
Abstract
Whole-exome sequencing (WES) is widely used to diagnose complex genetic diseases and rare conditions. The implementation of a robust and effective quality control system for sample identification and tracking throughout the WES process is essential. We established a multiplex panel that included 22 coding single-nucleotide polymorphism (cSNP) loci. The personal identification and paternity identification abilities of the panel were evaluated, and a preliminary validation of the practical feasibility of the panel was conducted in a clinical WES case. These results indicate that the cSNP panel could be a useful tool for sample tracking in WES.
Similar content being viewed by others


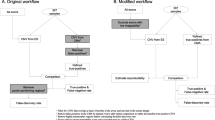
Background
In recent years, rapid advances in sequencing technologies have provided new options for diagnosing rare diseases [1, 2]. The remarkable success of WES in gene identification has led to its widespread integration into clinical practice [3,4,5]. However, the WES testing process involves a complex series of steps, making it prone to errors such as sample-labeling errors, sample mix-ups, and cross-contamination [6,7,8]. If such errors occur, it could potentially result in incorrect or delayed reporting of results, consequently affecting the decisions made by clinicians and the diagnosis and treatment of patients. Furthermore, errors can compromise the reliability of research findings based on this method. Therefore, sample tracking is crucial for the correct identity and accuracy of WES results. Current clinical quality control methods rely predominantly on automated equipment and the inclusion of internal quality control DNA. Genetic approaches have demonstrated efficacy in sample tracking, with multiple researchers advocating for the establishment of a quality control system for sample identification and tracking using WES to prevent testing errors [9,10,11,12]. Trio samples should be tested in parallel with WES to enhance the precision and interpretability of the quality control system results [13, 14].
Although molecular diagnostic technologies and relevant quality management are constantly improving, there is a lack of standardized methods for personal identification of WES samples. The implementation of fully automated instrumentation and information systems, although beneficial, does not entirely resolve challenges such as sample mix-ups, contamination, and the loss or deterioration of sample labels [15]. Therefore, clinical laboratories opting for genetic methods to establish personal identification, paternity tests, and sample tracking should consider independent testing methods, although creating an effective personal identification system that is practical and efficient is challenging [16]. Given the complexity and data-intensive nature of WES, and the relatively high testing costs, conducting two independent sequencing experiments simultaneously for sample identification is logistically difficult. Consequently, another genetic method should be applied to identify samples by comparing polymorphic genetic markers results. This approach allows for sample identity verification by comparing the results with the corresponding polymorphic biomarkers in the WES data, ensuring the comprehensive tracking and quality control of WES samples. Currently, the most common biomarker used for forensic personal identification and paternity testing is short tandem repeats (STRs). However, the primary location of STRs in intronic or intergenic regions makes them less compatible with the target regions of WES [17]. Furthermore, analyzing STR data within the context of WES can be challenging because of the nature of STR sequences.
In contrast, single-nucleotide polymorphisms (SNPs) are prevalent throughout the human genome, with a substantial number located in exonic (protein coding) regions [18]. SNPs involve single-base changes, and the genotyping accuracy obtained through next-generation sequencing (NGS) is superior to that of STRs [17, 19]. Thus, the development of a multiplex system using coding SNPs (cSNPs) for personal identification and parentage testing in WES offers a superior alternative to using STR. Although previous studies have made strides in this direction, challenges remain because of the need for two independent sequencing processes for comparison, and because the large number of SNPs may pose difficulties in paternity testing within trio samples [20].
Several studies have explored multiplex systems based on cSNP markers [21, 22]. Although valuable, these systems have limitations in terms of their discrimination power and complexity, often necessitating multiple experiments. Notably, established cSNP multiplex systems may not fully meet the requirements for personal identification and paternity testing in an increasing number of WES samples. In this context, the implementation of SNaPshot technology offers a cost-effective and efficient approach for detecting multiple loci simultaneously [23].
In this study, we devised a multiplex system comprising 22 cSNPs using SNaPshot technology. Through a meticulous comparison of the genotyping results of these 22 cSNPs from the WES data with those obtained using the established system, coupled with cumulative paternity index (CPI) calculations, we achieved initial sample identity alignment and confirmed parentage relationships within trio samples.
Methods
Sample collection and extraction
Blood samples were collected from 114 unrelated individuals, 12 parent–child pairs, and 9 trios (one of which had WES data) with their informed consent. DNA was extracted from the samples using the phenol–chloroform method [24], followed by quantification using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). The DNA extracts were stored at –20 °C.
cSNP selection and primer design
As the target region for WES is the exon region, we focused on screening candidate loci in this area. VCFtools [25] and BCFtools [26] were used to filter SNPs from the exon sequence data downloaded from the ExAC database (version r1). SNPs were further filtered by the following criteria: (1) SNPs minor allele frequency (MAF) ≥ 0.3 or allele amounts ≥ 3 were initially screened out in order to keep as many highly polymorphic SNP loci as possible; and (2) synonymous (silent) SNPs should be selected to avoid SNPs associated with diseases. Based on the primary candidate loci, SNPs were further optimized according to the following criteria: (1) biallelic SNPs with MAF ≥ 0.4 or triallelic SNPs with MAF ≥ 0.1; (2) SNPs with high specificity after screening the obtained ± 400 bp sequences of cSNPs to assess their specificity; (3) no SNPs in the selected cSNP ± 400 bp sequences in the genome; (4) the top six loci with the highest MAF on each autosomal chromosome and multi-alleles (more than two alleles) were selected; (5) the distance between two SNPs in the same chromosome is greater than 10 Mb.
For the selected cSNPs, amplification and sequencing primers were designed using Primer 3 online (http://primer3.ut.ee/) and SBEprimer software. All primers were synthesized by Thermo Fisher Scientific (Waltham, MA, USA) and verified for specificity by confirming their binding and dimerization using the AutoDimer software. Finally, SNPs with high polymorphisms and primer specificity were prioritized for inclusion in the system.
Multiplex amplification
In this study, we used SNaPshot, a common SNP detection technology, to establish a multiplex panel. The first step of the amplification reaction included 5 μL of 2 × Taq reaction mix (QIAGEN, Hilden, Germany), 2 μL of RNase-free water, 2 μL of primer mix, and 1 μL of DNA. The reaction conditions consisted of pre-denaturation at 95 °C for 15 min, followed by 30 cycles of 94 °C for 30 s, 58.5 °C for 90 s, 72 °C for 60 s, with a final extension at 60 °C for 30 min and then holding at 4 °C. The first amplification products were digested with 1 µL of shrimp alkaline phosphatase (SAP, 1U) (New England Biolabs, USA) and 0.6 µL of exonuclease I (EXO I, 4U) (New England Biolabs) to remove any dNTPs and primer sequences. The samples were incubated for 1 h at 37 °C, followed by 12 min at 80 °C. Subsequently, the purified products underwent single-base extension (SBE) reaction with a reaction volume of 5 µL, including 1.5 μL SNaPshot ready reaction mix (Applied Biosystems, Warrington, UK), 1.3 μL extension primers mix, 1.5 μL enzyme-treated PCR product, and 0.7 μL nuclease-free water. The reaction conditions for SBE consisted of 95 °C for 10 s, 53 °C for 5 s, and 60 °C for 30 s for 25 cycles, with a hold at 4 °C. The products were further purified by adding 1 μL SAP enzyme to remove any remaining dNTPs, incubated for 1 h at 37 °C, followed by 12 min at 80 °C. All reactions were performed using an Eppendorf 6331 Nexus Gradient Flexlid Thermal Cycler (Eppendorf, Hamburg, Germany).
Sensitivity study
To assess the sensitivity of the multiplex panel, we used varying amounts (10, 8, 6, 4, 2, 1, 0.5, and 0.1 ng) of DNA input. Each sample was subjected to the same conditions, with the only difference being the amount of DNA input.
Capillary electrophoresis and statistical analysis
The amplification products were analyzed using a 3130 Genetic Analyzer (Applied Biosystems). 1 µL product was added in a 9 µL mixture of Hi-Di formamide (Applied Biosystems) and Liz 120 (Applied Biosystems) internal standard. The resulting mixture was thoroughly mixed, and the injection voltage and time were set to 3kv and 10 s, respectively. The raw data were analyzed using GeneMapper software version 3.2 (Applied Biosystems) with a threshold value of 50 RFU. Exact tests for Hardy–Weinberg Equilibrium (HWE) and linkage disequilibrium were calculated using Arlequin statistical software version 3.5 [27]. P-values < 0.05 were indicative of deviation from the HWE. We used PowerStats v1.2 (Promega, Madison, WI, USA) to calculate the allele frequency, match probability (MP), discrimination power (DP), power of exclusion (PE), and paternity index (PI).
Extraction of cSNP genotyping from WES data
We used Basic Local Alignment Search Tool (BLAST) to select sequences that specifically represented each cSNP locus. Based on these sequences and a specific algorithm, we extracted the corresponding allele information from the trio's WES data. Subsequently, alleles with low read counts were filtered out to obtain the genotype of each locus in the sample, and the respective allele read counts. We used likelihood ratio (LR) method [28] to evaluate the identity of the samples. The LR is defined as the ratio of two conditional probabilities: 1) Hypothesis of prosecution (Hp): the probability that the genotype combination of the examinee matches that of the person of interest; and 2) Hypothesis of defense (Hd): the probability that the genotype combination of an unrelated random individual matches that of the person of interest.
Results
Marker selection and general information
A total of 23,782 SNPs located in exonic regions with MAF ≥ 0.3 were initially identified, of which 4,403 SNPs had allele counts ≥ 3. Subsequently, the top six cSNPs with the highest MAF on each chromosome were selected to yield a candidate database of 108 cSNPs. Further filtering based on one locus per chromosome resulted in the selection of 22 cSNPs for the multiplex panel. Details regarding the physical locations, mutation types, primer sequences, and other relevant information are summarized in Table 1. All markers were validated using polyacrylamide gel electrophoresis (PAGE) and Sanger sequencing.
Sensitivity study
A sensitivity test was performed using random samples diluted to serial DNA amounts of 10, 8, 6, 4, 2, 1, 0.5, and 0.1 ng. Full profiles were obtained when the DNA amount > 0.5 ng (Fig. 1). When the template decreased to 0.1 ng, a partial profile was obtained with 14 cSNPs, and the highest peak height was 283 RFU. The profiles of the other DNA amounts are shown in Additional file 1: Figure S1.

A full cSNP profile was obtained when the DNA input was 0.5 ng. X-axis—the detected genotypes of cSNP loci. Each peak corresponds to the allele of each locus, with the locus name abbreviated to the last four digits of its respective rs number
Statistical analysis
Using our multiplex panel of 22 cSNPs, we genotyped 114 unrelated individuals. The amplicon sizes ranged from 26 to 116 bp. Forensic parameters, including DP, PE, MP, polymorphism information content (PIC), and typical paternity index (TPI) were computed for the 22 cSNPs (Table 2). The cumulative matching probability of the panel was 3.314 × 10–10. Following the Bonferroni correction, a P-value threshold of 0.000216 was applied, with no deviations from the linkage equilibrium between pairwise loci. The average DP and PE values were 0.627 (range: 0.549–0.765) and 0.179 (range: 0.139–0.229), respectively. The combined DP and PE values were 0.99997 and 0.98726, respectively. Additionally, the observed and theoretical allele frequencies of the 22 cSNPs of the East Asia population are summarized in Table 3. The theoretical allele frequencies, DP, and PE of the 22 cSNPs in other populations are provided in Additional file 2: Table S1.
Paternity test
To validate the ability to establish parentage, we conducted tests on 12 duos and 8 trios, calculating the PI and CPI. Allele frequencies were calculated using PowerStats based on the genotyping results of 114 unrelated individuals. All parent–child pairs conformed to Mendel’s laws of inheritance. No mutations or recombinations were observed in any of the cSNP markers in the 12 parent–child pairs. The CPI averaged 66.696 (range: 2.905 to 313.697) for the 12 parent–child pairs and 950.022 (range: 63.383 to 2,908.424) for the 8 trio samples, using the 22 cSNP markers. Typically, a CPI exceeding 1,000 indicates strong support for a parent–child relationship. Moreover, the CPI tends to increase when analyzing trios simultaneously, making trio analysis useful for determining parent–child relationships.
cSNP genotyping from WES data
This cSNP extraction method was validated using the WES data from a trio family. The cSNP genotyping results for each extracted sample are shown in Table 4. The established cSNP system was used to analyze the trio samples in parallel. The genotyping results for each sample are shown in Fig. 2. The genotyping results obtained using the cSNP system for each sample in the trio were largely consistent with the extracted genotyping results. Finally, based on the frequency distribution of each cSNP locus in the tested population and using forensic genetic analysis methods, the CPIs for the father and mother were calculated to be 86.007, and 48.700, respectively; the MP for the child, father, and mother were calculated to be 1.289 × 10–10, 9.943 × 10–11, and 1.243 × 10–11, respectively; and the LRs for the child, father, and mother were calculated to be 775,526,885, 10,057,219,242, and 80,445,560,559, respectively.

The cSNP profiles of the trio sample. C = children, M = mother, F = father
Discussion
When using WES, confirming the sample identity is crucial for obtaining reliable results. The current methods and quality control protocols in clinical laboratories fall short of fully tracking samples throughout the testing process [29]. Although the establishment of medical testing centers and adoption of new equipment have been effective in reducing the number of manual errors, challenges persist, particularly in the pretest phase [15, 30]. For example, in the case of WES samples obtained from prenatal fetal sources (such as villi, amniotic fluid, and umbilical cord blood) or from miscarriage tissues, it is only after processing the sequencing data that one can determine whether the sample is singular or mixed. However, determining whether a single sample originates from the fetus or the mother is challenging. Despite the application of fully automated equipment and information systems, that streamline manual operations, the potential for technical error persists. Addressing issues such as sample exchange, contamination, and label loss or damage remains a challenge [15]. Even when quality control materials involve exogenous DNA sequences, they only allow quality control during the post-DNA extraction detection steps [31]. This does not guarantee that the DNA sample originated from the subject of interest, so it is unable to confirm the sample identity conclusively.
The commonly used forensic STR test kits primarily include the following core loci: 1) Combined DNA Index System (CODIS), including 13 STR loci: CSF1PO, FGA, TH01, TPOX, VWA, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, and D21S11, with a cumulative MP of 2.003 × 10–15; and 2) Expanded U.S. core loci, including the 13 CODIS loci mentioned above, and D1S1656, D2S441, D2S1338, D10S1248, D12S391, D19S433, and D22S1045, totaling 20 STR loci with a cumulative MP of 2.022 × 10–22 [32]. The CODIS system satisfies the basic requirements for personal identification and paternity testing. However, commercial kits available for forensic science, whether STR-based (including CODIS/expanded CODIS) or SNP-based, primarily target loci located in intronic or intergenic regions of the respective genes, rather than exons. The target region for WES is the exonic region, which cannot be directly assessed using existing forensic multiplex systems. Consequently, extracting the genotypes of these forensic loci from WES data is challenging [17]. Moreover, when samples are genotyped using methods for different target areas, comparability between the two results is compromised, rendering concordance tests unfeasible [3]. Consequently, the use of these kits for personal identification using WES, and parentage testing is limited.
The large number of SNPs in coding regions holds potential for their use in personal identification and sample tracking [33]. However, existing multiplex cSNP systems have drawbacks, including high cost, time inefficiencies, operational complexity, and inability to be applied to WES data analysis [34, 35]. Du et al. [36] developed an SNP panel comprising 74 genome-wide SNPs and introduced a user-friendly online validation tool. However, only a subset of the 74 SNPs were located in the exonic region, rendering many SNPs unsuitable for WES application. The sample consistency verification software NGSCheckMate, developed by Lee et al. [37], can be used to analyze 11,696 SNP genotypes corresponding to the same sequencing data type, the same sequencing data type but different data formats (such as FASTQ and BAM), and the same data type but different sequencing types (such as whole-genome sequencing, WES, and RNA sequencing). In addition, Wesrphal et al. [38] screened 6,000 SNPs in the human genome and devised SMaSH, a Bayesian framework adept at effectively discerning whether corresponding samples from different NGS datasets are congruent. Javed et al. [20] used approximately 60,000 SNPs based on the principle of linkage disequilibrium to establish CrosscheckFingerprints (Crosscheck), a tool capable of detecting the interchangeability of samples across diverse NGS data types. Pengelly et al. [22] identified 24 biallelic cSNPs with a theoretical cumulative MP of approximately 4.641 × 10–10 for CHB, which exhibited a DP significantly lower than that of CODIS, and proved inadequate for basic requirements in personal identification and paternity testing. Notably, Pengelly et al. did not establish a detection method for these cSNPs but merely suggested them as candidate biomarkers for laboratory use. Helsmoortel et al. [23] established a multiplex system containing 21 cSNPs using a high-resolution melting method with a total of eight reactions. However, this method requires multiple experimental operations and integration of results. Moreover, the DP of the 21 biallelic cSNPs falls short of the basic requirement for personal identification and paternity testing, particularly as the number of WES detection samples continues to increase. These results indicate that a method that screens for additional cSNP loci with high polymorphism is needed to construct an effective system for personal identification. Additionally, the development of software capable of analyzing the corresponding cSNP genotypes from WES data is a promising development.
In this study, we screened out 22 multi-allelic cSNPs to establish a multiplex panel for tracking WES samples. However, the observed frequency of these 22 cSNPs is lower than their theoretical frequency. This variation may be attributed to the study population. The cumulative discrimination power (CDP) and cumulative MP values are forensic parameters for evaluating the efficiency of the system for personal identification. A CDP closer to 1 suggests a higher probability that, when two unrelated individuals are randomly selected from the population, the genotyping results for the 22 cSNP will differ, signifying a robust differentiating ability based on these markers. The CDP of 22 cSNPs indicates that in a sufficiently large population, if we were to randomly select two unrelated individuals 100,000 times, the genotyping results of these 22 cSNPs will be different in more than 99,997 instances. The cumulative MP of the 22 cSNPs closer to 0 implies a reduced likelihood of a random individual matching the target individual in terms of genotyping results. These results suggest that our panel can effectively and reliably trace and identify individuals in WES sample tracing. In forensic statistics, the LR method [28] is commonly used to assess the strength of the evidence provided by genetic analysis. A higher LR value lends greater support to the prosecution's hypothesis, supporting the notion that the person of interest and the suspect are the same individual. In the WES data application, the LR values strongly support the three WES samples corresponding to the child, father, and mother.
These results signify that our panel has an extremely low likelihood of erroneously identifying samples in clinical applications. Although the MP of 22 cSNPs did not reach the level of STRs, this multiplex system provides an efficient and straightforward method for sample identity and tracking of WES samples. The multiplex panel data can be used for comparison with the corresponding cSNP genotypes in the WES data. During WES, it is possible to concurrently detect 22 cSNPs in the same sample using the capillary electrophoresis (CE) platform. On completion of the sequencing process, the genotyping results for the 22 cSNPs from both WES and the CE platform were compared to confirm the identity of the sample. This streamlined procedure saves time and is cost-effective, thereby eliminating the need for repetitive sequencing. These 22 cSNPs can be used for sample tracking throughout the WES process. Further research is needed to establish a multiplex detection system based on these 22 cSNPs and optimize the corresponding multiplex system. Additionally, more cSNP markers should be selected to improve the DP of the system. The CPI of the 22 cSNPs was lower than 10,000, because of the lower polymorphism compared with STRs. Therefore, a system that includes a greater number of multi-allelic cSNPs should be established to meet these needs. The preliminary results of the extraction method for WES data suggest that this approach can efficiently extract cSNP genotype results and perform comparisons.
In the future, we plan to develop software capable of directly, rapidly, and accurately extracting the genotyping data of all cSNP loci included in the established personal identification system, from WES data. This will enable personal identification and identity confirmation in WES samples as well as paternity tests in trio samples. Simultaneously, using the cSNP panel and WES data cSNP genotyping extraction software, we intend to develop a novel quality control framework for WES sample tracking. Ultimately, this will facilitate sample tracking throughout the WES testing process.
Conclusions
We successfully established a multiplex panel comprising 22 cSNPs based on SNaPshot technology and applied it to personal identification and paternity testing. This multiplex system not only enhances tracking of WES samples, but also increases the overall reliability of WES detection.
Availability of data and materials
All data generated or analyzed in this study are included in the article and supplementary material.
Abbreviations
- BLAST:
-
Basic local alignment search tool
- CDP:
-
Cumulative discrimination power
- CODIS:
-
Combined DNA Index System
- CPI:
-
Combined paternity index
- cSNP:
-
Coding single-nucleotide polymorphism
- DP:
-
Discrimination power
- EXO I:
-
Exonuclease I
- HWE:
-
Hardy–weinberg equilibrium
- LR:
-
Likelihood ratio
- MAF:
-
Minor allele frequency
- MP:
-
Match probability
- NGS:
-
Next-generation sequencing
- PAGE:
-
Polyacrylamide gel electrophoresis
- PE:
-
Probability of exclusion
- PI:
-
Paternity index
- PIC:
-
Polymorphism information content
- SAP:
-
Shrimp alkaline phosphatase
- SBE:
-
Single-base extension
- TPI:
-
Typical paternity index
- WES:
-
Whole-exome sequencing
References
Jung J, et al. Ultra-rare renal diseases diagnosed with whole-exome sequencing: Utility in diagnosis and management. BMC Med Genomics. 2021;14(1):177.
Kmoch S, Zeman J. Modern methods in diagnostics and research of molecular bases of rare diseases. Cas Lek Cesk. 2018;157(3):133–6.
Choy KW. Next-Generation Sequencing to Diagnose Suspected Genetic Disorders. N Engl J Med. 2019;380(2):200–1.
Lanillos J, et al. Clinical pharmacogenetic analysis in 5,001 individuals with diagnostic Exome Sequencing data. NPJ Genom Med. 2022;7(1):12.
Engelbrecht C, et al. Clinical Utility of Whole Exome Sequencing and Targeted Panels for the Identification of Inborn Errors of Immunity in a Resource-Constrained Setting. Front Immunol. 2021;12: 665621.
Hu H, et al. Evaluating information content of SNPs for sample-tagging in re-sequencing projects. Sci Rep. 2015;5:10247.
Sehn JK, et al. Occult Specimen Contamination in Routine Clinical Next-Generation Sequencing Testing. Am J Clin Pathol. 2015;144(4):667–74.
Pfeifer JD, Liu J. Rate of occult specimen provenance complications in routine clinical practice. Am J Clin Pathol. 2013;139(1):93–100.
Rehm HL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15(9):733–47.
Matthijs G, et al. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet. 2016;24(1):2–5.
Aziz N, et al. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch Pathol Lab Med. 2015;139(4):481–93.
Hume S, et al. CCMG practice guideline: laboratory guidelines for next-generation sequencing. J Med Genet. 2019;56(12):792–800.
Seaby EG, Pengelly RJ, Ennis S. Exome sequencing explained: a practical guide to its clinical application. Brief Funct Genomics. 2016;15(5):374–84.
Deignan JL, et al. Points to consider when assessing relationships (or suspecting misattributed relationships) during family-based clinical genomic testing: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2020;22(8):1285–7.
Bunnell M, Esfandiari N. Preimplantation genetic testing as a component of root cause analysis of errors and reassignment of embryos in IVF. Reprod Biomed Online. 2020;41(6):975–7.
Flanagan SP, Jones AG. The future of parentage analysis: From microsatellites to SNPs and beyond. Mol Ecol. 2019;28(3):544–67.
Mousavi N, et al. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res. 2019;47(15): e90.
Jiang R, et al. Genome-wide evaluation of the public SNP databases. Pharmacogenomics. 2003;4(6):779–89.
King JL, et al. Increasing the discrimination power of ancestry- and identity-informative SNP loci within the ForenSeq™ DNA Signature Prep Kit. Forensic Sci Int Genet. 2018;36:60–76.
Javed N, et al. Detecting sample swaps in diverse NGS data types using linkage disequilibrium. Nat Commun. 2020;11(1):3697.
Eno C, et al. Misattributed parentage as an unanticipated finding during exome/genome sequencing: current clinical laboratory practices and an opportunity for standardization. Genet Med. 2019;21(4):861–6.
Pengelly RJ, et al. A SNP profiling panel for sample tracking in whole-exome sequencing studies. Genome Med. 2013;5(9):89.
Helsmoortel C, Kooy RF, Vandeweyer G. Multiplexed High Resolution Melting Assay for Versatile Sample Tracking in a Diagnostic and Research Setting. J Mol Diagn. 2016;18(1):32–8.
Di Pietro F, et al. Genomic DNA extraction from whole blood stored from 15- to 30-years at -20 °C by rapid phenol-chloroform protocol: a useful tool for genetic epidemiology studies. Mol Cell Probes. 2011;25(1):44–8.
Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–93.
Excoffier L. and H.E. Lischer. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–7.
Caliebe A, et al. Likelihood ratio and posterior odds in forensic genetics: Two sides of the same coin. Forensic Sci Int Genet. 2017;28:203–10.
Xiao Y, et al. Problems and reflection on quality management of clinical molecular diagnosis. Chin J Lab Med. 2018;41(2):85–7.
Chen H, et al. Current situation and future prospect of regional medical laboratory center. Int J Lab Med. 2021;42(12):1409–13.
Kavlick MF. Development of a universal internal positive control. Biotechniques. 2018;65(5):275–80.
Alsafiah HM, et al. An evaluation of the SureID 23comp Human Identification Kit for kinship testing. Sci Rep. 2019;9(1):16859.
Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Kim JJ, et al. Development of SNP-based human identification system. Int J Legal Med. 2010;124(2):125–31.
Kim SM, et al. Identification of Korean-specific SNP markers from whole-exome sequencing data. Int J Legal Med. 2016;130(3):669–77.
Du Y, et al. A SNP panel and online tool for checking genotype concordance through comparing QR codes. PLoS ONE. 2017;12(9): e0182438.
Lee S, et al. NGSCheckMate: software for validating sample identity in next-generation sequencing studies within and across data types. Nucleic Acids Res. 2017;45(11): e103.
Westphal M, et al. SMaSH: Sample matching using SNPs in humans. BMC Genomics. 2019;20(Suppl 12):1001.
Acknowledgements
We are grateful to Dr. Qin Su for her assistance with genotype extraction and analysis of WES data.
Funding
This work was supported by the Sichuan Science and Technology Program (grant number: 2023NSFSC1482).
Author information
Authors and Affiliations
Contributions
YH and YYX performed the experiments and data analysis, and drafted the manuscript. QSQ, JMX, and LZ contributed to the analysis, editing, and proofreading of the manuscript. LW and WBL conceived and supervised the project and designed the study. All the authors have read and approved the final version of the manuscript. Yong Huang and Yuanyuan Xiao contributed equally to this work.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee at West China Second University Hospital, Sichuan University.
Written informed consent was obtained from all sample donors.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, Y., Xiao, Y., Qu, S. et al. Development of a coding SNP panel for tracking the origin of whole-exome sequencing samples. BMC Genomics 25, 142 (2024). https://doi.org/10.1186/s12864-024-10052-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10052-4