10.3389/fmars.2021.669976.
Wei F, Cui M, Huang W, Wang Y, Liu X, Zeng X. Ex situ reproduction and recruitment of scleractinian coral Galaxea fascicularis. Mar Biol. 2023;170(3):30.
Guo L, Accorsi A, He S, Guerrero-Hernández C, Sivagnanam S, McKinney S. An adaptable chromosome preparation methodology for use in invertebrate research organisms. BMC Biol. 2018;16(1):25.
Cleves PA, Shumaker A, Lee J, Putnam HM, Bhattacharya D. Unknown to known: advancing knowledge of Coral Gene function. Trends Genet. 2020;36(2):93–104.
Hagedorn M, Page CA, O’Neil KL, Flores DM, Tichy L, Conn T. Assisted gene flow using cryopreserved sperm in critically endangered coral. Proc Natl Acad Sci. 2021;118(38):e2110559118.
Allio R, Schomaker-Bastos A, Romiguier J, Prosdocimi F, Nabholz B, Delsuc F. MitoFinder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020;20(4):892–905.
Wang M, Sun J, Li J, Qiu J. wen.CompletemitochondrialgenomeofthebraincoralPlatygyracarnosus.MitochondrialDNA.2013;24(3):194–5.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3.
Hunt M, Silva ND, Otto TD, Parkhill J, Keane JA, Harris SR. Circlator: automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015;16(1):294.
Katoh K, Standley DM. MAFFT multiple sequence alignment Software Version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–90.
Wickham H. ggplot2:ElegantGraphicsforDataAnalysis[Internet].2016.Availablefrom:https://ggplot2.tidyverse.org.
Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8(1):28–36.
R Core Team.R:Alanguageandenvironmentforstatisticalcomputing.RFoundationforStatisticalComputing[Internet].Vienna,Austria.;2020.Availablefrom:https::www.R-project.org/.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10.
Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11(1):1432.
Coombe L, Li JX, Lo T, Wong J, Nikolic V, Warren RL. LongStitch: high-quality genome assembly correction and scaffolding using long reads. BMC Bioinformatics. 2021;22(1):534.
Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci. 2020;117(17):9451–7.
Sinclair CS, Richmond RH, Ostrander GK. Characterization of the telomere regions of scleractinian coral, Acropora surculosa. Genetica. 2007;129(3):227–33.
Zielke S, Bodnar A. Telomeres and telomerase activity in Scleractinian corals and Symbiodinium spp. Biol Bull. 2010;218(2):113–21.
Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24(5):637–44.
Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33(20):6494–506.
Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5(1):59.
Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–9.
Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9(1):R7.
Haas BJ, Zeng Q, Pearson MD, Cuomo CA, Wortman JR. Approaches to Fungal Genome Annotation. Mycology. 2011;2(3):118–41.
MacKnight NJ, Dimos BA, Beavers KM, Muller EM, Brandt ME, Mydlarz LD. Disease resistance in coral is mediated by distinct adaptive and plastic gene expression profiles. Sci Adv. 2022;8(39):eabo6153.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–40.
Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(Database issue):D309–14.
Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18(4):366–8.
Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: functional annotation, Orthology assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol. 2021;38(12):5825–9.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100.
Li H. New strategies to improve minimap2 alignment accuracy. Bioinformatics. 2021;37(23):4572–4.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10(2):giab008.
Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45.
Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R. Welcome to the Tidyverse. J Open Source Softw. 2019;4(43):1686.
Charif D, Lobry JR. SeqinR 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis. In: Bastolla U, Porto M, Roman HE, Vendruscolo M, editors. Structural Approaches to Sequence Evolution: Molecules, Networks, Populations [Internet]. Berlin, Heidelberg: Springer; 2007 [cited 2023 Aug 8]. p. 207–32. (Biological and Medical Physics, Biomedical Engineering). Available from: https://doi.org/10.1007/978-3-540-35306-5_10.
Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK. BEDOPS: high-performance genomic feature operations. Bioinformatics. 2012;28(14):1919–20.
Richard G, gtrichard/deepStats. deepStats0.3.1[Internet].Zenodo;2019[cited2023Jul21].Availablefrom:https://zenodo.org/record/3361799/export/csl.
Acknowledgements
The authors thank SECORE International and the Coral Ecology Unit at the Southeast Fisheries Science Center for field assistance during coral spawn collections in Key Largo (collections conducted under permit FKNMS-2018-163-A1). Coral fragments exposed to different biotic and abiotic perturbations were collected as corals of opportunity with permission by the Florida Fish and Wildlife Conservation Commission. We would like to thank the DNA Technologies and Expression Analysis Core Laboratory (UC Davis, California), specifically Noravit Chumchim and Dr. Ruta Sahasrabudhe, for their help in preparing and sequencing the DNA and RNA samples for this genome assembly. We would also like to thank Nicolas Locatelli for early use of the Colpophyllia natans in quality control of our assembly, as well as bioinformatic suggestions and recommendations. We would also like to thank IDSC at the University of Miami for computational resources used on Pegasus HCC. Finally, we thank the editor and reviewers for their time and effort in editing and reviewing the manuscript.
Funding
- FDEP C2002 to Muller, Traylor-Knowles, Rosales, and Studivan
- FDEP C1FB43 to Studivan, Rosales, Muller, and Williams
- OAR Omics to Studivan
- NSF Award #1951826 to Traylor-Knowles
- Revive and Restore Catalyst Award to Traylor-Knowles
Author information
Authors and Affiliations
Contributions
Study conception: BDY and MSS. Field work: OMW. Lab work: BDY and MSS. Data analysis BDY and NSK. Initial manuscript: BDY. Manuscript edits: BDY, MSS, NSK, OMW, NTK, LMI, NJM, EMM, SRM, SMS, SDW. Manuscript was approved by all authors before submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Gamete bundles collections were conducted under permit FKNMS-2018-163-A1. Fragments of Orbicella faveolata exposed to different biotic and abiotic perturbations were collected as corals of opportunity with permission by the Florida Fish and Wildlife Conservation Commission.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Young, B.D., Williamson, O.M., Kron, N.S. et al. Annotated genome and transcriptome of the endangered Caribbean mountainous star coral (Orbicella faveolata) using PacBio long-read sequencing. BMC Genomics 25, 226 (2024). https://doi.org/10.1186/s12864-024-10092-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10092-w