
Abstract
Background
Host genetics influences the development of infectious diseases in many agricultural animal species. Identifying genes associated with disease development has the potential to make selective breeding for disease tolerance more likely to succeed through the selection of different genes in diverse signaling pathways. In this study, four families of Pacific oysters (Crassostrea gigas) were identified to be segregating for a quantitative trait locus (QTL) on chromosome 8. This QTL was previously found to be associated with basal antiviral gene expression and survival to ostreid herpesvirus 1 (OsHV-1) mortality events in Tomales Bay, California. Individuals from these four families were phenotyped and genotyped in an attempt to find candidate genes associated with the QTL on chromosome 8.
Results
Genome-wide allele frequencies of oysters from each family prior to being planting in Tomales Bay were compared with the allele frequencies of oysters from respective families that survived an OsHV-1 mortality event. Six significant unique QTL were identified in two families in these genome-wide allele frequency studies, all of which were located on chromosome 8. Three QTL were assigned to candidate genes (ABCA1, PIK3R1, and WBP2) that have been previously associated with antiviral innate immunity in vertebrates.
Conclusion
The identification of vertebrate antiviral innate immunity genes as candidate genes involved in molluscan antiviral innate immunity reinforces the similarities between the innate immune systems of these two groups. Causal variant identification in these candidate genes will enable future functional studies of these genes in an effort to better understand their antiviral modes of action.
Similar content being viewed by others

Background
Ostreid herpesvirus 1 (OsHV-1) is a viral pathogen that causes disease and mortality in Pacific oysters (Crassostrea gigas) [1]. Since 2008, the emergence of virulent variants of OsHV-1 in Australia [2], France [3], New Zealand [4], and the United States [5] have led to the development of strategies to mitigate the negative economic effects from massive mortalities on oyster growers. One strategy that has been uniformly adopted by farmers in these countries is the use of oysters that are tolerant to OsHV-1 [6,7,8,9]. However, despite the success of breeding oysters with OsHV-1 tolerance, the genes and underlying mechanisms responsible for this increased tolerance have yet to be determined.
OsHV-1 is in the Herpesvirales order, which contains herpesviruses that infect and cause disease in a wide range of animal hosts spanning mammalian vertebrates, non-mammalian vertebrates, and invertebrates [10]. Genome-wide association studies (GWAS) in humans [11, 12], mice [13], rats [14], horses [15], pigs [16], chickens [17], common carp [18, 19], and Pacific oysters [20,21,22] have found significant regions in their respective genomes that are associated with herpesvirus disease severity. Many GWAS studies have been conducted in animals of agricultural importance because genetic loci associated with herpesvirus tolerance can be used for selective breeding to reduce herpesvirus-associated disease.
Selective breeding for a genetic locus controlling survival to OsHV-1, as well as antiviral gene expression, has recently been conducted in a Pacific oyster population in the United States [20]; however, the significance interval of this locus on chromosome 8 of the Pacific oyster genome spanned 6.8 Mb and contained 316 protein-coding genes. Identifying the genes responsible for the increased tolerance to OsHV-1 in this locus could provide insights into the mechanisms behind this trait. The objectives of the current study were to find candidate genes on chromosome 8 responsible for OsHV-1 tolerance in four biparental families (Table 1) from the Molluscan Broodstock Program (MBP) breeding population [23] by comparing the genome-wide allele frequencies of oysters that survived an OsHV-1 mortality event in Tomales Bay, California, with oysters that were collected prior to planting.
Results
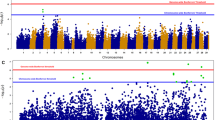
Six significant QTL were identified from the genome-wide allele frequency studies (GWAFS) in two families that mapped to unique positions on chromosome 8 in the genome (Table 2; Fig. 1). The GWAFS of family 30.058 identified three QTL on chromosome 8. The GWAFS of family 30.065 identified four unique QTL on chromosome 8, one of which mapped to the same genomic region as a QTL found in the GWAFS with family 30.058. The GWAFS of families 30.004 and 30.062 identified no QTL. Four candidate genes were assigned to QTL on chromosome 8 and included homologs to ABCA1 (phospholipid-transporting ATPase ABCA1) (Fig. 2), FRRS1 (ferric-chelate reductase 1), PIK3R1 (phosphatidylinositol 3-kinase regulatory subunit alpha) (Fig. 3), and WBP2 (WW domain-binding protein 2).

Manhattan plots showing the results of the genome-wide allele frequency studies (GWAFS) for survival to an OsHV-1-associated mortality event in Tomales Bay in four biparental families. GWAFS were performed with the post-mortality group as either oysters from all three replicate cages or oysters from one of the three replicate cages. Chromosomes 1 to 10 are indicated by different colors from left to right. Log-transformed p-values above and below zero represent those associated with the maternal and paternal haplotypes, respectively. The red horizontal line represents the genome-wide Bonferroni-corrected p-value significance threshold of 0.05

Genome-wide allele frequency study of the paternal haplotype of family 30.058 (replicate 1) at the a genome, b chromosome, and c sub-chromosome levels. The red horizontal line represents the genome-wide Bonferroni-corrected p-value significance threshold of 0.05. The gene highlighted in red is the candidate gene assigned to the QTL while the gray colored genes are other genes in the region

Genome-wide allele frequency study of the paternal haplotype of family 30.065 (all replicates) at the a genome, b chromosome, and c sub-chromosome levels. The red horizontal line represents the genome-wide Bonferroni-corrected p-value significance threshold of 0.05. The gene highlighted in red is the candidate gene assigned to the QTL while the gray colored genes are other genes in the region
GWAFS using post-mortality oysters from all replicates did not always find the same QTL as GWAFS using post-mortality oysters from individual replicates; for example, the GWAFS of family 30.058 using oysters from all replicates did not find the same QTL as the GWAFS of family 30.058 using oysters from only replicate 1 (Table 2). Additionally, different QTL were present or absent when using oysters from different replicate cages within the same family in a GWAFS; for example, significant QTL on chromosome 8 in family 30.065 were found in the GWAFS using oysters from replicate 3 (Fig. 1p) but not when oysters from replicates 1 and 2 were used (Fig. 1n, o). The presence of QTL in a GWAFS using oysters from a replicate was not always determined by the percentage of oysters surviving in that replicate; for example, 30.062 (replicate 2) and 30.065 (replicate 3) had 55% and 53% survival in the field, respectively, but the GWAFS with the former found no QTL (Fig. 1k) while the GWAFS with the latter found four QTL (Fig. 1p).
Discussion
The QTL found in the GWAFS were all located on one end of chromosome 8 where an antiviral QTL was previously identified in a GWAS with oyster families from MBP cohorts 27 and 29 [20]. With the greater mapping resolution available in the current study, it is evident that there are multiple QTL segregating on chromosome 8 rather than a single QTL segregating in the MBP population. In the field study, there was an unexpected amount of variability in the presence and absence of QTL in GWAFS with oysters from different replicate cages. Because there was a limit to how many oysters were able to be put into a single replicate cage without density-dependent mortality, oysters from a single family were distributed among multiple replicate cages in order to increase the sample size in the GWAFS. We found that GWAFS with oysters from different replicates from the same family that experienced similar levels of mortality did not necessarily identify the same QTL despite the replicates being planted very close to each other; for example, the three replicate cages of family 30.058 were planted approximately 1 m from each other and experienced between 64 and 72% mortality but a QTL was only detected in the GWAFS with replicate 1. Factors other than host genetics, such as size [24, 25], energy reserves [26], and opportunistic bacteria [27], have been shown to play a role in survival to OsHV-1. Additionally, significant micro-scale spatial effects on survival have previously been observed during an OsHV-1 mortality event that have been associated with OsHV-1 load heterogeneity [28, 29]. We believe that these factors were likely responsible for the presence/absence variability of QTL in the GWAFS. Ideally, oysters within cages would have been monitored daily to obtain a continuous survival phenotype, e.g., time to death, as well as other phenotypes, such as size, to better understand the influence of other potentially interacting factors; however, the planting site in this study was intertidal and accessible only at low tide with a boat, which made trips to the site for long-term, short-interval sampling impractical for the company hosting the study site.
Three candidate genes (ABCA1, PIK3R1, WBP2) identified among the QTL have previously been associated with the pathogenesis of herpesviruses or other viruses. ABCA1 is a regulator of cholesterol and has been associated with the severity of herpes simplex virus 2 in a GWAS in humans [30]. Additionally, ABCA1 gene expression has been shown to be upregulated after infection with Marek's disease virus (gallid alphaherpesvirus 2) in chicken fibroblasts [31]. PIK3R1 is part of the PI3K/Akt/mTOR signaling pathway, which is involved in innate immunity [32], and mutations in PIK3R1 have been associated with severe, recurrent, or persistent infections of herpesviruses in humans [33]. WBP2 is a gene involved in many signaling pathways, including the PI3K/Akt/mTOR signaling pathway [34], and has been shown to be downregulated in pig macrophages after infection with porcine reproductive and respiratory syndrome virus [35].
The identification of candidate genes in the Pacific oyster associated with survival to an OsHV-1 mortality event that have previously been implicated in the pathogenesis of vertebrate herpesviruses reinforces the parallels previously identified between the innate immune systems of molluscs and vertebrates [36, 37]. It also suggests that OsHV-1 could potentially be used as a model to study vertebrate herpesviruses, some of which share the genomic structure of OsHV-1 [10], and that insights from vertebrate herpesviruses could potentially be used to better understand OsHV-1. Despite identifying candidate genes for some of the QTL, it was not possible to identify candidate causal variants related to these candidate genes with the reduced-representation sequencing method used in this study, i.e., GBS. We anticipate that future studies utilizing whole genome sequencing will enable the identification of candidate causal variants for these genes and that these variants will motivate future functional studies of these genes.
Methods
Genotyping
Families in cohort 30 of the Molluscan Broodstock Program (MBP) were reared, planted in Tomales Bay, California, and phenotyped for survival as previously described by Divilov et al. [20]. Briefly, cohort 30 (n = 79 biparental families) was spawned in the MBP hatchery at the Hatfield Marine Science Center (HMSC) in Newport, Oregon, on 18 March 2021 and planted in Tomales Bay, California (38°12′17″N 122°56′05″W) on 8 June 2021. Oysters were checked every two weeks until the detection of significant mortality. Oyster mortality counts were taken on 19 October 2021. Individuals in four of these families, namely families 30.004, 30.058, 30.062, 30.065 (pedigree provided in Fig. S1), were chosen for pre-planting and post-mortality genotyping. Individuals in these families were chosen because their parents were heterozygous for a SNP at position 9,719,736 on chromosome 8, which has been associated with survival in Tomales Bay and basal antiviral gene expression [20]. Whole animal tissues of spat (juvenile oysters) from the four families obtained from the same pool of spat that were chosen for planting in Tomales Bay were stored in 95% ethanol and are referred to as the pre-planting samples (Table 1). These four families were planted in Tomales Bay in triplicate with a density of 150 oysters per replicate cage that was concurrent with the planting date and location of cohort 30 in Tomales Bay described above. The replicate cages for this additional planting were planted adjacent to each other in a row. Mantle tissue from oysters that survived the mortality event in each replicate cage from the four families were sampled in a biosafety level 2 laboratory, stored in 95% ethanol, and are referred to as the post-mortality samples (Table 1).
DNA from the pre-planting and post-mortality samples as well as the parents of the four families were extracted using the MagMAX DNA Multi-Sample Ultra 2.0 Kit (Thermo Fisher Scientific) and quantified using the Quant-iT Broad Range dsDNA Assay Kit (Thermo Fisher Scientific) on a Synergy LX Multi-Mode Microplate Reader (BioTek). Barcoded 96-plex genotyping-by-sequencing (GBS) libraries were constructed as described by Elshire et al. [38] using the ApeKI restriction enzyme and sequenced on an Illumina NextSeq 2000 (1 × 100 bp) at Oregon State University’s Center for Quantitative Life Sciences.
Reads were demultiplexed using Axe [39] and adapter trimmed using fastp [40]. A 5’ to 3’ sliding window of 4 bp was also used in fastp to drop all base pairs after the mean quality dropped below 20. Afterward, all reads less than 50 bp were removed. Filtered reads were then aligned to the Crassostrea gigas NCBI RefSeq genome [41] using bwa [42], and biallelic SNPs were called using bcftools [43] with a minimum mapping quality of 40 and a minimum base quality of 30. SNPs were then phased and imputed using AlphaFamImpute [44] with the genotype calling threshold set to 0.9 after filtering for minimum read depth (< 5), maximum read depth (> 100), minor allele frequency (< 0.15), and SNP missingness rate (> 0.8) (Table 1). Parentage assignment of offspring in each of the four families was confirmed using the apparent algorithm [45].
Genome-wide allele frequency study (GWAFS)
Fisher’s exact test [46] was used to test for SNP allele frequency differences between the pre-planting and post-mortality samples within a family. Tests were conducted using SNPs within maternal and paternal haplotypes in a family. Additionally, tests were performed with the post-mortality samples being either oysters from all three replicate cages of a family or oysters from one of the three replicate cages of a family. A Bonferroni-corrected p-value of 0.05 was used as the genome-wide significance threshold. The R packages fastman [47] and LocusZoom-like [48] were used to generate Manhattan plots, and the RefSeq genome annotation was used to identify candidate genes.
Availability of data and materials
The raw GBS reads of the oysters used in this study have been deposited at NCBI (BioProject PRJNA873124). The code used to run the analyses is available at https://github.com/kdivilov/BMC_Genomics_2023.
References
Fuhrmann M, Pathirana E, de Kantzow M, Hick P. Ostreid herpesvirus disease. In: Aquaculture Pathophysiology. Academic Press; 2022. https://www.sciencedirect.com/book/9780323954341/aquaculture-pathophysiology.
Trancart S, Tweedie A, Liu O, Paul-Pont I, Hick P, Houssin M, Whittington RJ. Diversity and molecular epidemiology of Ostreid herpesvirus 1 in farmed Crassostrea gigas in Australia: Geographic clusters and implications for “microvariants” in global mortality events. Virus Res. 2023;323: 198994.
Segarra A, Pépin JF, Arzul I, Morga B, Faury N, Renault T. Detection and description of a particular Ostreid herpesvirus 1 genotype associated with massive mortality outbreaks of Pacific oysters, Crassostrea gigas, in France in 2008. Virus Res. 2010;153(1):92–9.
Keeling SE, Brosnahan CL, Williams R, Gias E, Hannah M, Bueno R, McDonald WL, Johnston C. New Zealand juvenile oyster mortality associated with ostreid herpesvirus 1 – an opportunistic longitudinal study. Dis Aquat Org. 2014;109(3):231–9.
Burge CA, Friedman CS, Kachmar ML, Humphrey KL, Moore JD, Elston RA. The first detection of a novel OsHV-1 microvariant in San Diego, California, USA. J Invertebr Pathol. 2021;184: 107636.
Dégremont L, Nourry M, Maurouard E. Mass selection for survival and resistance to OsHV-1 infection in Crassostrea gigas spat in field conditions: response to selection after four generations. Aquaculture. 2015;446:111–21.
Divilov K, Schoolfield B, Cortez DM, Wang X, Fleener GB, Jin L, Dumbauld BR, Langdon C. Genetic improvement of survival in Pacific oysters to the Tomales Bay strain of OsHV-1 over two cycles of selection. Aquaculture. 2021;543: 737020.
Gutierrez AP, Symonds J, King N, Steiner K, Bean TP, Houston RD. Potential of genomic selection for improvement of resistance to ostreid herpesvirus in Pacific oyster (Crassostrea gigas). Anim Genet. 2020;51(2):249–57.
Kube P, Dove M, Cunningham M, Kirkland P, Gu X, Hick P, O’Connor W, Elliot N. Genetic selection for resistance to Pacific oyster mortality syndrome. FRDC and Seafood CRC Project 2012/760; 2018.
Knipe DM, Howley PM. Fields Virology. Lippincott Williams & Wilkins; 2013.
Crosslin DR, Carrell DS, Burt A, Kim DS, Underwood JG, Hanna DS, Comstock BA, Baldwin E, De Andrade M, Kullo IJ, Tromp G. Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun. 2015;16(1):1–7.
Urayama KY, Jarrett RF, Hjalgrim H, Diepstra A, Kamatani Y, Chabrier A, Gaborieau V, Boland A, Nieters A, Becker N, Foretova L. Genome-wide association study of classical Hodgkin lymphoma and Epstein-Barr virus status–defined subgroups. J Natl Cancer Inst. 2012;104(3):240–53.
Norose K, Yano A, Zhang XM, Blankenhorn E, Heber-Katz E. Mapping of genes involved in murine herpes simplex virus keratitis: identification of genes and their modifiers. J Virol. 2002;76(7):3502–10.
Abdelmagid N, Bereczky-Veress B, Atanur S, Musilová A, Zídek V, Saba L, Warnecke A, Khademi M, Studahl M, Aurelius E, Hjalmarsson A. Von Willebrand factor gene variants associate with herpes simplex encephalitis. PLoS ONE. 2016;11(5): e0155832.
Brosnahan MM, Al Abri MA, Brooks SA, Antczak DF, Osterrieder N. Genome-wide association study of equine herpesvirus type 1-induced myeloencephalopathy identifies a significant single nucleotide polymorphism in a platelet-related gene. Vet J. 2019;245:49–54.
Reiner G, Melchinger E, Kramarova M, Pfaff E, Büttner M, Saalmüller A, Geldermann H. Detection of quantitative trait loci for resistance/susceptibility to pseudorabies virus in swine. J Gen Virol. 2002;83(1):167–72.
Li DF, Lian L, Qu LJ, Chen YM, Liu WB, Chen SR, Zheng JX, Xu GY, Yang N. A genome-wide SNP scan reveals two loci associated with the chicken resistance to Marek’s disease. Anim Genet. 2013;44(2):217–22.
Palaiokostas C, Robledo D, Vesely T, Prchal M, Pokorova D, Piackova V, Pojezdal L, Kocour M, Houston RD. Mapping and sequencing of a significant quantitative trait locus affecting resistance to koi herpesvirus in common carp. G3 (Bethesda). 2018;8(11):3507–13.
Tadmor-Levi R, Hulata G, David L. Multiple interacting QTLs affect disease challenge survival in common carp (Cyprinus carpio). Heredity. 2019;123(5):565–78.
Divilov K, Merz N, Schoolfield B, Green TJ, Langdon C. Marker-assisted selection in a Pacific oyster population for an antiviral QTL conferring increased survival to OsHV-1 mortality events in Tomales Bay. Aquaculture. 2023;567: 739291.
Gutierrez AP, Bean TP, Hooper C, Stenton CA, Sanders MB, Paley RK, Rastas P, Bryrom M, Matika O, Houston RD. A genome-wide association study for host resistance to ostreid herpesvirus in Pacific oysters (Crassostrea gigas). G3 (Bethesda). 2018;8(4):1273–80.
Sauvage C, Boudry P, De Koning DJ, Haley CS, Heurtebise S, Lapegue S. QTL for resistance to summer mortality and OsHV-1 load in the Pacific oyster (Crassostrea gigas). Anim Genet. 2010;41(4):390–9.
de Melo CMR, Durland E, Langdon C. Improvements in desirable traits of the Pacific oyster, Crassostrea gigas, as a result of five generations of selection on the West Coast, USA. Aquaculture. 2016;460:105–15.
Dégremont L. Size and genotype affect resistance to mortality caused by OsHV-1 in Crassostrea gigas. Aquaculture. 2013;416:129–34.
Hick PM, Evans O, Rubio A, Dhand NK, Whittington RJ. Both age and size influence susceptibility of Pacific oysters (Crassostrea gigas) to disease caused by Ostreid herpesvirus-1 (OsHV-1) in replicated field and laboratory experiments. Aquaculture. 2018;489:110–20.
Petton B, Alunno-Bruscia M, Mitta G, Pernet F. Increased growth metabolism promotes viral infection in a susceptible oyster population. Aquac Environ Interact. 2023;15:19–33.
de Lorgeril J, Lucasson A, Petton B, Toulza E, Montagnani C, Clerissi C, Vidal-Dupiol J, Chaparro C, Galinier R, Escoubas J-M, Haffner P, Dégremont L, Charrière GM, Lafont M, Delort A, Vergnes A, Chiarello M, Faury N, Rubio T, Leroy MA, Pérignon A, Régler D, Morga B, Alunno-Bruscia M, Boudry P, Le Roux F, Destoumieux-Garzόn D, Gueguen Y, Mitta G. Immune-suppression by OsHV-1 viral infection causes fatal bacteraemia in Pacific oysters. Nat Commun. 2018;9(1):4215.
Paul-Pont I, Dhand NK, Whittington RJ. Spatial distribution of mortality in Pacific oysters Crassostrea gigas: reflection on mechanisms of OsHV-1 transmission. Dis Aquat Org. 2013;105(2):127–38.
Pernet F, Lagarde F, Jeannée N, Daigle G, Barret J, Le Gall P, Quere C, D’orbcastel ER. Spatial and temporal dynamics of mass mortalities in oysters is influenced by energetic reserves and food quality. PLoS ONE. 2014;9(2): e88469.
Kleinstein SE, Shea PR, Allen AS, Koelle DM, Wald A, Goldstein DB. Genome-wide association study (GWAS) of human host factors influencing viral severity of herpes simplex virus type 2 (HSV-2). Genes Immun. 2019;20(2):112–20.
Boodhoo N, Kamble N, Behboudi S. De novo cholesterol biosynthesis and its trafficking in LAMP-1-positive vesicles are involved in replication and spread of Marek’s disease virus. J Virol. 2020;94(24):e01001-e1020.
Weichhart T, Säemann MD. The PI3K/Akt/mTOR pathway in innate immune cells: emerging therapeutic applications. Ann Rheum Dis. 2008;67(Suppl 3):70–4.
Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kδ and primary immunodeficiencies. Nat Rev Immunol. 2016;16(11):702–14.
Tabatabaeian H, Rao A, Ramos A, Chu T, Sudol M, Lim YP. The emerging roles of WBP2 oncogene in human cancers. Oncogene. 2020;39(24):4621–35.
Genini S, Delputte PL, Malinverni R, Cecere M, Stella A, Nauwynck HJ, Giuffra E. Genome-wide transcriptional response of primary alveolar macrophages following infection with porcine reproductive and respiratory syndrome virus. J Gen Virol. 2008;89(Pt 10):2550.
Green TJ, Raftos D, Speck P, Montagnani C. Antiviral immunity in marine molluscs. J Gen Virol. 2015;96(9):2471–82.
Iwama RE, Moran Y. Origins and diversification of animal innate immune responses against viral infections. Nat Ecol Evol. 2023;7(2):182–93.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6(5): e19379.
Murray KD, Borevitz JO. Axe: rapid, competitive sequence read demultiplexing using a trie. Bioinformatics. 2018;34(22):3924–5.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Peñaloza C, Gutierrez AP, Eöry L, Wang S, Guo X, Archibald AL, Bean TP, Houston RD. A chromosome-level genome assembly for the Pacific oyster Crassostrea gigas. Gigascience. 2021;10(3):giab020.
Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. https://arxiv.org/abs/1303.3997.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008.
Whalen A, Gorjanc G, Hickey JM. AlphaFamImpute: high-accuracy imputation in full-sib families from genotype-by-sequencing data. Bioinformatics. 2020;36(15):4369–71.
Melo AT, Hale I. ‘apparent’: a simple and flexible R package for accurate SNP-based parentage analysis in the absence of guiding information. BMC Bioinformatics. 2019;20(1):1–10.
Fisher RA. Statistical methods for research workers. 5th ed. Edinburgh: Oliver & Boyd; 1934.
Paria SS, Rahman SR, Adhikari K. fastman: A fast algorithm for visualizing GWAS results using Manhattan and QQ plots. 2022. https://www.biorxiv.org/content/10.1101/2022.04.19.488738v1.
Major TJ, Takei R. LocusZoom-like Plots for GWAS Results (v2.1). Zenodo. 2021. https://doi.org/10.5281/zenodo.5154379.
Acknowledgements
We thank Antonia Barela and William Schoeneck for their assistance in the hatchery and field. We thank Hog Island Oyster Co. for providing the planting site and personnel. We thank Ling Jin for access to a biosafety level 2 laboratory in Corvallis, Oregon. We thank Katie Carter for preparing GBS libraries. We thank Michael Banks for providing computational resources at the Center for Quantitative Life Sciences and Oregon State University for hosting those resources.
Funding
Funding for this study was provided by the United States Department of Agriculture—Agricultural Research Service (USDA-ARS) project “Identifying Genetic Factors Associated with the Expression and Regulation of Economically Important Traits in Cultured Pacific Oysters” (Project No. 2076–63000-005–002-S) and the Pacific States Marine Fisheries Commission (PSMFC) project “Preparing for Future Challenges – Threats from Ocean Acidification, Vibrio coralliilyticus and OsHV-1 µvar to West Coast Oyster Farmers” (Award No. NA18NMF4720007).
Author information
Authors and Affiliations
Contributions
KD, TJG, and CL conceived and designed the study. KD, NM, and BS performed the experiments. KD conducted the statistical analysis and wrote the draft manuscript. All authors read, edited, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Crassostrea gigas is not a protected invertebrate.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig. S1. Pedigree of families 30.004, 30.058, 30.062, and 30.065. Ancestry prior to cohort 22 is not shown as families in these cohorts were not spawned using single pair matings.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Divilov, K., Merz, N., Schoolfield, B. et al. Genome-wide allele frequency studies in Pacific oyster families identify candidate genes for tolerance to ostreid herpesvirus 1 (OsHV-1). BMC Genomics 24, 631 (2023). https://doi.org/10.1186/s12864-023-09744-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09744-0