
Abstract
Background
The lesser grain borer (Rhyzopertha dominica), a worldwide primary pest of stored grain, causes serious economic losses and threatens stored food safety. R. dominica can respond to changes in temperature, especially the adaptability to heat. In this study, transcriptome analysis of R. dominica exposed to different temperatures was performed to elucidate differences in gene expression and the underling molecular mechanism.
Results
Isoform-sequencing generated 17,721,200 raw reads and yielded 20,416 full-length transcripts. A total of 18,880 (92.48%) transcripts were annotated. We extracted RNA from R. dominica reared at 5 °C (cold stress), 15 °C (cold stress), 27 °C (ambient temperature) and 40 °C (heat stress) for RNA-seq. Compared to those of control insects reared at 27 °C, 119, 342, and 875 differentially expressed genes (DEGs) were identified at 5 °C, 15 °C, and 40 °C, respectively. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed that pathways associated with “fatty acid metabolism”, “fatty acid biosynthesis”, “AMPK signaling pathway”, “neuroactive ligand receptor interaction”, and “longevity regulating pathway-multiple species” were significantly enriched. The functional annotation revealed that the genes encoding heat shock proteins (HSPs), fatty acid synthase (FAS), phospholipases (PLA), trehalose transporter (TPST), trehalose 6-phosphate synthase (TPS), and vitellogenin (Vg) were most likely involved in temperature regulation, which was also validated by RT-qPCR. Seven candidate genes (rdhsp1, rdfas1, rdpla1, rdtpst1, rdtps1, rdvg1, and rdP450) were silenced in the RNA interference (RNAi) assay. RNAi of each candidate gene suggested that inhibiting rdtps1 expression significantly decreased the trehalose level and survival rate of R. dominica at 40 °C.
Conclusions
These results indicated that trehalose contributes to the high temperature resistance of R. dominica. Our study elucidates the molecular mechanisms underlying heat tolerance and provides a potential target for the pest management in R. dominica.
Similar content being viewed by others


Background
The lesser grain borer, Rhyzopertha dominica (Fabricius) (Coleoptera: Bostrychidae), is one of the most devastating pests of stored grain. R. dominica displays remarkable adaptability, attributed to its resistance to desiccation, heat, and hypoxia [1,2,3,4]. Both the adults and larvae of R. dominica cause damage inside the grain, resulting in a decrease in grain quality [5,6,7]. In addition, the frass produced by insects can also promote populations of secondary stored product pests and microorganisms [8]. The grains contaminated by R. dominica emit a severe amount of off-odor, rendering them unfit for human consumption [9]. Therefore, R. dominica causes serious economic losses and constitutes a grave threat to food safety.
Temperature is one of the most prominent abiotic factors influencing the ecological distribution, life cycle, growth, development, reproduction, phylogenetic evolution, and immune response of organisms [10, 11]. However, insects have various mechanisms to adapt to changes in ambient temperature [12,13,14,15]. Heat shock proteins (HSPs) are critical factors in the response to temperature stress. For example, the expression of HSP genes significantly differed under temperature stress in Glyphodes pyloalis and Callosobruchus chinensis [15, 16]. In Galeruca daurica, the genes encoding cuticle protein, P450, clock protein, fatty acid synthase, and fatty acyl-CoA reductase were upregulated under cold stress [17]. In addition, regulating the level of trehalose is one of the mechanisms by which insects counteract temperature changes [18, 19]. Trehalose, a ubiquitous nonreducing disaccharide, acts as a metabolic substance in the stress response of many organisms. Its levels change in response to external conditions such as heat, cold, desiccation and anoxia [20,21,22,23,24,25]. Many insect species, including Monochamus alternatus and Helicoverpa armigera, use trehalose as a protective agent to enhance cold resistance [26, 27]. Studies have shown that trehalose regulates the viscosity of the cytoplasm during thermal stimulation to maintain cytoplasmic homeostasis and stabilize the reaction rate in cells [28, 29]. Trehalose is predominantly synthesized via the trehalose-6-phosphate synthase (TPS) and trehalose-6-phosphate phosphatase (TPP) pathways in insects. In this pathway, uridine diphosphate glucose (UDP-glucose) and 6-phosphate glucose can generate 6-phosphate trehalose under the action of TPS, while 6-phosphate trehalose can detach the phosphate group and ultimately produce trehalose under the action of TPP [30]. In addition, trehalose can be synthesized from some insect species by TPS alone. This type of TPS has an N-terminal TPS (glycosyltransferase family 20) domain that catalyzes the production of trehalose-6-phosphate using glucose 6-phosphate and UDP-glucose as substrates, whereas the C-terminal TPS (trehalose-phosphatases) domain then dephosphorylates trehalose-6-phosphate, generating trehalose and orthophosphate [31, 32]. Therefore, TPSs play a vital role in trehalose synthesis and in the regulatory pathway of the temperature response.
The rapid development of sequencing technology has made it possible to analyze differentially expressed genes (DEGs) that may be involved in temperature response mechanisms. Given that the genome sequence was unavailable during our study, full-length isoform sequencing (ISO-Seq) facilitated the profiling of the genome-independent transcriptome. Short-read RNA-Seq using high-throughput next-generation sequencing (NGS) technology enables the analysis of DEGs [33]. RNA interference (RNAi) is a widely used technology in which target genes are silenced through double-stranded DNA (dsDNA). RNAi is commonly employed in gene function research and a potential new method for pest control [34, 35]. In this study, we constructed a full-length transcriptomic database and identified DEGs related to temperature regulation in R. dominica. The DEGs were further validated using RT-qPCR and RNAi. Taken together, our findings demonstrated that the accumulation of trehalose contributes to resistance to high temperature stress, and the trehalose 6-phosphate synthase gene rdtps1 positively regulates trehalose levels during thermal acclimation in R. dominica.
Results
Rhyzopertha dominica adapted to a wide range of temperature conditions
To determine the mortality rate of R. dominica at different temperatures, we reared the insects at a suitable temperature (27 °C), low temperatures (5 and 15 °C) and a high temperature (40 °C) for 7 days. R. dominica kept at 27 °C were used as the control group (Rd-ck), whereas R. dominica kept at 5 °C, 15 °C, and 40 °C were the treatment groups (Rd-5, Rd-15, and Rd-40). The mortality rates of Rd-ck, Rd-5, and Rd-15 were less than 10% after seven days, while Rd-ck, the group reared at ambient temperature for R. dominica growth and development, had the highest mortality rate among these three groups. Although 40 °C is a relatively high temperature for most stored grain pests and the mortality rate of Rd-40 reached 82% on day 7, the mortality of the pests did not exceed 10% until day 3 (Fig. 1A). In addition, we measured the weight of the adults before and after temperature treatment (Fig. 1B). The average weight of Rd-40 decreased after 7 days, while that of the Rd-ck and Rd-5 did not significantly change. Since the mortality rate at different temperatures did not change significantly during the first 48 h, the time point was used for transcriptome sequencing.

Rhyzopertha dominica adapted to a wide range of temperature conditions. (A) Mortality of R. dominica at different temperatures. (B) The average weight of R. dominica reared at different temperatures for 7 days. Rd-ck, Rd-5, Rd-15, and Rd-40 indicate R. dominica reared at 27 °C, 5 °C, 15 °C, and 40 °C, respectively. Different letters (a to d) above the bars indicate a significant difference (P < 0.05) by the Turky test
Full-length transcriptome sequencing provides a reference for differentially expressed genes analysis
Since there was no available genome sequence during this study, the full-length transcriptome was sequenced to obtain full-length transcripts as reference sequences. Isoform sequencing generated 17,721,200 raw reads. After clustering, polishing, and removing redundant isoforms, we obtained 20,416 full-length transcripts. To characterize the functional information of the full-length transcripts, we aligned the transcript sequences with public databases, including Swiss-Prot, NCBI nucleotide sequences (NT), NCBI non-redundant protein sequences (NR), Clusters of orthologous groups for Eukaryote (KOG), Kyoto Encyclopedia of Genes and Genomes (KEGG) KEGG, and Gene Ontology (GO). In total, 18,880 (92.48%) of the non-redundant high-quality transcripts (also known as unigenes) could be functionally annotated using Swiss-Prot (16,075), NT (6,795), NR (18,746), KOG (14,663), KEGG (13,201), and GO (11,795) (Fig. S1A). The four databases with the most annotations that provided 18,824 transcript annotations were NR, Swiss-Prot, KOG, and KEGG. Some of the transcripts were mapped to more than one database, and 11,510 transcripts were simultaneously mapped to the NR, Swiss-Prot, KOG, and KEGG databeases (Fig. S1B). According to the KEGG Orthology database (KO) of KEGG, 2,135 (14.88%), 2,137 (14.90), 2,460 (17.15%), 3,687 (25.70%), and 3,927 (27.37%) of the isoforms were associated with cellular processes (A), environmental information processing (B), genetic information processing (C), metabolism (D), and organismal systems (E), respectively (Fig. S1C). Additionally, GO annotation incorporates three categories, namely, biological process (19,619 isoforms, 45.32%), cellular component (14,338 isoforms, 33.12%), and molecular function (9,330 isoforms, 21.56%) (Fig. 2). To further profile the functional annotations of all the isoforms, we annotated 14,663 transcripts with 16,550 KOG functions in 4 categories and 25 subcategories (Fig. S1D). The four categories were “cellular processes and signaling” (6,468 isoforms, 39.08%), “information storage and processing” (3,256 isoforms, 19.67%), “metabolism” (3,465 isoforms, 20.94%), and “poorly characterized” (3,361 isoforms, 20.31%).

GO classification comprised biological process, molecular function and cellular component of all identified isoforms from the reference transcriptome of Rhyzopertha dominica
Identification and functional profiles of differentially expressed genes under temperature stress
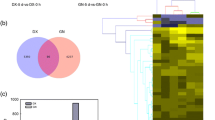
To characterize the functional genes of R. dominica in response to temperature changes, Illumina libraries of Rd-ck, Rd-5, Rd-15, and Rd-40 were generated (Table 1). Compared to Rd-ck, there were 119, 342, and 875 DEGs identified in Rd-5, Rd-15, and Rd-40, respectively. Thirty-six genes were up-regulated and 83 genes were down-regulated at 5 °C (Fig. 3A). 107 genes were up-regulated and 235 genes were down-regulated at 15 °C (Fig. 3B). 264 genes were up-regulated and 611 genes were down-regulated at 40 °C (Fig. 3C). A total of 1142 DEGs were identified and used for heatmap analysis (Fig. 3D). The results demonstrated significant intergroup differences, particularly with respect to the different expression patterns observed for Rd-40.

Differentially expressed genes (DEGs) of Rhyzopertha dominica exposed to different temperatures. (A-C) DEGs of Rd-5, Rd-15, and Rd-40 compared to Rd-ck. The x-axis represents the log2 (fold-change) values under the mean normalized expression of all isoforms (y-axis). Red dots indicate the upregulated DEGs, and green dots represent the downregulated DEGs. (D) Heatmap of DEGs in Rd-5, Rd-15, and Rd-40 compared to Rd-ck. Rd-ck, Rd-5, Rd-15 and Rd-40 indicate R. dominica reared at 27 °C, 5 °C, 15 °C, and 40 °C, respectively
Metabolic pathways were systematically analyzed to construct a network of gene expression information via KEGG analysis [36]. The top 20 most reliable enrichment pathways are shown in Fig. 4. Under cold stress, 124 and 179 pathways were enriched in Rd-5 and Rd-15, respectively. The primary components were “fatty acid metabolism”, “fatty acid biosynthesis”, “AMPK signaling pathway”, “insulin signaling pathway”, and “starch and sucrose metabolism” (Fig. 4A-B). Under heat stress, “neuroactive ligand-receptor interaction”, “longevity regulating pathway-multiple species”, “influenza A”, “spliceosome”, and “fatty acid metabolism” were the primary components (Fig. 4C). Thirty-four genes were coregulated among Rd-5, Rd-15, and Rd-40, which accounted for 3% of the total DEGs (Fig. S2). These genes included mainly the genes encoding fatty acid synthase (FAS) and vitellogenin (Vg), and their expression levels were inhibited under temperature stress. Genes with high expression under 40 °C conditions were primarily associated with heat shock proteins (HSPs), ATP-binding cassettes, and phospholipases, while genes with decreased expression were mainly related to serine protease activity regulation. A total of 117 genes (7.6%) were coregulated between Rd-15 and Rd-40, including genes related to the trehalose pathway, vitellogenin, and cytochrome P450 monooxygenase (P450s). Forty-six genes (1.4%) were coregulated between Rd-5 and Rd-40, including scavenger receptors and uncharacterized proteins (Fig. S2).

The top 20 significantly enriched KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways among Differentially expressed genes (DEGs) in A (5 °C vs. 27 °C), B (15 °C vs. 27 °C) and C (40 °C vs. 27 °C). The X axis indicates the top 20 terms enriched in KEGG, and the Y axis represents the number of DEGs annotated under that term
Validation of RNA-seq gene expression levels
To validate the RNA-Seq results, a subset of DEGs was randomly selected for RT-qPCR. The correlation coefficient R2 was 0.8782, as determined by linear regressions of fold change differences, which indicated that the RT-qPCR and RNA-seq results were consistent (Fig. S3). The RNA-Seq results suggested that certain genes, including those coding for HSPs, FAS, phospholipases, trehalose transporter (TPST), trehalose-6-phosphate synthase (TPS), and Vg, were differentially expressed under temperature stress. Hence, we evaluated the expression levels of two HSPs (rdhsp1, rdhsp2), two fatty acid synthesis (rdfas1, rdfas2), two phospholipases (rdpla1, rdpla2), one trehalose transporter (rdtpst1), one trehalose-6-phosphate synthase (rdtps1), and two vitellogenin (rdvg1, rdvg2) using RT-qPCR analysis with rps3 (a ribosomal protein S3 gene) as a reference gene. The expression of the genes encoding HSPs, phospholipases, and trehalose-6-phosphate synthase increased significantly under heat stress. Specifically, FAS and Vg were down-regulated under both heat and cold stress (Fig. 5). These results confirmed the different regulatory mechanisms that the lesser grain borers undergo when exposed to low and high temperature conditions.

Expression of representative selected genes was determined in various samples of Rhyzopertha dominica. Different letters (a to c) above the bars indicate a significant difference (P < 0.05) by the Tukey test. The expression level of each gene in control Rd-ck was mathematically designated as 1 in each reaction
Silencing rdtps1 significantly reduced the survival rate of Rhyzopertha dominica at high temperature
To further investigate the function of the DEGs, RNAi assay was performed on seven candidate genes (rdhsp1, rdfas1, rdpla1, rdtpst1, rdtps1, rdvg1, and a P450 encoding gene) at different temperatures (Fig. 6A). The results indicated that interference with the expression of rdtps1 had a significant impact on pest mortality under high temperature conditions. Then, we determined the silencing efficiency of RNAi by examining the relative expression level of rdtps1 via RT-qPCR. The results showed that compared to the negative control (dsGFP), the dsTPS treatment effectively reduced the rdtps1 expression even with the elevated rdtps1 expression at 40 °C (Fig. 6B). Since rdtps1 is a predicted trehalose synthetase, we measured the trehalose level in each sample. Among the dsGFP controls, the trehalose level in the Rd-40 increased significantly compared with Rd-ck, while that of Rd-5 and Rd-15 did not change significantly. When the insects were reared at 40 °C with dsTPS, the trehalose level decreased significantly (Fig. 6C). These results showed that Rdtps1 is an important trehalose synthase that responds to high temperature stress. (Fig. 6D). However, we did not observe a reduced survival rate in the other dsTPS treated groups at normal or low temperature (Fig. 6D). These results demonstrated that rdtps1 responded to heat stress and expression level of this gene was crucial for pest survival at high temperature.

The rdtps-1 Silencing impact on the survival rate of Rhyzopertha dominica at high temperature. (A) The effect of silencing 7 candidate genes on the survival rates after 7 days of temperature treatments. (B) Effect of RNAi-mediated silencing on rdtps1 expression levels determined in various samples by using RT-qPCR. (C) The trehalose content of each sample was tested by anthrone colorimetric method. The insects were fed on dsGFP or dsTPS for two days and then raised at different temperatures for two days. (D) Survival rates after 7 days of temperature treatments were recorded and calculated. Different letters (a to d) above the bars indicate a significant difference (P < 0.05) by the Turky test
Discussion
Rhyzopertha dominica is one of the most temperature-adaptive insects among stored product pests. Transcriptome analysis, as an important means of studying the molecular mechanisms by which pests resist temperature stress, has also been applied multiple times in stored grain pests, such as in the studies of Tribolium castaneum [13] and C. chinensis [15]. In this study, our transcriptome analysis revealed that R. dominica regulated numerous genes to respond to temperature stress. The average length of the transcripts was 2,806 bp, which indicated that the data were good enough to represent full-length transcripts [37]. Using the RT-qPCR analysis and RNAi assay, we characterized thatthe trehalose-phosphate synthase encoding gene rdtps1 positively regulates the high temperature resistance of R. dominica. Trehalose is an important carbohydrate in insect hemolymph, and trehalose-phosphate synthase (TPS) is responsible for the synthesis of trehalose [38]. Thermal acclimation led to an increase in trehalose level, suggesting the involvement of trehalose in temperature response regulation in R. dominica. Furthermore, rdtps1 was highly expressed at elevated temperature, indicating its role in trehalose synthesis. When the gene expression of rdtps1 was repressed by RNAi, both the trehalose content and survival rate decreased significantly. This highlights the importance of rdtps1 as an important regulatory factor for R. dominica in response to high temperature stress. Additionally, under moderate or low temperature stress conditions, interfering with the expression of rdtps1 did not alter trehalose levels and the survival rate of pests, indicating that R. dominica have different regulatory pathways in response to high and low temperature stress.
In the RNAi assay, we silenced seven DEGs, including rdhsp1, rdfas1, rdpla1, rdtpst1, rdtps1, rdvg1, and a P450 encoding gene (rdP450). However, silencing only rdtps1 resulted in increased insect mortality. These findings suggested that rdtps1 is more important for the pest heat resistance compared to the other six genes. Alternatively, it is also possible that the function of these genes can be compensated by other metabolic pathways. Further validation is required to elucidate the functions of the other genes. When we silenced rdtps1, we observed a 50% increase in mortality, although trehalose level of the insects only reduced 10%. This observation suggested that the heat resistance of R. dominica requires a high level of trehalose. However, we cannot exclude the possibility that reduced rdtpst1 gene expression has a systemic impact on the metabolism of the pest under high temperature conditions. Further research is needed to determine the effect of reduced gene expression. In addition, transcriptome sequencing revealed that approximately 30 other genes were putative TPS genes. Although these predicted TPS genes did not exhibit upregulated expression under high-temperature conditions, they may compensate for the significant reduction in rdtps1 expression and maintenance of the trehalose levels in the pests. Future studies of these genes are also necessary.
At 40 °C, the average weight of the pests significantly decreased 31% after being fed for 7 days. This may be due to the intensified metabolism caused by high temperature, which results in the consumption of fat by pests. For instance, the gene expression level of the putative helicase (B3-1_1-10k_transcript/13,726) was upregulated at 40 °C in the transcriptome data. The B3-1_1-10k_transcript/13,726, which catalyzes the unwinding of double-stranded nucleic acids using energy from ATP, is in the spliceosome pathway (ko03040). Aldehyde dehydrogenase (B3-1_1-10k_transcript/14,337) participants in fatty acid degradation and produces a large amount of ATP (ko00071). Moreover, the expression of fatty acid synthase (ko00061) and acyl-CoA delta (11) desaturase (ko01212), which are responsible for synthesizing fat, was down-regulated, leading to the inhibition of fat synthesis. Consequently, the average weight of pests decreased significantly under high temperature conditions. Although low temperature also resulted in the down-regulation of fatty acid synthase expression, it caused pests to become unconscious and suppressed metabolism, leading to a reduction in fat consumption.
Previous studies have shown that several other genes are involved in temperature regulation, although little is known about this topic in R. dominica. Numerous studies have indicated that the expression of HSPs significantly changes under temperature stress to ensure the configuration stability of proteins and to repair DNA damage [39,40,41,42,43,44,45,46]. As such, HSPs are also vital for the temperature response of insects. In our study, the expression of HSPs, including HSP70, HSP90, and small HSPs, was upregulated in Rd-40 (40 °C), which indicated that the HSPs of R. dominica play an important role in the response to high temperature. In addition to HSPs, heat stress also led to high expression of phospholipases encoding genes rdpla1 and rdpla2 in R. dominica, and phospholipases are essential enzymes for cell membrane stability [47]. In Apolygus lucorum, phospholipase regulates trehalose in 20-hydroxyecdysone-induced fecundity [48], and our transcriptome analysis and RT-qPCR results showed that the coding genes of vitellogenin (rdvg1, rdvg2) were also significantly downregulated under temperature stress conditions. Vitellogenin, the precursor of vitellin, is an indicator of oviposition ability and reproduction in most oviparous species including insects [49,50,51,52]. Vitellogenin is stored in eggs to provide a source of nutrients for the early development of embryos and larvae. We hypothesized that reducing the expression of vitellogenin by pests is also a strategy for helping the lesser grain borers cope with temperature stress, reducing energy consumption while preventing offspring from facing adverse environmental conditions. These results indicate that the lesser grain borers employed multiple strategies to respond to temperature stress.
Conclusions
We thoroughly explored the regulatory genes involved in the response of lesser grain borers to temperature changes through transcriptome sequencing. The results showed that the gene expression profiles of insects reared at high temperature changed dramatically compared to those reared at ambient temperature. The metabolic pathways co-regulated by temperature stresses (5 °C, 15 and 40 °C) were mainly enriched in “fatty acid metabolism”, “fatty acid biosynthesis”, “starch and sucrose metabolism”, “neuroactive ligand-receptor interaction” and “protein digestion and absorption”. The DEGs under cold stress ((5 °C, 15 °C) mainly encoded fatty acid synthase and protein kinases. The DEGs under heat stress (40 °C) were associated primarily with heat shock proteins (HSPs), ATP-binding cassettes, and phospholipases. Thirty-four genes, which are mainly encoding genes of FASs and Vgs, were co-regulated under both cold and heat stress. RNAi assay of seven candidate genes (rdhsp1, rdfas1, rdpla1, rdtpst1, rdtps1, rdvg1, and rdP450) revealed that the coding gene of trehalose synthase (rdtps1) played a crucial role in regulating the heat resistance of R. dominica. This study provides insights into the molecular biology of R. dominica and contributes to the identification of a novel target gene for heat resistant pest control.
Methods
Insects and samples
Rhyzopertha dominica were collected from the granary of Guangzhou (23.13 N 117.19 E) in southern China. The insects were maintained in the laboratory and reared on wheat at 27 ± 0.1 °C, 65 ± 0.5% relative humidity (RH) in an incubator (Memmert, Germany) for at least three years.
We selected two-week-old adults for the assays. All the individuals were maintained at the same relative humidity (65% RH). We set four temperature conditions, including 5 °C, 15 °C, 27 °C, and 40 °C. R. dominica kept at 27 °C was used as the control group (Rd-ck), whereas R. dominica kept at 5 °C, 15 °C, and 40 °C were the treatment groups (Rd-5, Rd-15, and Rd-40).
Laboratory bioassays
For the mortality assay, 30 individuals for each treatment of Rd-ck, Rd-5, Rd-15, and Rd-40 were reared for 7 days. The number of dead insects was recorded each day and used to calculate the mortality rate of R. dominica. The experiments were repeated five independent times.
To measure the effects of different temperatures on the average weight over a period of 7 days, we weighed 30 individuals of R. dominica for each temperature condition. Each individual was fed a clean grain of wheat in a separate well on a 24-well plate for 7 days. The individuals’ weight was measured by an analytical balance (ME204E, Mettler Toledo, China) to assess weight change. The experiments were repeated three independent times.
Sample preparation
For ISO-Seq, 10 adults of the Rd-ck were selected, and then immediately frozen in liquid nitrogen. For RNA-Seq, 3 samples of each treatment group, Rd-ck, Rd-5, Rd-15, and Rd-40, were processed, and each sample included 10 adults. The insects were reared for 2 days and then frozen in liquid nitrogen. Total RNA was extracted using an RNeasy Plus Mini Kit (Qiagen, Germany), following the manufacturer’s instructions. RNA degradation and contamination were assessed on agarose gels. To determine the concentration and purity of the total RNA, we added 1 µL of RNA to a NanoDrop 2000 Spectrophotometer (Thermo Fisher, USA). RNA integrity was evaluated using an Agilent Technologies 2100 Bioanalyzer (Agilent, USA).
Library construction, sequencing, and data quality control for PacBio full-length ISO-Seq
Third-generation Isoform-sequencing sequencing via PacBio can be used to obtain high-quality single full-length transcriptome sequences without assembly. The average read length is 8–15 kb, making it easy to cross the full transcript. It can accurately identify isomers and perform precise analysis of variable splicing, fusion genes, homologous genes, superfamily genes, and allele expression. We used magnetic beads with an oligo(dT) primer to enrich the mRNA of the samples. Then, full-length cDNA was synthesized using a SMARTer™ PCR cDNA Synthesis Kit (Clontech, USA). The cDNA was amplified and enriched by PCR. To screen full-length cDNA fragments, we used BluePippin (Sage Science, USA) and constructed libraries based on the length of the insert fragments. Sequencing was performed on a PacBio Sequel (Pacific Biosciences, USA), and the raw data output was 20 Gb.
To obtain full-length transcripts, the raw data were filtered to remove linker sequences, fragments < 300 bp, and sequences with an accuracy < 0.9. Then, the raw reads with the number of full passes ≥ 1 and sequence accuracy > 0.9 were screened to obtain the raw reads. The transcripts were subsequently obtained through classification, clustering, and correction processes. Since there may be many repeated transcripts, we clustered the transcripts using an isoform-level clustering (ICE) algorithm. Similar sequences were clustered into a cluster, and each cluster contained a consistent transcript. Combined with non-full-length sequences, the polish program was used to correct the raw reads in each cluster. We obtained high-quality transcripts (HQ, high-quality isoforms) and low-quality transcripts (LQ, low-quality) transcripts with an accuracy > 99%. The data from raw RNA-Seq (Illumina NGS) were used to correct LQ full-length transcript isoforms using proofreading software. For the full-length transcriptome, CD-HIT software was used to capture de-redundant sequences in the high-quality full-length transcriptome (parameters: C = 0.99, T = 6, G = 1, U = 10, S = 0.999, D = 40, P = 1) [53], resulting in a non-redundant high-quality transcriptome, which is the full-length transcripts. The transcriptome raw data were deposited to the NCBI SRA database (accession number: PRJNA733562).
Annotation of transcripts
Functional annotation was performed via comparison with public databases. The NCBI nucleotide sequences (NT) is the nucleotide database of NCBI (https://www.ncbi.nlm.nih.gov/), which includes the nucleotide sequences in GenBank, the European Molecular Biology Laboratory (EMBL), and the DNA Data Bank of Japan (DDBJ). The NCBI non-redundant protein sequences (NR) is the protein sequence database of NCBI. Swiss-Prot (http://www.ebi.ac.uk/uniprot/) is a manually annotated and reviewed protein sequence database of protein sequences sorted and studied by biologists. Gene Ontology (GO) (http://geneontology.org/) is an internationally standardized gene function classification system that provides a set of dynamically updated controlled terms to comprehensively describe the attributes of genes and gene products in organisms. The Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.genome.jp/kegg/) is a database that systematically analyses metabolic pathways and functions of gene products and compounds in cells. Clusters of orthologous groups for Eukaryote (KOG) (https://ftp.ncbi.nih.gov/pub/COG/KOG/) possesses clusters of orthologous groups of proteins in eukaryotes according to their phylogenetic classification.
Library construction, sequencing, and data quality control for RNA-Seq
For NGS, libraries were generated using the VAHTS Stranded mRNA-seq Library Prep Kit for Illumina V2 (Vazyme Biotech, China). The libraries were subsequently sequenced on an Illumina NovaSeq platform to generate 150-bp paired-end reads. The raw data of RNA-Seq were first processed through primary quality control. Clean reads were obtained by removing read pairs. The data filtering procedure was as follows: (1) When N > 3 in any end sequencing read, this pair of paired reads was removed; (2) when the proportion of bases with quality value < 5 was > = 20% at any end, this pair of paired reads was removed; (3) when the adapter sequence was removed, it was necessary to match at least 8 bp. The RNA-Seq transcriptome raw reads were deposited to the NCBI SRA database (accession number: PRJNA734384).
Differentially expressed genes analysis
Using the full-length transcript (ISO-Seq data) as a reference sequence, we mapped the clean reads (RNA-Seq data) of each sample to the reference sequence by RSEM [54]. In this process, the mismatch parameter of Bowtie2 [55] is 0. RSEM was used to analyzlyse the comparison results obtained with Bowtie software, and the read counts of each gene were further obtained for each sample. Next, we converted the read count into FPKM (the expected number of fragments per kilobase of transcript sequence per million base pairs sequenced) to analyze the expression level of each gene. To analyze the DEGs in each sample, we used the package edgeR [56] of RStudio [57], and differentially expressed P and FDR values were calculated. FDR is the corrected P value. The smaller the FDR value is the more significant the difference in gene expression. The screening conditions were as follows: FDR < 0.05 and log2|fold-change| > 1.
Reverse transcription quantitative real-time PCR (RT-qPCR)
HiScript® III RT SuperMix for qPCR (Vazyme Biotech, China) was used to synthesize cDNA. Reverse transcription quantitative real-time PCR (RT-qPCR) was performed using 2 ng of cDNA and AceQ® Universal SYBR® qPCR Master Mix (Vazyme Biotech, China) on a Bio-Rad CFX real-time PCR system. Each 20 µL reaction consisted of 10 µL of 2×ChamQ Universal SYBR qPCR Master Mix, 5 µL of 1:100 diluted cDNA, 0.4 µL for forward and reverse primers (10 µM), and 4.2 µL of RNase-free ddH2O. The RT-qPCR reaction cycle consisted of a denaturation step at 95 °C for 30 s, amplified with 40 cycles of 95 °C for 10 s, 60 °C for 30 s, and ending with a melting-curve step. 10 DEGs genes were selected and the relative levels of gene expression were determined using the threshold cycle 2−ΔΔCT method, with the ribosomal protein S3 gene (rps3) serving as the reference gene [58]. In this study, RT-qPCR primers were designed with Primer 5 (Table 2). Three technical replicates were used each time.
RNAi assay
A 486 bp fragment of the rdtps1 gene was selected for RNAi assay. Double-stranded RNA (dsRNA) was synthesized using the T7 RiboMAX™ Express RNAi System (P1700, Promega, USA). The primers dsTPS-F and dsTPS-R were used to synthesize the dsTPS. To eliminate any potential effects from experimental procedures or nanocarriers, a 428bp fragment of the enhanced green fluorescent protein gene (gfp) was separately constructed [59], and dsGFP-F and dsGFP-R were used to synthesize the negative control dsGFP (Table 2). The dsRNA was mixed with the nanocarrier at the recommended mass ratio of 1:1 in RNase-free water. Then, the dsRNA solution was added to wheat flour to make feed for the adults of R. dominica. Adults were fed on the feed with dsRNA at 27 °C for 2 days and then were incubated at 5, 15, 27 or 40 °C for 2 days. The silence efficiency was examined by RT-q-PCR of the primers was evaluated via RT-q-PCR using rps3 as the reference gene [58]. Three technical replicates were used each time. The trehalose content of each treatment was determined using 30 individuals by Trehalose Content Assay Kit (BC0355, Solarbio, China). Three technical replicates were used each time. After 7 days of treatment at different temperatures, 30 individuals were observed. and the survival rate was calculated. Five technical replicates were used each time.
Statistical analysis
The means and standard deviations of the experimental results were calculated using Excel. Tukey test was performed using the Statistical Package for the Social Sciences (SPSS) version 27.0 (IBM SPSS Statistics, Armonk, NY, USA).
Data availability
The sequencing reads are available from the Sequence Read Archive (SRA) under the BioProject accession numbers PRJNA733562 and PRJNA734384. For additional data and custom scripts, please contact the corresponding author.
Abbreviations
- DEGs:
-
Differentially expressed genes
- TPS:
-
Trehalose-6-phosphate synthase
- TPP:
-
Trehalose-6-phosphate phosphatase
- HSPs:
-
Heat shock proteins
- FAS:
-
Fatty acid synthase Vg:vitellogenin
- P450s:
-
Cytochrome P450 monooxygenase
References
Fields PG. The control of stored-product insects and mites with extreme temperatures. J Stored Prod Res. 1992;28(2):89–118. https://doi.org/10.1016/0022-474X(92)90018-L.
Beckett SJ, Morton R, Darby JA. The mortality of Rhyzopertha dominica (F.) (Coleoptera: Bostrychidae) and Sitophilus oryzae (L.) (Coleoptera: Curculionidae) at moderate temperatures. J Stored Prod Res. 1998;34(4):363–76. https://doi.org/10.1016/S0022-474X(98)00022-8.
Rahim M. Biological activity of azadirachtin-enriched neem kernel extracts against Rhyzopertha dominica (F.) (coleoptera: Bostrichidae) in stored wheat. J Stored Prod Res. 1998;34(2–3). https://doi.org/10.1016/S0022-474X(97)00058-1
Mbata GN, Phillips TW. Effects of temperature and exposure time on mortality of stored-product insects exposed to low pressure. J Econ Entomol. 2001;94(5):1302–7. https://doi.org/10.1603/0022-0493-94.5.1302.
Potter C. The biology and distribution of Rhizopertha dominica (Fab). Ecol Entomol. 1935;83(4):449–82. https://doi.org/10.1111/j.1365-2311.1935.tb02995.x
Edde PA. A review of the biology and control of Rhyzopertha dominica (F.) the lesser grain borer. J Stored Prod Res. 2012;48:1–18. https://doi.org/10.1016/j.jspr.2011.08.007.
Kavallieratos NG, Athanassiou CG, Arthur FH, Throne JE. Lesser grain borers, Rhyzopertha dominica, select rough rice kernels with cracked hulls for reproduction. J Insect Sci. 2012;12:38. https://doi.org/10.1673/031.012.3801.
Shah JA, Vendl T, Aulicky R, Stejskal V. Frass produced by the primary pest Rhyzopertha dominica supports the population growth of the secondary stored product pests Oryzaephilus surinamensis, Tribolium castaneum, and T. confusum. Bull Entomol Res. 2020;1–7. https://doi.org/10.1017/S0007485320000425
Hubert J, Stejskal V, Athanassiou CG, Throne JE. Health hazards associated with arthropod infestation of stored products. Annu Rev Entomol. 2018;63:553–73. https://doi.org/10.1146/annurev-ento-020117-043218.
Abdelghany AY, Awadalla SS, Abdel-Baky NF, El-Syrafi HA, Fields PG. Effect of high and low temperatures on the drugstore beetle (Coleoptera: Anobiidae). J Econ Entomol. 2010;103(5):1909–14. https://doi.org/10.1603/ec10054.
Colinet H, Sinclair BJ, Vernon P, Renault D. Insects in fluctuating thermal environments. Annu Rev Entomol. 2015;60:123–40. https://doi.org/10.1146/annurev-ento-010814-021017.
Ju RT, Xiao YY, Li B. Rapid cold hardening increases cold and chilling tolerances more than acclimation in the adults of the sycamore lace bug, Corythucha ciliata (say) (Hemiptera: Tingidae). J Insect Physiol. 2011;57(11):1577–82. https://doi.org/10.1016/j.jinsphys.2011.08.012.
Lü J, Huo M, Kang Y. Transcript-level analysis in combination with real-time PCR elucidates heat adaptation mechanism of Tribolium castaneum (Herbst) (Coleoptera: Tenebrionidae) Larvae. J Econ Entomol. 2019;112(6):2984–92. https://doi.org/10.1093/jee/toz239.
Wang B, Hao X, Xu J, Ma Y, Ma L. Transcriptome-based analysis reveals a crucial role of BxGPCR17454 in low temperature response of pine wood nematode (Bursaphelenchus xylophilus). Int J Mol Sci. 2019;20(12). https://doi.org/10.3390/ijms20122898
Zhang C, Wang H, Zhuang G, Zheng H, Zhang X. Comparative transcriptome analysis of Callosobruchus chinensis (L.) (Coleoptera: Chrysomelidae-Bruchinae) after heat and cold stress exposure. J Therm Biol. 2023;112:103479. https://doi.org/10.1016/j.jtherbio.2023.103479.
Liu Y, Su H, Li R, Li X, Xu Y, Dai X, Zhou Y, Wang H. Comparative transcriptome analysis of Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) reveals novel insights into heat stress tolerance in insects. BMC Genomics. 2017;19(181):974. https://doi.org/10.1186/s12864-017-4355-5
Zhou XR, Shan YM, Tan Y, Zhang ZR, Pang BP. Comparative analysis of transcriptome responses to cold stress in Galeruca daurica (Coleoptera: Chrysomelidae). J Insect Sci. 2019;19(6):8. https://doi.org/10.1093/jisesa/iez109
Benoit JB, Lopez-Martinez G, Elnitsky MA, Lee RE Jr, Denlinger DL. Dehydration-induced cross tolerance of Belgica antarctica larvae to cold and heat is facilitated by trehalose accumulation. Comp Biochem Physiol A Mol Integr Physiol. 2009;152(4):518–23. https://doi.org/10.1016/j.cbpa
Wen X, Wang S, Duman JG, Arifin JF, Juwita V, Goddard WA, Rios A, Liu F, Kim SK, Abrol R, DeVries AL, Henling LM. Antifreeze proteins govern the precipitation of trehalose in a freezing-avoiding insect at low temperature. Proc Natl Acad Sci U S A. 2016;14;113(24):6683-8. https://doi.org/10.1073/pnas.1601519113
Crowe JH, Crowe LM, Chapman D. Preservation of membranes in anhydrobiotic organisms: the role of trehalose. Science. 1984;223(4637):701–3. https://doi.org/10.1126/science.223.4637.701.
Behm CA. The role of trehalose in the physiology of nematodes. Int J Parasitol. 1997;27(2): 215– 29. https://doi.org/10.1016/s0020-7519(96)00151-8
Jorge JA, Polizeli ML, Thevelein JM, Terenzi HF. Trehalases and trehalose hydrolysis in fungi. FEMS Microbiol Lett. 1997;154(2):165–71. https://doi.org/10.1111/j.1574-6968.1997.tb12639.x.
Elbein AD, Pan YT, Pastuszak I, Carroll D. New insights on trehalose: a multifunctional molecule. Glycobiology. 2003;13(4):17R–27R. https://doi.org/10.1093/glycob/cwg047
Crowe JH. Trehalose as a chemical chaperone: fact and fantasy. Adv Exp Med Biol. 2007; 594,143– 58. https://doi.org/10.1007/978-0-387-39975-1_13
Iordachescu M, and Imai. R. Trehalose biosynthesis in response to abiotic stresses. J Integr Plant Biol. 2008;50(10):1223–9. https://doi.org/10.1111/j.1744-7909.2008.00736.x.
Xu J, Bao B, Zhang ZF, Yi YZ, Xu WH. Identification of a novel gene encoding the trehalose phosphate synthase in the cotton bollworm. Helicoverpa armigera Glycobiology. 2009;19(3):250–7. https://doi.org/10.1093/glycob/cwn127
Zhang B, Zhao L, Ning J, Wickham JD, Tian H, Zhang X, Yang M, Wang X, Sun J. Mir-31-5p regulates cold acclimation of the wood-boring beetle Monochamus alternatus via ascaroside signaling. BMC Biol. 2020;18(1):184. https://doi.org/10.1186/s12915-020-00926-w
Persson LB, Ambati VS, Brandman O. Cellular control of viscosity counters changes in temperature and energy availability. Cell. 2020;183(6):1572–85. https://doi.org/10.1016/j.cell.2020.10.017.
Wang G, Gou Y, Guo S, Zhou JJ, Liu C. RNA interference of trehalose-6-phosphate synthase and trehalase genes regulates chitin metabolism in two color morphs of Acyrthosiphon pisum Harris. Sci Rep. 2021;11(1):948. https://doi.org/10.1038/s41598-020-80277-2.
Leloir LF, Cabib E. The enzymic synthesis of trehalose phosphate. J Am Chem Soc. 1953;75(21):5445–6. https://doi.org/10.1021/ja01117a528.
Tang B, Wang S, Wang SG, Wang HJ, Zhang JY, Cui SY. Invertebrate trehalose-6-phosphate synthase gene: genetic architecture, biochemistry, physiological function, and potential applications. Front Physiol. 2018;9:30. https://doi.org/10.3389/fphys.2018.00030.
Chen J-X, Lyu Z-H, Wang C-Y, Cheng J, Lin T. RNA interference of a trehalose-6-phosphate synthase gene reveals its roles in the biosynthesis of chitin and lipids in Heortia vitessoides (Lepidoptera: Crambidae). Insect Sci. 2020;27:212–23. https://doi.org/10.1111/1744-7917.12650.
Hu Z, Zhang Y, He Y, Cao Q, Zhang T, Lou L, Cai Q. Full-length transcriptome assembly of Italian ryegrass root integrated with RNA-Seq to identify genes in response to plant cadmium stress. Int J Mol Sci. 2020;21(3). https://doi.org/10.3390/ijms21031067.
San Miguel K, Scott JG. The next generation of insecticides: dsRNA is stable as a foliar-applied insecticide. Pest Manag Sci. 2016;72(4):801–9. https://doi.org/10.1002/ps.4056.
Flynt AS. Insecticidal RNA interference, thinking beyond long dsRNA. Pest Manag Sci. 2021;77(5):2179–87. https://doi.org/10.1002/ps.6147.
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–4. https://doi.org/10.1093/nar/gkm882D480-4.
Minoche AE, Dohm JC, Schneider J, Holtgrawe D, Viehover P, Montfort M, Sorensen TR, Weisshaar B, Himmelbauer H. Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biol. 2015;16:184. https://doi.org/10.1186/s13059-015-0729-7.
Tellis MB, Kotkar HM, Joshi RS. Regulation of trehalose metabolism in insects: from genes to the metabolite window. Glycobiology. 2023;33(4):262–73. https://doi.org/10.1093/glycob/cwad011.
Ashburner M, Bonner JJ. The induction of gene activity in drosophilia by heat shock. Cell. 1979;17(2):241–54. https://doi.org/10.1016/0092-8674(79)90150-8.
Doring K, Ahmed N, Riemer T, Suresh HG, Vainshtein Y, Habich M, Riemer J, Mayer MP, O’Brien EP, Kramer G, Bukau B. Profiling ssb-nascent chain interactions reveals principles of Hsp70-assisted folding. Cell. 2017;170(2):298–311. https://doi.org/10.1016/j.cell.2017.06.038.
Li R, Wang YT, Jiang GF. The transcriptome analysis of the bamboo grasshopper provides insights into hypothermic stress acclimation. Int J Biol Macromol. 2019;134:237–46. https://doi.org/10.1016/j.ijbiomac.2019.05.002.
Wang XR, Wang C, Ban FX, Zhu DT, Liu SS, Wang XW. Genome-wide identification and characterization of HSP gene superfamily in whitefly (Bemisia tabaci) and expression profiling analysis under temperature stress. Insect Sci. 2019;26(1):44–57. https://doi.org/10.1111/1744-7917.12505.
Dubrez L, Causse S, Borges Bonan N, Dumetier B, Garrido C. Heat-shock proteins: chaperoning DNA repair. Oncogene. 2020;39(3):516–29. https://doi.org/10.1038/s41388-019-1016-y.
Sadura I, Libik-Konieczny M, Jurczyk B, Gruszka D, Janeczko A. HSP transcript and protein accumulation in brassinosteroid barley mutants acclimated to low and high temperatures. Int J Mol Sci. 2020;21(5). https://doi.org/10.3390/ijms21051889.
Song P, Jia Q, Xiao X, Tang Y, Liu C, Li W, Li T, Li L, Chen H, Zhang W, Zhang Q. HSP70-3 interacts with phospholipase Dδ and participates in heat stress defense. Plant Physiol. 2020;185(3):1148–65. https://doi.org/10.1093/plphys/kiaa083.
Tiwari LD, Khungar L, Grover A. AtHsc70-1 negatively regulates the basal heat tolerance in Arabidopsis thaliana through affecting the activity of HsfAs and Hsp101. Plant J. 2020;103(6):2069–83. https://doi.org/10.1111/tpj.14883.
Aloulou A, Rahier R, Arhab Y, Noiriel A, Abousalham A. Phospholipases: an overview. Methods Mol Biol. 2018;1835, 69–105. https://doi.org/10.1007/978-1-4939-8672-9_3
Tan YA, Zhao XD, Sun HJ, Zhao J, Xiao LB, Hao DJ, Jiang YP. Phospholipase C gamma (PLCgamma) regulates soluble trehalase in the 20E-induced fecundity of Apolygus lucorum. Insect Sci. 2021;28(2):430–44. https://doi.org/10.1111/1744-7917.12772.
Tufail M, Takeda M. Molecular characteristics of insect vitellogenins. J Insect Physiol. 2008;54(12):1447–58. https://doi.org/10.1016/j.jinsphys.2008.08.007.
Tufail M, Takeda M. Insect vitellogenin/lipophorin receptors: molecular structures, role in oogenesis, and regulatory mechanisms. J Insect Physiol. 2009;55(2):87–103. https://doi.org/10.1016/j.jinsphys.2008.11.007.
Husain M, Tufail M, Mehmood K, Rasool KG, Aldawood AS. Transcriptome analysis of the almond moth, Cadra cautella, female abdominal tissues and identification of reproduction control genes. BMC Genomics. 2019;20(1):1–14. https://doi.org/10.1186/s12864-019-6130-2
Husain M, Rasool KG, Tufail M, Sutanto KD, Alwaneen WS, Aldawood AS. Ultra Violet (UV-B) radiation intrudes Cadra cautella reproductive biology by influencing vitellogenin expression. J King Saud University-Science. 2022;34(8):102290. https://doi.org/10.1016/j.jksus.2022.102290.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 2012; 28(23), 3150–3152. https://doi.org/10.1093/bioinformatics/bts565.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. https://doi.org/10.1186/1471-2105-12-323.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. https://doi.org/10.1038/nmeth.1923.
McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–97. https://doi.org/10.1093/nar/gks042.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. https://doi.org/10.1093/bioinformatics/btp616.
Xue D, Chen T, Wu Y, Stability evaluation of candidate reference genes for real-time qPCR normalization in Rhyzopertha dominica (Coleoptera: Bostrycidae), J Econ Entomol. 2024; toae004. https://doi.org/10.1093/jee/toae004.
Qiu L, Hou L, Zhang B, Liu L, Li B, Deng P, Ma W, Wang X, Fabrick JA, Chen L,. Cadherin is involved in the action of Bacillus thuringiensis toxins Cry1Ac and Cry2Aa in the beet armyworm. Spodoptera exigua J Invertebr Pathol. 2015;127:47–53. https://doi.org/10.1016/j.jip.2015.02.009
Acknowledgements
We would like to express our gratitude to Professor Shen Jie from China Agricultural University, Beijing, for generously providing us with the nanocarriers needed for the RNAi assay.
Funding
This research was funded by the Optional Research Project of Academy of National Food and Strategic Reserves Administration (JY2306).
Author information
Authors and Affiliations
Contributions
DX and YW designed the study. DX contributed to the sequencing. DX, YY and SW carried out the experimental procedure. DX and LF performed the analyses. DX wrote the manuscript. LF and YW reviewed the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xue, D., Yang, Y., Fang, L. et al. Trehalose 6-phosphate synthase gene rdtps1 contributes to thermal acclimation in Rhyzopertha dominica. BMC Genomics 25, 172 (2024). https://doi.org/10.1186/s12864-024-10028-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10028-4