
Abstract
Human endogenous retroviruses (HERVs) are the germline embedded proviral fragments of ancient retroviral infections that make up roughly 8% of the human genome. Our understanding of HERVs in physiology primarily surrounds their non-coding functions, while their protein coding capacity remains virtually uncharacterized. Therefore, we applied the bioinformatic pipeline “hervQuant” to high-resolution ribosomal profiling of healthy tissues to provide a comprehensive overview of translationally active HERVs. We find that HERVs account for 0.1–0.4% of all translation in distinct tissue-specific profiles. Collectively, our study further supports claims that HERVs are actively translated throughout healthy tissues to provide sequences of retroviral origin to the human proteome.
Similar content being viewed by others


Background
Human endogenous retroviruses (HERVs) persist within the genome as the legacy of ancient retroviral infections that integrated into the germline [1, 2]. Germline embedded retroviruses then transmit vertically where over time they then accumulate mutations or deletions that prohibit infectious particle formation. Once a retrovirus no longer produces infectious particles, they are deemed “endogenous” [1]. Endogenization is not an instantaneous process, but instead occurs through complex transgenerational invasion of genomic sequences by a retrovirus, as demonstrated by the active endogenization events occurring in Koala species [3]. Once retrovirus has invaded the germline of a host specific, endogenization can then be driven by a multitude of factors, such as xenotropic restriction [4], mutations [1, 2], host-antiviral responses [5], and recombination events [6]. Collectively, HERVs make up about 8% of human genetic material [2, 7, 8], and have therefore substantially impacted the genome. While HERVs are mostly inactive [9] and none are replication competent like the ERVs of other mammals [10], many do display spatiotemporal activity in somatic [11,12,13,14] and developing cells [15,16,17,18,19,20,21] alike. Since their endogenization, many HERV elements have been coopted to accomplish molecular tasks in which are observable throughout reproduction [22, 23], immune responses [24, 25], and cell type specific transcription [11, 17, 19, 26]. Our current understanding of HERVs is primarily derived from their genomic and transcriptomic functions while little is known about their protein encoding capabilities.
Here, we performed the first large-scale characterization of HERV translation in healthy tissues by analyzing publicly available ribosomal profiling (RiboSeq) datasets [27]. RiboSeq quantifies the translatome by sequencing the short fragments (~ 25-35 bps) of ribosomal protected RNA, therefore providing a ‘snapshot’ of protein production [28]. By applying the bioinformatic pipeline ‘hervQuant’ [29] to publicly available RiboSeq data, we quantify the translational abundance of over 3000 annotated HERV proviruses [30] across an atlas of healthy tissue and cell types by aligning ribosomal protected short RNA sequencing fragments to full length proviruses. Collectively, this approach provides the first comprehensive characterization of actively translated HERV proviruses under healthy conditions. We term the collective of HERV proteins undergoing translation as the “endoretrotranslatome” (ERT) and suggest further investigation into the ERT as an understudied component of human health.
Results and discussion
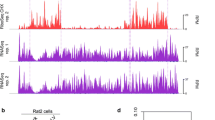
Despite characterizations of multiple uniquely identified HERV proteins [20, 23, 31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50], HERV-derived peptides as neoantigens in cancers [29, 51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67], and the identification of open reading frames (ORFs) embedded within HERV loci [2, 30, 47, 68,69,70,71,72,73,74,75,76,77,78,79], little is known regarding whether or not HERVs are readily translated in healthy tissues specifically. For this purpose, we aligned ribosomal embedded mRNA fragments from the RiboSeq atlas [27] with an annotation of proviral sequences [30] using hervQuant [29], a biologically validated pipeline that accurately depicts HERV translation. Using this approach, we provide the first large-scale examination of the ERT in brain, liver, and fat tissues, as well as within cell types such as vascular smooth muscle cells (VSMCs), embryonic stem cells (ESCs), human aorta endothelial cells (HA_ECs), human coronary artery endothelial cells (HCA_ECs), human umbilical vein endothelial cells (HUVECs), and primary human atrial fibroblasts (PHAFs). (Fig. 1A; Table S1). We found that all samples display detectable translation of HERV products ranging from 0.08% (brain) to 0.39% (ESCs) of all translation (Fig. 1B; Table S2). We next quantified the number of HERV proviruses that contain ≥1 read per million (RPM) and found that fat tissue displayed the most diverse expression profiles with an average of 533 HERV proviruses surpassing this threshold per sample, while HCA_ECs displayed the least diverse expression profile averaging only 105 distinct HERV proviruses that surpass this threshold (Fig. 1C). Principal component analysis (PCA) plots based on HERV protein production alone demonstrate that ESCs can be distinguished from other sample types by the ERT alone, while somatic sample types are indistinguishable from one another (Fig. 1D). This ESC-specific profile is largely attributed to high translation of the HERVHF superfamily (Fig. S1; Table S3), a large HERV clade whose activity coordinates early embryonic development [16, 80]. Heatmaps of HERV proviral transcript abundances organized in descending order of RPM abundance showcase discrete changes in the ERT between tissue and cell types (Fig. 1E-F). Collectively, these data demonstrate that HERVs are translated throughout healthy tissue types with-site specific translational profiles.

Ribosomal profiling reveals active translation of HERV proviruses in healthy tissue and cell types. a Schematic overview of workflow for profiling HERV proviral abundances from RiboSeq data. b HERV-aligned reads as a percentage of all filtered sequencing reads per sample. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± standard error of the mean (SEM). c Sum number of HERV proviruses possessing ≥1RPM per sample. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. d PCA plot of all tissue and cell types based on HERV-aligned ribosomal profiling reads alone. e Individual sample RPM abundances of all HERV proviruses per sample clustered per cell or tissue type. HERVs are listed in descending order by average RPM abundance. f Average RPM abundances of all HERV proviruses per cell or tissue type. HERVs are listed in descending order by average RPM abundance
Next, we profiled the ERT based on phylogeny. Summary data demonstrates the average RPM (Fig. 2A) and proportional (Fig. 2B) abundances of all HERV superfamilies across tissue and cell types. Collectively, the HML (HML1 through HML10, including HML2), HERVW9, HERVIDADP, and HERVHF superfamilies were most translationally active in healthy tissues (Fig. S2). Next, analysis of RPM abundances amongst HERV superfamilies in HA_ECs (Fig. 2C), HCA_ECs (Fig. 2D), ESCs (Fig. 2E), brain (Fig. 2F), fat (Fig. 2G), PHAFs (Fig. 2H), hepatocytes (Fig. 2I), HUVECs (Fig. 2J), and VSMCs (Fig. 2K) suggests that sample type is the dominant factor in determining HERV translation, while interindividual discrepancies are a secondary determinant. Proportional abundances of HERV superfamilies further supports this conclusion (Fig. S3). Next, we identify the 10 most highly translated HERVs in HA_ECs (Fig. 2L), HCA_ECs (Fig. 2M), ESCs (Fig. 2N), brain (Fig. 2O), fat (Fig. 2P), PHAFs (Fig. 2Q), hepatocytes (Fig. 2R), HUVECs (Fig. 2S), and VSMCs (Fig. 2T) demonstrate distinct changes in translational abundances (Table S1). Proviruses HERV_4295 (Fig. S4A) and HERV_4184 (Fig. S4B) possess conserved RPM abundances, suggesting conserved roles in ubiquitous processes. Meanwhile, highly translated proviruses such as HERV_1844 (Fig. S4C), HERV_4378 (Fig. S4D), and HERV_4231 (Fig. S4E) contain differential RPM abundances and may instead contribute to specialized roles indicative of a local environment.

Profiling of the endoretrotranslatome. a HERV superfamily RPM abundance averages per tissue or cell type. RPM values are calculated based on the total number of filtered reads per sample. b HERV superfamily abundance averages per sample type as a proportion of all HERV-aligned reads. c HERV superfamily RPM abundances per sample in HA_ECs. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 6). d HERV superfamily RPM abundances per sample in HCA_ECs. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 5) e HERV superfamily RPM abundances per sample in ESCs. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 6) f HERV superfamily RPM abundances per sample in brain tissue. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 5) g HERV superfamily RPM abundances per sample in fat tissue. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 6) h HERV superfamily RPM abundances per sample in PHAFs. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 10) i HERV superfamily RPM abundances per sample in hepatocytes. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 5) j HERV superfamily RPM abundances per sample in HUVECs. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 3) k HERV superfamily RPM abundances per sample in VSMCs. Dots indicate individual biological replicates with the graphed mean. Error bars indicate ± SEM. (n = 11) l Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in HA_ECs. (n = 6) m Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in HCA_ECs. (n = 5) n Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in ESCs. (n = 6). o Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in brain tissue. (n = 5). p Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in fat tissue. (n = 6). q Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in PHAFs.(n = 10). r Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in hepatocytes. (n = 5). s Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in HUVECs. (n = 3). t Heatmap displaying RPM abundances of the top 10 most highly translated HERVs in VSMCs. (n = 11)
Our analyses demonstrate that HERV-provirus aligned reads make up a surprising portion of the human translatome, encompassing roughly between 0.1–0.4% of all translation in a site-specific manner. Unsurprisingly, the ERT displays substantial diversity across tissue sites. As the expression of HERVs at the RNA level is tightly regulated by an excessive complement of epigenetic modifications [9], their translation with little interindividual discrepancies suggests that their expression at the protein level is likely by design and not inadvertent. Post-translation, HERV protein stability and function may be rapidly compromised by the host via post translational modifications [81] or by the targeted clearance of dysfunctional protein aggregates [82, 83], and therefore a limitation of this study pertains to their unknown half-life. In example, our results find that paraneoplastic Ma antigen 1 (PNMA1), a domesticated LTR retrotransposon capsid containing a neuronal autoantigen associated with paraneoplastic neurological pathologies [84], is translated throughout all tissue types tested (Fig. S5). Therefore, going forward considering the rate of transcription, translation, and degradation would provide the most comprehensive determination of HERV activity [85].
Conclusions
In this study, we demonstrate that HERVs, acquired via ancient retroviral infections, are translationally active elements. Previous misconceptions suggested that HERVs were merely inert or parasitic sequences, however it is now appreciated that HERVs innervate host physiology [86], regulate transcriptional networks [87, 88], contribute to the transcriptome [11,12,13], and provide retroviral motifs that propagate immunity [24, 25]. Here, we demonstrate that HERVs are translated in greater than anticipated proportions, and that HERV proteins are a reservoir of poorly defined macromolecules that may impact human health and disease. Previous studies have shown that a diverse profile of HERVs are expressed that the RNA level throughout various tissue sites, and that HERV RNAs make up roughly 0.19–1.91% of all polyadenylated RNA in site-specific manners [12]. Additionally, the authors demonstrate HERV RNA activity is sensitive to confounding variables, such as background and age [12]. Transcriptional activity of the HML and HERHF superfamilies, which we found to be most abundant in the ERT, has previously been detected in fully differentiated somatic tissues [12, 13, 89, 90]. Additionally, in ESCs many HERV elements are derepressed, and HERVH elements are highly active and contribute to cellular ESC cell specific processes [16, 91]. Therefore, it is unsurprising that we see the highest proportions of HERV translation globally and from the HERVHF family in ESCs.
In accordance with previous observations of HERV activity in the transcriptome and genome, we now demonstrate that HERV RNAs can be found in the ribosome of healthy human tissues. While ribosomal RNA content does not perfectly equate to stable protein levels, as demonstrated by the translational abundances of PNMA1 which is absent in the protein content of healthy cells [92], it does suggest that HERV elements are participating in the intricacies cellular biology than previously considered. We emphasize that future studies which investigate the translational efficiency and stability of HERV proteins, and whether pre- or post-translational modifications contributing to their clearance go awry in diseases associated with HERV protein abundance, are of the utmost importance, and continued characterization of the ERT will provide valuable insight into the mysterious mechanisms by which ancient retroviral genes underlie cellular processes as potentially viable and unstudied protein coding genes. These results also suggest reassessment of previous nomenclature that, while lowly abundant in the translatome, might have considered HERVs to be non-coding genes.
Methods
Data and code availability
All original code utilized for this study can be found at https://github.com/nixonlab/te_riboseq_atlas. The code for quantifying HERV-provirus aligning reads was adapted from the previously developed hervQuant pipeline [29] which can be found at https://unclineberger.org/vincentlab/resources/. Post hoc visualization of HERV provirus loci was performed with Integrated Genomics Viewer (IGV) [93] desktop application available at https://software.broadinstitute.org/software/igv/. Scatter plots and heatmaps were generated with GraphPad Prism version 9.3.1 available at https://www.graphpad.com/scientific-software/prism/. Biplots displaying PCA differentiation of samples were generated using PCATools available at https://github.com/kevinblighe/PCAtools.
Quantification of HERV provirus aligned reads from RiboSeq datasets
Quantification of HERVs from the RiboSeq atlas [27] was accomplished using modified methods for the hervQuant pipeline [29]. Briefly, an annotated reference was generated using full-length HERV provirus sequences within hg19 [30]. FASTQ files from were first filtered to remove rRNA reads with Ribodetector [94]. FASTQ reads were then filtered to retain only sequences between 25 and 35 bps in length using SeqKit [95]. Next, known tRNA and rRNA sequences were removed using Bowtie2 v2.5.1 [96]. Post hoc analysis in IGV v2.12.3 [93] demonstrated 5 highly abundant sequences within HERV proviruses that possess high-homology to common RNA contaminants of RiboSeq data [28, 97] based on query searches with BLAST [98] and RNAcentral [99]. These 5 sequences were manually added to the tRNA and rRNA annotation before reanalysis. The final FASTA file of contaminant RNAs removed can be found at https://github.com/nixonlab/te_riboseq_atlas/blob/main/custom_databases/tRNA_rRNA_hg19_ND.fa. Filtered FASTQ files were then aligned to the HERV reference annotation using STAR v2.7.9a [100] (multimaps ≤3 and mismatches ≤1). Next, SAM file outputs were filtered to generate BAM files containing only HERV aligned reads with SAMtools v1.14 [101] before quantification with Salmon v0.8.2 [102] (quant mode − 1 a). For quality assurance, SAMtools v1.14 [101] sorted BAM files merged per tissue type were visualized in IGV v2.12.3 [93]. HERV_1613, HERV_2322, HERV_2740, HERV_4231, HERV_4596, and HERV_5896 were removed from analyses due to RNA contaminant alignment. For RPM abundances, all samples were normalized to filtered reads (Table S2). HERV superfamily annotations were gathered from the supplemental information provided by the original description of hervQuant pipeline [29] and HERV annotations [30]. PCA was performed using PCAtools v2.6.0 on DESEQ2 v1.34.0 [103] transformed objects from raw count matrices. Characterization of translated ORFs was performed using BLAST [98], clustal omega [104], and ORFFinder [105] tools. Statistical analysis was performed using GraphPad Prism v9.3.1. Degree of significance was demonstrated using the following key: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Availability of data and materials
The datasets analyzed during the current study are available in the Sequence Read Archive (SRA), https://www.ncbi.nlm.nih.gov/bioproject/PRJNA756018. All data was collected from previously published works [27, 106] and can be accessed through the SRA Run Selector under the BioProject number “PRJNA756018”.
References
de Parseval N, Heidmann T. Human endogenous retroviruses: from infectious elements to human genes. Cytogenet Genome Res. 2005;110:318–32.
Jern P, Coffin JM. Effects of retroviruses on host genome function. Annu Rev Genet. 2008;42:709–32.
Tarlinton RE, Meers J, Young PR. Retroviral invasion of the koala genome. Nature. 2006;442:79–81.
Levy JA. Xenotropic viruses: murine leukemia viruses associated with NIH Swiss, NZB, and other mouse strains. Science. 1973;182:1151–3.
Yu P, Lübben W, Slomka H, Gebler J, Konert M, Cai C, et al. Nucleic acid-sensing toll-like receptors are essential for the control of endogenous retrovirus viremia and ERV-induced tumors. Immunity. 2012;37:867–79.
Hughes JF, Coffin JM. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc Natl Acad Sci U S A. 2004;101:1668–72.
Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. The complete sequence of a human genome. Science. 2022;376:44–53.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921.
Dopkins N, O’Mara MM, Lawrence E, Fei T, Sandoval-Motta S, Nixon DF, et al. A field guide to endogenous retrovirus regulatory networks. Mol Cell. 2022;82:3763–8.
Blanco-Melo D, Gifford RJ, Bieniasz PD. Reconstruction of a replication-competent ancestral murine endogenous retrovirus-L. Retrovirology. 2018;15:34.
He J, Babarinde IA, Sun L, Xu S, Chen R, Shi J, et al. Identifying transposable element expression dynamics and heterogeneity during development at the single-cell level with a processing pipeline scTE. Nat Commun. 2021;12:1456.
She J, Du M, Xu Z, Jin Y, Li Y, Zhang D, et al. The landscape of hervRNAs transcribed from human endogenous retroviruses across human body sites. Genome Biol. 2022;23:231.
Burn A, Roy F, Freeman M, Coffin JM. Widespread expression of the ancient HERV-K (HML-2) provirus group in normal human tissues. PLoS Biol. 2022;20:e3001826.
Seifarth W, Frank O, Zeilfelder U, Spiess B, Greenwood AD, Hehlmann R, et al. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J Virol. 2005;79:341–52.
Xiang Y, Liang H. The regulation and functions of endogenous retrovirus in embryo development and stem cell differentiation. Stem Cells Int. 2021;2021:6660936.
Lu X, Sachs F, Ramsay L, Jacques P-É, Göke J, Bourque G, et al. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat Struct Mol Biol. 2014;21:423–5.
Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487:57–63.
Wang J, Xie G, Singh M, Ghanbarian AT, Raskó T, Szvetnik A, et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature. 2014;516:405–9.
Göke J, Lu X, Chan Y-S, Ng H-H, Ly L-H, Sachs F, et al. Dynamic transcription of distinct classes of endogenous retroviral elements marks specific populations of early human embryonic cells. Cell Stem Cell. 2015;16:135–41.
Fuchs NV, Loewer S, Daley GQ, Izsvák Z, Löwer J, Löwer R. Human endogenous retrovirus K (HML-2) RNA and protein expression is a marker for human embryonic and induced pluripotent stem cells. Retrovirology. 2013;10:115.
Grow EJ, Flynn RA, Chavez SL, Bayless NL, Wossidlo M, Wesche DJ, et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature. 2015;522:221–5.
Bjerregaard B, Holck S, Christensen IJ, Larsson L-I. Syncytin is involved in breast cancer-endothelial cell fusions. Cell Mol Life Sci. 2006;63:1906–11.
Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature. 2000;403:785–9.
Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016;351:1083–7.
Lima-Junior DS, Krishnamurthy SR, Bouladoux N, Collins N, Han S-J, Chen EY, et al. Endogenous retroviruses promote homeostatic and inflammatory responses to the microbiota. Cell. 2021;184:3794–3811.e19.
Wu F, Liufu Z, Liu Y, Guo L, Wu J, Cao S, et al. Species-specific rewiring of definitive endoderm developmental gene activation via endogenous retroviruses through TET1-mediated demethylation. Cell Rep. 2022;41:111791.
Chothani SP, Adami E, Widjaja AA, Langley SR, Viswanathan S, Pua CJ, et al. A high-resolution map of human RNA translation. Mol Cell. 2022;82:2885–2899.e8.
Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23.
Smith CC, Beckermann KE, Bortone DS, De Cubas AA, Bixby LM, Lee SJ, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J Clin Invest. 2018;128:4804–20.
Vargiu L, Rodriguez-Tomé P, Sperber GO, Cadeddu M, Grandi N, Blikstad V, et al. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology. 2016;13:7.
Perron H, Garson JA, Bedin F, Beseme F, Paranhos-Baccala G, Komurian-Pradel F, et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis. Proc Natl Acad Sci U S A. 1997;94:7583–8.
Denne M, Sauter M, Armbruester V, Licht JD, Roemer K, Mueller-Lantzsch N. Physical and functional interactions of human endogenous retrovirus proteins Np9 and rec with the Promyelocytic leukemia zinc finger protein. J Virol. 2007;81:5607–16.
Li W, Lee M-H, Henderson L, Tyagi R, Bachani M, Steiner J, et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med. 2015;7:307ra153.
Rigogliuso G, Biniossek ML, Goodier JL, Mayer B, Pereira GC, Schilling O, et al. A human endogenous retrovirus encoded protease potentially cleaves numerous cellular proteins. Mob DNA. 2019;10:36.
Liu X, Liu Z, Wu Z, Ren J, Fan Y, Sun L, et al. Resurrection of endogenous retroviruses during aging reinforces senescence. Cell. 2023;186:287–304.e26.
Blaise S, de Parseval N, Bénit L, Heidmann T. Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc Natl Acad Sci U S A. 2003;100:13013–8.
Heidmann O, Béguin A, Paternina J, Berthier R, Deloger M, Bawa O, et al. HEMO, an ancestral endogenous retroviral envelope protein shed in the blood of pregnant women and expressed in pluripotent stem cells and tumors. Proc Natl Acad Sci U S A. 2017;114:E6642–51.
Aagaard L, Villesen P, Kjeldbjerg AL, Pedersen FS. The approximately 30-million-year-old ERVPb1 envelope gene is evolutionarily conserved among hominoids and Old World monkeys. Genomics. 2005;86:685–91.
Kjeldbjerg AL, Villesen P, Aagaard L, Pedersen FS. Gene conversion and purifying selection of a placenta-specific ERV-V envelope gene during simian evolution. BMC Evol Biol. 2008;8:266.
Blanco-Melo D, Gifford RJ, Bieniasz PD. Co-option of an endogenous retrovirus envelope for host defense in hominid ancestors. eLife. 2017;6:e22519.
Wu B, Gan Y, Xu Y, Wu Z, Xu G, Wang P, et al. Identification of the novel Np17 oncogene in human leukemia. Aging (Albany NY). 2020;12:23647–67.
Curzio DD, Gurm M, Turnbull M, Nadeau M-J, Meek B, Rempel JD, et al. Pro-inflammatory signaling upregulates a neurotoxic Conotoxin-like protein encrypted within human endogenous retrovirus-K. Cells. 2020;9:1584.
Löwer R, Tönjes RR, Korbmacher C, Kurth R, Löwer J. Identification of a rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J Virol. 1995;69:141–9.
Magin C, Löwer R, Löwer J. cORF and RcRE, the rev/rex and RRE/RxRE homologues of the human endogenous retrovirus family HTDV/HERV-K. J Virol. 1999;73:9496–507.
Yang J, Bogerd HP, Peng S, Wiegand H, Truant R, Cullen BR. An ancient family of human endogenous retroviruses encodes a functional homolog of the HIV-1 rev protein. Proc Natl Acad Sci U S A. 1999;96:13404–8.
Baldwin ET, Götte M, Tchesnokov EP, Arnold E, Hagel M, Nichols C, et al. Human endogenous retrovirus-K (HERV-K) reverse transcriptase (RT) structure and biochemistry reveals remarkable similarities to HIV-1 RT and opportunities for HERV-K–specific inhibition. Proc Natl Acad Sci U S A. 2022;119:e2200260119.
Lindeskog M, Mager DL, Blomberg J. Isolation of a human endogenous retroviral HERV-H element with an open env Reading frame. Virology. 1999;258:441–50.
Sutkowski N, Conrad B, Thorley-Lawson DA, Huber BT. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity. 2001;15:579–89.
Dewannieux M, Blaise S, Heidmann T. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses. J Virol. 2005;79:15573–7.
Sugimoto J, Sugimoto M, Bernstein H, Jinno Y, Schust D. A novel human endogenous retroviral protein inhibits cell-cell fusion. Sci Rep. 2013;3:1462.
Saini SK, Ørskov AD, Bjerregaard A-M, Unnikrishnan A, Holmberg-Thydén S, Borch A, et al. Human endogenous retroviruses form a reservoir of T cell targets in hematological cancers. Nat Commun. 2020;11:5660.
Sauter M, Schommer S, Kremmer E, Remberger K, Dölken G, Lemm I, et al. Human endogenous retrovirus K10: expression of gag protein and detection of antibodies in patients with seminomas. J Virol. 1995;69:414–21.
Tubb VM, Schrikkema DS, Croft NP, Purcell AW, Linnemann C, Freriks MR, et al. Isolation of T cell receptors targeting recurrent neoantigens in hematological malignancies. J Immunother Cancer. 2018;6:70.
Bonaventura P, Alcazer V, Mutez V, Tonon L, Martin J, Chuvin N, et al. Identification of shared tumor epitopes from endogenous retroviruses inducing high-avidity cytotoxic T cells for cancer immunotherapy. Sci Adv. 2022;8:eabj3671.
Bonté P-E, Arribas YA, Merlotti A, Carrascal M, Zhang JV, Zueva E, et al. Single-cell RNA-seq-based proteogenomics identifies glioblastoma-specific transposable elements encoding HLA-I-presented peptides. Cell Rep. 2022;39:110916.
Merlotti A, Sadacca B, Arribas YA, Ngoma M, Burbage M, Goudot C, et al. Noncanonical splicing junctions between exons and transposable elements represent a source of immunogenic recurrent neo-antigens in patients with lung cancer. Sci Immunol. 2023;8:eabm6359.
Burbage M, Rocañín-Arjó A, Baudon B, Arribas YA, Merlotti A, Rookhuizen DC, et al. Epigenetically controlled tumor antigens derived from splice junctions between exons and transposable elements. Sci Immunol. 2023;8:eabm6360.
Takahashi Y, Harashima N, Kajigaya S, Yokoyama H, Cherkasova E, McCoy JP, et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J Clin Invest. 2008;118:1099–109.
Cherkasova E, Scrivani C, Doh S, Weisman Q, Takahashi Y, Harashima N, et al. Detection of an immunogenic HERV-E envelope with selective expression in clear cell kidney Cancer. Cancer Res. 2016;76:2177–85.
Cherkasova E, Malinzak E, Rao S, Takahashi Y, Senchenko VN, Kudryavtseva AV, et al. Inactivation of the von Hippel-Lindau tumor suppressor leads to selective expression of a human endogenous retrovirus in kidney cancer. Oncogene. 2011;30:4697–706.
Mullins CS, Linnebacher M. Endogenous retrovirus sequences as a novel class of tumor-specific antigens: an example of HERV-H env encoding strong CTL epitopes. Cancer Immunol Immunother. 2012;61:1093–100.
Schiavetti F, Thonnard J, Colau D, Boon T, Coulie PG. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. 2002;62:5510–6.
Rycaj K, Plummer JB, Yin B, Li M, Garza J, Radvanyi L, et al. Cytotoxicity of human endogenous retrovirus K-specific T cells toward autologous ovarian cancer cells. Clin Cancer Res. 2015;21:471–83.
Rezaei SD, Hayward JA, Norden S, Pedersen J, Mills J, Hearps AC, et al. HERV-K gag RNA and protein levels are elevated in malignant regions of the prostate in males with prostate Cancer. Viruses. 2021;13:449.
Tatkiewicz W, Dickie J, Bedford F, Jones A, Atkin M, Kiernan M, et al. Characterising a human endogenous retrovirus(HERV)-derived tumour-associated antigen: enriched RNA-Seq analysis of HERV-K(HML-2) in mantle cell lymphoma cell lines. Mob DNA. 2020;11:9.
Ng KW, Boumelha J, Enfield KSS, Almagro J, Cha H, Pich O, et al. Antibodies against endogenous retroviruses promote lung cancer immunotherapy. Nature. 2023:1–11. https://doi.org/10.1038/s41586-023-05771-9.
Laumont CM, Vincent K, Hesnard L, Audemard É, Bonneil É, Laverdure J-P, et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci Transl Med. 2018;10:eaau5516.
Subramanian RP, Wildschutte JH, Russo C, Coffin JM. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology. 2011;8:90.
Jern P, Lindeskog M, Karlsson D, Blomberg J. Full-length HERV-H elements with env SU open Reading frames in the human genome. AIDS Res Hum Retrovir. 2002;18:671–6.
de Parseval N, Casella J-F, Gressin L, Heidmann T. Characterization of the three HERV-H proviruses with an open envelope Reading frame encompassing the immunosuppressive domain and evolutionary history in Primates. Virology. 2001;279:558–69.
Mayer J, Meese E, Mueller-Lantzsch N. Multiple human endogenous retrovirus (HERV-K) loci with gag open reading frames in the human genome. Cytogenet Genome Res. 1997;78:1–5.
Barbulescu M, Turner G, Seaman MI, Deinard AS, Kidd KK, Lenz J. Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans. Curr Biol. 1999;9:861–S1.
Ueda MT, Kryukov K, Mitsuhashi S, Mitsuhashi H, Imanishi T, Nakagawa S. Comprehensive genomic analysis reveals dynamic evolution of endogenous retroviruses that code for retroviral-like protein domains. Mob DNA. 2020;11:29.
Boso G, Fleck K, Carley S, Liu Q, Buckler-White A, Kozak CA. The oldest co-opted gag Gene of a human endogenous retrovirus shows placenta-specific expression and is upregulated in diffuse large B-cell lymphomas. Mol Biol Evol. 2021;38:5453–71.
Villesen P, Aagaard L, Wiuf C, Pedersen FS. Identification of endogenous retroviral reading frames in the human genome. Retrovirology. 2004;1:32.
Nakagawa S, Takahashi MU. gEVE: a genome-based endogenous viral element database provides comprehensive viral protein-coding sequences in mammalian genomes. Database (Oxford). 2016;2016:baw087.
Blond JL, Besème F, Duret L, Bouton O, Bedin F, Perron H, et al. Molecular characterization and placental expression of HERV-W, a new human endogenous retrovirus family. J Virol. 1999;73:1175–85.
Jern P, Sperber GO, Ahlsén G, Blomberg J. Sequence variability, gene structure, and expression of full-length human endogenous retrovirus H. J Virol. 2005;79:6325–37.
Sandmann C-L, Schulz JF, Ruiz-Orera J, Kirchner M, Ziehm M, Adami E, et al. Evolutionary origins and interactomes of human, young microproteins and small peptides translated from short open reading frames. Mol Cell. 2023;83(6):994–1011.
Barakat TS, Halbritter F, Zhang M, Rendeiro AF, Perenthaler E, Bock C, et al. Functional dissection of the enhancer repertoire in human embryonic stem cells. Cell Stem Cell. 2018;23:276–288.e8.
Walsh CT, Garneau-Tsodikova S, Gatto GJ. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed. 2005;44:7342–72.
Hyttinen JMT, Amadio M, Viiri J, Pascale A, Salminen A, Kaarniranta K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res Rev. 2014;18:16–28.
Kesner JS, Chen Z, Shi P, Aparicio AO, Murphy MR, Guo Y, et al. Noncoding translation mitigation. Nature. 2023;617:395–402.
Dalmau J, Gultekin SH, Voltz R, Hoard R, DesChamps T, Balmaceda C, et al. Ma1, a novel neuron- and testis-specific protein, is recognized by the serum of patients with paraneoplastic neurological disorders. Brain. 1999;122:27–39.
Hausser J, Mayo A, Keren L, Alon U. Central dogma rates and the trade-off between precision and economy in gene expression. Nat Commun. 2019;10:68.
Dopkins N, Nixon DF. Activation of human endogenous retroviruses and its physiological consequences. Nat Rev Mol Cell Biol. 2023:1–11. https://doi.org/10.1038/s41580-023-00674-z.
Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, et al. Ten things you should know about transposable elements. Genome Biol. 2018;19:199.
Fueyo R, Judd J, Feschotte C, Wysocka J. Roles of transposable elements in the regulation of mammalian transcription. Nat Rev Mol Cell Biol. 2022;23:481–97.
Li F, Sabunciyan S, Yolken RH, Lee D, Kim S, Karlsson H. Transcription of human endogenous retroviruses in human brain by RNA-seq analysis. PLoS One. 2019;14:e0207353.
Nevalainen T, Autio A, Hurme M. Human endogenous retroviruses of the HERV-K (HML-2) family are expressed in the brain of healthy individuals and modify the composition of the brain-infiltrating immune cells. Heliyon. 2023;9:e21283.
Zhang Y, Li T, Preissl S, Amaral ML, Grinstein JD, Farah EN, et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat Genet. 2019;51:1380–8.
Jiang S-H, He P, Ma M-Z, Wang Y, Li R-K, Fang F, et al. PNMA1 promotes cell growth in human pancreatic ductal adenocarcinoma. Int J Clin Exp Pathol. 2014;7:3827–35.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Deng Z-L, Münch PC, Mreches R, McHardy AC. Rapid and accurate identification of ribosomal RNA sequences via deep learning. Nucleic Acids Res. 2022;50:e60.
Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11:e0163962.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–9.
Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7:1534–50.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
The RNAcentral Consortium, Petrov AI, Kay SJE, Kalvari I, Howe KL, Gray KA, et al. RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 2017;45:D128–34.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Sievers F, Higgins DG. Clustal omega for making accurate alignments of many protein sequences: Clustal omega for many protein sequences. Protein Sci. 2018;27:135–45.
Wheeler DL. Database resources of the National Center for biotechnology. Nucleic Acids Res. 2003;31:28–33.
Chothani S, Schäfer S, Adami E, Viswanathan S, Widjaja AA, Langley SR, et al. Widespread translational control of fibrosis in the human heart by RNA-binding proteins. Circulation. 2019;140:937–51.
Acknowledgements
Not applicable.
Funding
These works are supported by US NIH grants NCI CA260691 (DFN) and NIAID UM1AI164559 (DFN).
Author information
Authors and Affiliations
Contributions
N.D conceptualized the hypotheses with aid from B.S, S.M, P.Z, M.S, C.F, and D.F.N. N.D wrote the manuscript with aid from B.S, S.M, J.L.M, T.F, N.C, M.L.B, and D.F.N. N.D conducted data analysis with the help of B.S and S.M. N.D interpreted the data and curated figures with help from B.S, S.M, P.Z, M.S, C.F, and D.F.N. Original code for the paper was developed by N.D and B.S with help from P.Z and M.S. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dopkins, N., Singh, B., Michael, S. et al. Ribosomal profiling of human endogenous retroviruses in healthy tissues. BMC Genomics 25, 5 (2024). https://doi.org/10.1186/s12864-023-09909-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09909-x