
Abstract
Background
Protists of the family Trypanosomatidae (phylum Euglenozoa) have gained notoriety as parasites affecting humans, domestic animals, and agricultural plants. However, the true extent of the group's diversity spreads far beyond the medically and veterinary relevant species. We address several knowledge gaps in trypanosomatid research by undertaking sequencing, assembly, and analysis of genomes from previously overlooked representatives of this protistan group.
Results
We assembled genomes for twenty-one trypanosomatid species, with a primary focus on insect parasites and Trypanosoma spp. parasitizing non-human hosts. The assemblies exhibit sizes consistent with previously sequenced trypanosomatid genomes, ranging from approximately 18 Mb for Obscuromonas modryi to 35 Mb for Crithidia brevicula and Zelonia costaricensis. Despite being the smallest, the genome of O. modryi has the highest content of repetitive elements, contributing nearly half of its total size. Conversely, the highest proportion of unique DNA is found in the genomes of Wallacemonas spp., with repeats accounting for less than 8% of the assembly length. The majority of examined species exhibit varying degrees of aneuploidy, with trisomy being the most frequently observed condition after disomy.
Conclusions
The genome of Obscuromonas modryi represents a very unusual, if not unique, example of evolution driven by two antidromous forces: i) increasing dependence on the host leading to genomic shrinkage and ii) expansion of repeats causing genome enlargement. The observed variation in somy within and between trypanosomatid genera suggests that these flagellates are largely predisposed to aneuploidy and, apparently, exploit it to gain a fitness advantage. High heterogeneity in the genome size, repeat content, and variation in chromosome copy numbers in the newly-sequenced species highlight the remarkable genome plasticity exhibited by trypanosomatid flagellates. These new genome assemblies are a robust foundation for future research on the genetic basis of life cycle changes and adaptation to different hosts in the family Trypanosomatidae.
Similar content being viewed by others


Background
The kinetoplastid family Trypanosomatidae comprises parasitic flagellates that infect a diverse range of hosts, encompassing vertebrates, arthropods, leeches, plants, and even ciliated protists [1, 2]. For decades, trypanosomatid research was focused on the species causing diseases in humans, domestic animals, and agricultural plants, effectively neglecting the rest of the group. This resulted in significant knowledge gaps, with the first issue being a near absence of genomic data for monoxenous (one-host) members of the family, which predominantly infect insects [3]. Yet, these data are indispensable for understanding the evolutionary transitions from monoxeny to dixeny, which occurred at least three times independently in the evolution of Trypanosomatidae, in the vertebrate-parasitic Leishmania, Trypanosoma, and plant-infecting Phytomonas [4]. In addition, monoxenous trypanosomatids are characterized by an impressive adaptability to various insect hosts worldwide, even though the genetic background of such plasticity is not well understood. The situation has improved in recent years with reports of reference genome sequences for several insect parasites, including representatives of the genera Blastocrithidia, Crithidia, Herpetomonas, Leptomonas, Novymonas, Paratrypanosoma, and Vickermania [5,6,7,8,9,10,11,12]. In-depth analysis of these genomes has proven to be instrumental for shedding light on various aspects of trypanosomatid, and, more generally, eukaryotic biology. For instance, it led to the identification of novel virulence factors in human parasites of the genus Leishmania [7, 13], understanding the metabolic cooperation between trypanosomatids and their bacterial endosymbionts [11, 14, 15], as well as the discovery of novel mechanisms enabling stop-to-sense codon reassignment [12]. Despite recent progress in mitigating the bias towards practically relevant pathogens, the knowledge about several monoxenous genera is still restricted to formal taxonomic descriptions with sequencing data confined to some common phylogenetic markers, such as genes for 18S rRNA, glycosomal glyceraldehyde phosphate dehydrogenase, and spliced leader RNA [16, 17]. Consequently, the exact phylogenetic position of some trypanosomatid genera, e.g., Sergeia, Wallacemonas, and Jaenimonas [18,19,20] remains to be established using whole-genome data.
The second significant gap in trypanosomatid research pertains to the paucity of information on the diversity and biology of dixenous trypanosomatids that do not infect humans. The bulk of Trypanosoma research is focused on salivarian species (Trypanosoma brucei, T. congolense, and T. vivax), as well as the T. cruzi complex, causing severe diseases in humans and domestic animals [21,22,23]. Yet, the genus Trypanosoma is very species-rich, with its members isolated from a variety of sources, including amphibians, birds, fishes, mammals, and reptiles [1].
Lastly, obtaining genome sequences for the closest relatives of medically-, veterinary-, and agriculturally-significant species can provide insight into host switches and life cycle changes in trypanosomatids. Despite the fact that several genomes of these flagellates are currently available, they are not always ideally suitable for comparative analysis. For instance, the closest known relative of the dixenous genus Leishmania, is the monoxenous trypanosomatid Novymonas esmeraldas, whose gene content and metabolism appear to be affected by the presence of an endosymbiotic bacterium [11].
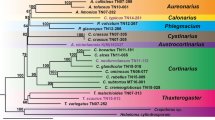
In this study, we aim to address the gaps mentioned above by presenting genome assemblies for twenty-one species of the family Trypanosomatidae, including sixteen monoxenous representatives (Fig. 1). We report the genomes of monoxenous Zelonia costaricensis and Borovskyia barvae, close relatives of dixenous Leishmania [24, 25], as well as that of Obscuromonas modryi, a member of the genus sister to Blastocrithidia spp., which have all three stop codons reassigned as sense [12, 26, 27]. Additionally, we assembled the genomes of five non-human infective Trypanosoma spp. of four subgenera (Haematomonas, Squamatrypanum, Trypanomorpha and Trypanosoma). These data will provide insight into the phylogenetic relationships between these dixenous parasites and highlight genetic changes associated with host switches. We also report several genomes for representatives of the monoxenous genera Crithidia and Wallacemonas, including C. thermophila, a species capable of withstanding elevated temperatures [28] and Wallacemonas sp. TrypX, which was isolated from a rodent host [29]. The genomes of two representatives of the genus Herpetomonas, the closest known relatives of the plant-infecting Phytomonas spp. can shed light on the origin of this peculiar dixenous genus. For Sergeia podlipaevi and Wallacemonas spp., the presented genomes will be instrumental in ascertaining their phylogenetic position, while in the case of Jaenimonas drosophilae it is necessary for the scrutiny of host-parasite interactions in the Jaenimonas-Drosophila experimental model [20].

A cladogram depicting the phylogenetic relationships among trypanosomatids based on the available literature. The number of species within each genus with publicly available genomes is indicated, with the numbers in red representing the genomes sequenced in this study. The dixenous genera are displayed in boxes. Subfamilies are highlighted with a yellow background and their names are shown on the right. Not resolved relationships are indicated by dashed lines
Results
Genome assemblies for monoxenous trypanosomatids and Trypanosoma spp.
The family Trypanosomatidae comprises over twenty recognized genera [1]. However, the majority of sequenced genomes belong to just two of them encompassing medically relevant species of the genera Leishmania and Trypanosoma [30]. We have obtained genome assemblies for representatives of ten trypanosomatid genera, including five (Borovskyia, Jaenimonas, Sergeia, Wallacemonas, and Zelonia), for which no such data were previously available (Table 1). For most species, the isolates used in this work have been obtained from insect hosts: such as Hemiptera (B. barvae, C. thermophila, H. samuelpessoai, O. modryi, W. rigidus, W. collosoma, Wallacemonas sp. Wsd and Z. costaricensis) and Diptera (C. brevicula, J. drosophilae, S. podlipaevi, V. spadyakhi, Wallacemonas sp. 195SL and MBr04). However, Wallacemonas sp. strain TrypX was isolated from a rodent [29], and therefore, might represent another example of a monoxenous trypanosomatid adapted to survival at elevated temperatures of a mammal body in addition to Leptomonas seymouri and Crithidia thermophila [28, 31]. It remains to be elucidated what is the molecular basis of this adaptation. We also sequenced genomes of several trypanosomes from non-human hosts, such as lizards, a toad, a ray, and a bird (Table 1).
These assemblies are based solely on Illumina data, although two different strategies were employed to ensure optimal results (see Materials and Methods for details). The shortest assembly is that of O. modryi (18.2 Mb), and the longest ones are those of C. brevicula (35.4 Mb) and Z. costaricensis (35.3 Mb) (Table 2). In each case, a significant proportion of the assembly is transcribed, ranging from 71 to almost 100% for B. barvae and T. scelopori, respectively (Additional file 1). Regardless of the size, almost all assemblies have BUSCO scores comparable to those for the reference trypanosomatid genomes (Fig. 2; Additional file 1). Only the assemblies of O. modryi and C. brevicula have slightly higher percentages of missing BUSCOs than other species and the reference genomes: 5.4% and 2.3%, respectively, when using Euglenozoa database (Fig. 2; Additional file 1). The high level of completeness of the assemblies and a high level of coherence between the genome assemblies and the reads used to produce them are supported by the results of the k-mer analysis (Fig. 3). The proportion of reads included into the final assembly ranges from 96.74% to 99.22% for H. takarana and W. rigidus, respectively (Fig. 3; Additional file 1). The percentage of genomic reads mapping back to the assembly ranges from ~ 86% to 100% for C. thermophila and H. samuelpessoai, respectively (Table 2; Additional file 1). The assembly error content is minimal as estimated based on the number of homozygous SNPs per 100 kb of genomic sequence ranging from 0.32 for Wallacemonas sp. TrypX to 1.49 for V. spadyakhi (Additional file 1).

Genome assembly completeness. Presence of Benchmarking Universal Single-Copy Orthologs (BUSCOs) from two reference databases: specific Euglenozoa_odb10 (panel A) and more general Eukaryota_odb10 (panel B) is shown

Spectra copy number plots for the genome assemblies. The multiplicity and number of distinct k-mers are plotted on the X and Y axes, respectively. The colors indicate the number of times each read is found in the genome assembly
Importantly, although all the cultures except that of B. barvae were axenic, as a precautionary measure, we screened all genome assemblies for contamination. A few potential contaminating sequences were filtered out from the final assemblies of B. barvae, O. modryi, and all trypanosomes (Additional file 2). In the latter case, a contamination by vertebrate DNA can be explained by cultivation on blood agar (see Materials and Methods).
Analysis of repetitive elements
It is widely recognized that trypanosomatids possess a highly similar repertoire of protein-coding genes, and demonstrate a striking conservation of gene order, known as synteny [32]. An intriguing characteristic that is both remarkably divergent and significantly understudied across trypanosomatid genomes is the extent to which repetitive elements contribute to genome size [33]. To investigate whether there is a discernible pattern across trypanosomatids regarding the prevalence of repetitive elements, we examined this characteristic in all assembled genomes.
Among analyzed assemblies, repetitive DNA is the least abundant in those of W. collosoma and W. rigidus, with repeats representing 2.88% and 3.24% of the total length, respectively (Fig. 4A; Additional file 3). In general, genome assemblies of Wallacemonas spp. are characterized by low repetitive content, with the maximal value of 7.59% documented in the MBr04 strain. The highest proportion of this content is represented by simple and unclassified repeats. In contrast, despite the smallest assembly size, O. modryi contains nearly 44% repetitive content, of which (among categorized repetitive content) retroelements (mainly LTRs and LINEs) are the most frequent (~ 12%).

Content of repetitive DNA and single nucleotide polymorphisms in the genome assemblies. A Bar plot showing proportions of repetitive (dark yellow) and unique DNA (gray). The assembly size in megabases is also indicated for each species on X axis. B Total number of SNPs identified for each genome assembly. Bar charts were produced with the R v. 4.3 package ggplot2
The repetitive content varies greatly among Crithidia spp. The reference assembly of C. fasciculata contains almost 20% of the repeats, followed by C. brevicula with ~ 9% of them, and, finally, C. thermophila demonstrating only ~ 5%. Herpetomonas genomes also differ: H. tarakana possesses a shorter assembly and a higher repetitive content compared to H. samuelpessoai, with most differences originating in proportions of simple repeats (Fig. 4A; Additional file 3). The genomes of Trypanosoma spp. assembled in this study show medium repeat content, ranging from 8.80% to 16.37% in T. platydactyli and T. avium, respectively (Fig. 4A; Additional file 3). Repetitive elements in the genome assemblies of T. avium and T. scelopori consist of mostly simple repeats, while in T. platydactyli retroelements (LTRs and LINEs) are predominant (Fig. 4A; Additional file 3).
The analysis of repeat content of the ten longest scaffolds for each species revealed a relatively even distribution of repeats (Additional file 4). The majority of the repeats identified on both the ten longest scaffolds and the entire assembly are transcribed (Additional files 3 and 4. The proportion of transcribed repeats varies, ranging from approximately 73% in Wallacemonas sp. 195SL to nearly 100% in T. scelopori (Additional file 3).
Single nucleotide polymorphisms and chromosome copy number variation
Variations in ploidy and heterozygosity are recognized as significant factors contributing to genome plasticity across various eukaryotes, including such distantly related organisms as trypanosomatids and fungi [34, 35]. Yet, the extent to which ploidy and heterozygosity drive genetic diversity in Trypanosomatidae is relatively well understood only for human pathogens [34, 36, 37]. Thus, we opted to investigate these features in the species sequenced in this study.
Variant calling allowed estimation of intragenomic variation across the assemblies produced here. The highest total SNP numbers, 289,423 and 254,508, were documented in the monoxenous representatives of the subfamily Leishmaniinae, C. brevicula and Z. costaricensis, respectively (Fig. 4B; Additional file 1). Conversely, all genomes of dixenous trypanosomatids sequenced herein demonstrated the lowest SNP content, with the smallest numbers being 5,037 and 7,177 for T. scelopori and T. platydactyli, respectively (Fig. 4B; Additional file 1). This correlates with the presence of only a single peak corresponding to homozygous content on the spectra copy number plots for the dixenous trypanosomatids, with the heterozygous peak being either entirely absent or barely visible (Fig. 3).
We conducted coverage-based estimation of somy for the 50 longest scaffolds (used as chromosome proxies) for each species. Our analysis assumed that the median genome coverage reflects a disomic state. Only five out of twenty-one species with the assembled genomes appear to be diploid: Z. costaricensis, J. drosophilae, V. spadyakhi, S. podlipaevi, and Wallacemonas sp. MBr04 (Fig. 5; Additional files 5 and 6). Other genomes exhibit variable levels of aneuploidy, with the second most frequent state (after disomy) being trisomy. Aneuploidy is especially pronounced in B. barvae as well as in most Trypanosoma and Wallacemonas spp., where up to 25% of scaffolds exhibit notably altered coverage, in some cases consistent with tetra- and, even, pentasomy (Fig. 5). Importantly, in contrast to the aneuploidy in other species, the genomes of O. modryi and T. mega do not possess supernumerary chromosomes and their assemblies feature three and one scaffolds with reduced somy levels, respectively (Fig. 5; Additional files 5 and 6). In order to check if there is a correlation between somy levels and repeat content, we conducted a comparison between the proportion of repeats identified in disomic scaffolds and scaffolds with other somy levels (Additional file 5). However, we did not observe any statistically significant differences between the two groups (p-value < 0.01).

Stacked barplots showing somy estimates for 50 longest scaffolds for each trypanosomatid species. Numerical values in the plot indicate the number of scaffolds with each somy level. Somy estimation was performed based on the ratio of median-of-means coverage for 1 kb windows to the median genome coverage. Somy values are color coded: blue – monosomy, gray – disomy, dark yellow – trisomy, orange – tetrasomy, brown – pentasomy
An independent approach for ploidy estimation, based on the analysis of heterozygous k-mer pairs, further supports our assumption that disomy is the prevailing state for each species (Additional file 7). This method considers all genomic information in trimmed sequencing reads, not just the largest scaffolds, and reveals the presence of k-mers indicative of aneuploidy even in species where diploidy was initially inferred using coverage analysis of the 50 largest scaffolds (Fig. 5, Additional files 5, 6 and 7. To fully comprehend whether the respective k-mers originate from chromosomes exhibiting states other than disomy or, for example, from segmental duplications, chromosome-level assemblies are necessary.
Discussion
The assemblies obtained in this study cover a substantial number of monoxenous trypanosomatid genera with no previously available sequencing data, as well as several Trypanosoma spp. from non-human hosts (Fig. 1; Table 1). These data provide further evidence of the previously observed high genome size variability in trypanosomatids apparently reflecting distinct life strategies. The difference in size between the smallest (O. modryi) and largest (C. brevicula and Z. costaricensis) genome assemblies was almost twofold (Table 2). While we do not know much about the biology of the latter species, we suggest that one of the factors explaining the size difference between the remaining two can be the host specificity. Indeed, O. modryi (referred to as TU6/7 C before the formal description) is restricted to the heteropteran family Alydidae [26, 38]. In contrast, C. brevicula has a very wide host range and can successfully establish infections at least in phytophagous (Miridae) and predatory (Nabidae and Gerridae) true bugs, various flies (Antomyidae, Calliphoridae, Heleomyzidae, Muscidae, Sepsidae), as well as in mosquitoes (Culicidae) [19, 39,40,41]. Undoubtedly, the host range cannot be the only factor explaining these differences and a thorough analysis of the genomic composition should better clarify the underlying reasons.
Another highly variable characteristic of trypanosomatid genomes is the proportion of repetitive DNA [33]. We observe an approximately 15-fold span in estimates of repetitive DNA content among the assemblies obtained in this study: from 3% in W. collosoma to 44% in O. modryi (Fig. 4A; Additional file 3). The situation with the latter species is paradoxical: it has the highest proportion of repeats and the smallest genome size (Fig. 4A). In general, genome shrinkage can be stipulated by the growing dependence of an endosymbiont (including endoparasites) on the host or simplification of the lifestyle due to stable conditions (relevant also for free-living organisms) [42, 43]. Natural selection supports genome reduction in such conditions, because this allows faster genome replication [44]. Repetitive DNA is one of usual targets for genomic reduction, as demonstrated by studies elucidating the factors contributing to genome size variation in microsporidians, a large group of fungi-related unicellular eukaryotes infecting a wide range of hosts [45]. In O. modryi, on the contrary, we observe the expansion of repeats. This suggests the existence of an underlying strong evolutionary force competing with the one leading to genome shrinkage. Previously it has been argued that expansion of repetitive DNA and, in particular, simple repeats can enhance genome plasticity [46, 47] and here it also can be a case. A slightly higher proportion of missing BUSCOs compared to other assemblies indicates that the competing evolutionary forces driving O. modryi genome shrinkage are also quite prominent, which is evidenced by the loss of some protein-coding genes, which are typically highly conserved throughout Euglenozoa (Fig. 2). Repetitive DNA of trypanosomatids includes members of multigene families (e.g., variant surface glycoproteins, trans-sialidases, mucins), transposable elements, and satellite DNA repeats [33]. Before this work, Trypanosoma spp. were considered to have the highest proportions of repetitive DNA. For example, it comprises 51.3% of the T. cruzi genome and 20.7% (or ~ 26%, as estimated in our study) of that of T. brucei TREU927 [33]. Repeats in these species mainly consist of multigene families and retroelements [33]. Although our analyses did not include multigene families except for those of non-protein coding genes [48], we find that repetitive content of assemblies of Trypanosoma spp. of non-human hosts shows almost two-fold differences, from 9% in T. platydactyli to 16% in T. avium (Additional file 3). Furthermore, a substantial portion of the annotated content consists of simple repeats (up to 11% of the assembly in T. avium) and retroelements (up to 3% T. platydactyli) (Additional file 3). Although the proportion of repetitive content represented by multigene families has yet to be determined for the genomes sequenced in this study, our analyses suggest that the content of interspersed repeats and low complexity DNA sequences represents one of the factors responsible for intrageneric variations in trypanosomatid genome sizes. Most of the repeats identified in the analyzed assemblies are transcribed and appear to be evenly distributed along the longest scaffolds (Additional files 3 and 4). Of note, repeats, especially those exceeding the length of sequencing reads, can negatively affect assembly quality and lead to various artefacts in the subsequent analyses [49]. Therefore, the values we obtained for proportions of repetitive elements may be underestimated for our short-reads-based Illumina data, since some repeats might have been collapsed and/or not assembled.
Previous studies detected aneuploidy in Leishmania spp. [36, 50, 51], T. cruzi [52], Leptomonas pyrrhocoris [7], Angomonas deanei [53], Endotrypanum spp. and Porcisia spp. [54], as well as in T. brucei brucei, but not in the infective to humans T. brucei gambiense and T. brucei rhodesiense [55, 56]. However, it has never been systematically investigated for the whole family. Here we revealed aneuploidy in sixteen out of twenty-one genomes representing multiple lineages of Trypanosomatidae (Fig. 5) and, therefore, argue that this phenomenon is widespread among these flagellates. Trypanosomatids are predisposed to aneuploidy because, with only a few exceptions, they lack gene-specific transcriptional regulation for the majority of protein-coding genes [57]. It remains to be investigated further what mechanisms of tolerance to aneuploidy they have evolved, since the main problem to solve in this case is the meiotic segregation [58]. Whatever the solution can be, the advantage of chromosome copy number variation is a simple way to permanently change (increase or decrease, in case of poly- and monosomy, respectively) the expression of multiple genes, such as those coding, for example, for virulence factors, in an adaptation to changing environmental conditions or new hosts [59,60,61]. Nevertheless, the analysis of coverage using 50 largest scaffolds, led to the identification of several truly diploid species in our study (Z. costaricensis, J. drosophilae, V. spadyakhi, S. podlipaevi, and Wallacemonas sp. MBr04). Similarly to T. brucei, they may represent exceptions, rather than a rule within the family [62]. It remains to be elucidated why such species are diploid and how aneuploidy can affect sexual process in trypanosomatids. Of note, an independent approach to ploidy estimation, based on the analysis of heterozygous k-mer pairs, indicates that the putatively diploid species mentioned above show some indications of aneuploidy and/or other types of sequence duplication (Additional file 7).
Heterozygosity, the presence of two distinct alleles at a specific locus, is influenced by a number of factors including mode of reproduction [63] and ploidy [35]. In aneuploid fungal pathogens, loss of heterozygosity can stem from the chromosome gain with the subsequent loss of the heterozygous homolog [35]. Although we noticed that analyzed trypanosomes with lowest number of SNPs (T. scleropori and T. platydactyli) also have a relatively high aneuploidy level (with 8 out of 50 analyzed scaffolds showing somy levels different from disomy) (Fig. 5), we do not see a clear correlation between aneuploidy levels and loss of heterozygosity in trypanosomatids as observed in fungi [64]. This indicates that other factors in addition to ploidy variations define heterozygosity levels in these parasitic protists.
The assemblies generated in the course of this study do much to fill veritable canyons in the genetic and taxonomic record of trypanosomatids. However, important work remains to be done. The scaffolds assembled here are proxies for actual chromosomes. Assembly of full chromosomes will be desirable in the future to accurately infer genetic details such as chromosome copy number variations. Confirmation that most chromosomes in each genome are disomic will also validate our ploidy estimates for each species. However, until we or others generate such assemblies, the current work provides rich resources for many other types of biological comparisons within the richly varied trypanosomatids.
Conclusions
In this work, we present genome assemblies for twenty-one trypanosomatid species, including overlooked monoxenous species and dixenous Trypanosoma spp. parasitizing non-human hosts. As judged from multiple standard metrics, our assemblies are highly contiguous and complete, making them valuable resource for various future analyses. We revealed relatively high intra- and intergeneric genome diversity in trypanosomatids in terms of size, repeat content, and ploidy. The new assemblies will be instrumental for establishing the molecular basis of tolerance to vertebrate host temperatures, elucidating impacts of life cycle changes and host switches on the genome, studying the origin of stop codon reassignment in Blastocrithidiinae, and many other phenomena that can now be approached using genomic data.
Materials and methods
DNA and RNA isolation
The sources of trypanosomatid cultures used in this work are specified in Table 1. All monoxenous species were cultivated at 23 °C in Schneider’s Drosophila medium (SDM) (Merck, St. Louis, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, USA), 100 μg/ml of streptomycin and 100 Units/ml of penicillin (Merck). Trypanosoma spp. were cultivated on biphasic blood agar overlaid with supplemented SDM.
Total genomic DNA was isolated from 10 ml of a culture either using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, or by the standard phenol–chloroform method. RNA isolation was performed using the RNeasy minikit (Qiagen) following the manufacturer’s protocol. Species identity was confirmed as in [65].
Genome and transcriptome sequencing
DNA and RNA libraries were prepared and sequenced using Illumina instruments at Macrogen Europe (Amsterdam, Netherland), Institute of Applied Biotechnologies (Praha – Strašnice, Czech Republic), or Biomarker Technologies BMKGene (Münster, Germany) (Additional file 1). Depending on species, this yielded 14–128 million of 100 or 150 nt long paired-end genomic reads (Additional file 1). To facilitate future genome annotation process 16–93 million Illumina paired-end transcriptomic reads were produced for the same species.
Genome assembly
Raw Illumina sequencing reads were adapter- and quality trimmed using Fastp v.0.20.1 [66] or BBDuk v.38.98 from BBMap package [67], and only paired-end reads with a minimum length of 75 and 50 nt were retained for further analysis in the case of genomic and transcriptomic data, respectively (Additional file 8, example command lines). Read quality and adapter content were assessed before and after the trimming with FastQC v.0.11.9 [68]. Genomic reads were subjected to a multiple sequence alignment-based error correction procedure using Karect [69]. The results of the error correction step were assessed with Karect ‘-align’ and ‘-eval’ on the preliminary assemblies. The read correction procedure resulted in the reduction of the area under the curve corresponding to the low-frequency k-mers estimated using KAT v.2.4.2 [70] with default settings.
Trimmed genomic reads were assembled de novo using two strategies and the best result was preserved. The Spades-Platanus strategy included using SPAdes v.3.13.0 [71] to assemble into contigs and Platanus v.1.2.4 [72] for scaffolding in two rounds intercalated with GapCloser v.1.12 module from SOAPde-novo2 for gap filling [73]. It showed better results for J. drosophilae, H. tarakana, B. barvae, T. avium, T. scelopori, T. boissoni, T. mega, T. platydactyli, W. rigidus, W. collosoma, Wallacemonas sp. 195SL, and O. modryi. The Platanus-solo strategy consisted in using Platanus for assembly and two-round scaffolding with gap filling in GapCloser. This strategy worked better for C. thermophila, C. brevicula, Z. costaricensis, H. samuelpessoai, V. spadyakhi, S. podlipaevi, Wallacemonas spp. MBr04, Wsd, and TrypX.
Overall, the selection of an assembly strategy for each species was carried out based on estimating the following assembly parameters: i) N50 as a measure of contiguity; ii) percentage of missing universal single-copy orthologs as a measure of completeness (BUSCO v.5 and Euglenozoa_odb10 and Eukaryota_odb10 as reference databases) [74]; iii) completeness estimated using k-mer analysis results produced using KAT v.2.4.2 [70]; iv) total gap length; v) percentage of homozygous SNPs as measure of accuracy; vi) size of the largest scaffold estimated using QUAST v.5.0.2 [75]; vii) percentage of sequencing reads mapping back to the assembly.
Trimmed transcriptomic reads were mapped to the assemblies using Bwa-mem2 [76], and the resulting alignments were sorted with SAMtools v.1.16.1 [77]. For estimating the transcribed portion of the genome, GTF files produced using Cufflinks v.2.2.1 [78] were analyzed with SeqKit v.0.16.1 [79].
Assembly decontamination
The genome assemblies were checked for potential contamination with BlobTools v.1.1.1 [80]. The scaffolds satisfying the following criteria were discarded: 1) shorter than 500 nucleotides; 2) showing high-quality BLASTN hits (i.e., nucleotide sequence identity > 95% and query coverage > 85%) to non-euglenozoan sequences in NCBI nucleotide (nt) database (download date: 2022–05-08). Scaffolds with non-euglenozoan hits below the removal threshold were verified either using DIAMOND v.2.0.15 [81] in the sensitive mode or by BLASTX and kept in the final assembly if euglenozoan sequences were retrieved as best hits. The BLAST package v.2.13.0 was used for the homology searches mentioned above [82].
Different scaffold filtering criteria were applied to the genome assembly of B. barvae, since this species cannot be cultivated without accompanying yeast [83]. A preliminary assembly was produced, and the following scaffolds were removed: 1) with fungal sequences as best hits; 2) unannotated sequences demonstrating genomic read coverage below 63 × and grouping with fungal sequences according to the BlobTools analysis (Additional file 2). The genome was then re-assembled as described above using the reads mapping to the remaining scaffolds.
Repeat analysis
The final genome assemblies were submitted to RepeatModeler v.2.0.3 [84] with the LTRStruct parameter for long terminal repeat (LTR) retroelements search. RepeatMasker v.4.1.2 [48] with sensitive slow search was used for repeats’ identification and soft masking using the database built with RepeatModeler. Statistical analysis of repeat content between disomic and other somy level scaffolds was performed using two-tailed t-test with a significance level of 0.01 for the species where at least two scaffolds demonstrated somy level distinct from 2n based on the coverage analysis.
For calculating the proportion of transcribed repeats, a GFF file with the repeat coordinates was used as an input for featureCounts v.2.0.1 [85] along with the BAM file containing transcriptomic reads mapped on the genome assembly. The read pairs mapping to the same scaffold and strand only once were counted. A repeat was considered transcribed if at least one read mapped to it.
J-Circos2 v.1.0 interface for Circos plot was used for visualization of repetitive content of the ten largest scaffolds for each species [86]. The GC skew was calculated for 1 kb non-overlapping windows using GCcalc [87], and the transcriptomic coverage track data was generated using bamCoverage v.3.5.2 implemented in deepTools2 software [88].
Ploidy analysis
For each scaffold, mean read depths were calculated in successive non-overlapping 1 kb windows using Mosdepth v.0.3.3 with default settings [89] and then served to obtain a median-of-means (MOM) estimate. The median genome coverage was calculated based on those of the 50 largest scaffolds for each species. The ratio (R) between the scaffold’s MOM coverage and the median genome coverage was used to define somy: 0.25 ≥ R < 0.75 – monosomic; 0.75 ≥ R ≤ 1.25 – disomic; 1.25 > R ≤ 1.75 – trisomic, etc. The somy of each scaffold was inferred assuming that most of the scaffolds/chromosomes are in the disomic state. To provide a more detailed visualization of scaffold/chromosome copy number variation, the copy number was estimated for each of the 1 kb windows using the same strategy as above, but now dividing mean coverage within each 1 kb window by the genome median coverage (Additional file 6). The results were visualized using R packages ggplot2 v.3.4.2 and dplyr v.1.0.8 [49, 90].
In another approach to ploidy estimation, trimmed and corrected sequencing reads were used for k-mer analysis by KMC v.3.1.1 [91] with the subsequent ploidy inference using Smudgeplot v.0.2.5 [92].
Variant calling
Trimmed and corrected genomic reads were mapped back to the assemblies using Bwa-mem2 [76], and the resulting alignments were sorted with SAMtools v.1.16.1 [77]. The mapping rates and median insert size were assessed with ‘stats’ from BamTools v.2.4.1 [93].
After mapping, genomic read duplicates were removed with MarkDuplicates, and the reads were locally realigned using IndelRealigner tools of GATK v.4.2 [94] with the default settings, except for REMOVE_DUPLICATES = true. Variant calling was performed using Platypus v.0.8.1 [95] with the default settings. Identified variants were extracted using GATK VariantsToTable.
Availability of data and materials
Raw sequencing reads were deposited in the NCBI database under BioProject accessions PRJNA949447 and PRJNA543408. The assemblies are deposited in NCBI under the following accession numbers: JASAOP000000000 for C. thermophila, JASAOD000000000 for C. brevicula, JASAOO000000000 for Z. costaricensis, JASAON000000000 for J. drosophilae, JASAOC000000000 for H. samuelpessoai, JASAOB000000000 for H. tarakana, JASAOA000000000 for V. spadyakhi, JASANZ000000000 for S. podlipaevi, JASAOM000000000 for B. barvae, JASANY000000000 for T. avium, JASANX000000000 for T. scelopori, JASANW000000000 for T. boissoni, JASANV000000000 for T. mega, JASANU000000000 for T. platydactyli, JASAOL000000000 for W. rigidus, JASANT000000000 for W. collosoma, JASANS000000000 for Wallacemonas sp. 195SL, JASANR000000000 for Wallacemonas sp. MBr04, JASANQ000000000 for Wallacemonas sp. Wsd, JASANP000000000 for Wallacemonas sp. Trypx, and JAKVQF000000000 for O. modryi. All other data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Kostygov AY, Karnkowska A, Votýpka J, Tashyreva D, Maciszewski K, Yurchenko V, Lukeš J. Euglenozoa: taxonomy, diversity and ecology, symbioses and viruses. Open Biol. 2021;11(3):200407.
Lukeš J, Butenko A, Hashimi H, Maslov DA, Votýpka J, Yurchenko V. Trypanosomatids are much more than just trypanosomes: clues from the expanded family tree. Trends Parasitol. 2018;34(6):466–80.
Maslov DA, Votýpka J, Yurchenko V, Lukeš J. Diversity and phylogeny of insect trypanosomatids: all that is hidden shall be revealed. Trends Parasitol. 2013;29(1):43–52.
Lukeš J, Skalický T, Týč J, Votýpka J, Yurchenko V. Evolution of parasitism in kinetoplastid flagellates. Mol Biochem Parasitol. 2014;195(2):115–22.
Runckel C, DeRisi J, Flenniken ML. A draft genome of the honey bee trypanosomatid parasite Crithidia mellificae. PLoS ONE. 2014;9(4):e95057.
Skalický T, Dobáková E, Wheeler RJ, Tesařová M, Flegontov P, Jirsová D, Votýpka J, Yurchenko V, Ayala FJ, Lukeš J. Extensive flagellar remodeling during the complex life cycle of Paratrypanosoma, an early-branching trypanosomatid. Proc Natl Acad Sci U S A. 2017;114(44):11757–62.
Flegontov P, Butenko A, Firsov S, Kraeva N, Eliáš M, Field MC, Filatov D, Flegontova O, Gerasimov ES, Hlaváčová J, et al. Genome of Leptomonas pyrrhocoris: a high-quality reference for monoxenous trypanosomatids and new insights into evolution of Leishmania. Sci Rep. 2016;6:23704.
Schmid-Hempel P, Aebi M, Barribeau S, Kitajima T, du Plessis L, Schmid-Hempel R, Zoller S. The genomes of Crithidia bombi and C. expoeki, common parasites of bumblebees. PLoS One. 2018;13(1):e0189738.
Sloan MA, Brooks K, Otto TD, Sanders MJ, Cotton JA, Ligoxygakis P. Transcriptional and genomic parallels between the monoxenous parasite Herpetomonas muscarum and Leishmania. PLoS Genet. 2019;15(11):e1008452.
Opperdoes FR, Butenko A, Zakharova A, Gerasimov ES, Zimmer SL, Lukeš J, Yurchenko V. The remarkable metabolism of Vickermania ingenoplastis: Genomic predictions. Pathogens. 2021;10(1):68.
Zakharova A, Saura A, Butenko A, Podešvová L, Warmusová S, Kostygov AY, Nenarokova A, Lukeš J, Opperdoes FR, Yurchenko V. A new model trypanosomatid, Novymonas esmeraldas: Genomic perception of its “Candidatus Pandoraea novymonadis” endosymbiont. mBio. 2021;12(4):e0160621.
Kachale A, Pavlíková Z, Nenarokova A, Roithová A, Durante IM, Miletínová P, Záhonová K, Nenarokov S, Votýpka J, Horáková E, et al. Short tRNA anticodon stem and mutant eRF1 allow stop codon reassignment. Nature. 2023;613(7945):751–8.
Ishemgulova A, Kraeva N, Hlaváčová J, Zimmer SL, Butenko A, Podešvová L, Leštinová T, Lukeš J, Kostygov A, Votýpka J, et al. A putative ATP/GTP binding protein affects Leishmania mexicana growth in insect vectors and vertebrate hosts. PLoS Negl Trop Dis. 2017;11(7):e0005782.
Morales J, Kokkori S, Weidauer D, Chapman J, Goltsman E, Rokhsar D, Grossman AR, Nowack EC. Development of a toolbox to dissect host-endosymbiont interactions and protein trafficking in the trypanosomatid Angomonas deanei. BMC Evol Biol. 2016;16(1):247.
Zakharova A, Tashyreva D, Butenko A, Morales J, Saura A, Svobodová M, Poschmann G, Nandipati S, Zakharova A, Noyvert D, et al. A neo-functionalized homolog of host transmembrane protein controls localization of bacterial endosymbionts in the trypanosomatid Novymonas esmeraldas. Curr Biol. 2023;33(13):2690-2701.e5.
d’Avila-Levy CM, Boucinha C, Kostygov A, Santos HL, Morelli KA, Grybchuk-Ieremenko A, Duval L, Votýpka J, Yurchenko V, Grellier P, et al. Exploring the environmental diversity of kinetoplastid flagellates in the high-throughput DNA sequencing era. Mem Inst Oswaldo Cruz. 2015;110(8):956–65.
Maslov DA, Opperdoes FR, Kostygov AY, Hashimi H, Lukeš J, Yurchenko V. Recent advances in trypanosomatid research: genome organization, expression, metabolism, taxonomy and evolution. Parasitology. 2019;146(1):1–27.
Svobodová M, Zídková L, Čepička I, Oborník M, Lukeš J, Votýpka J. Sergeia podlipaevi gen. nov., sp. nov. (Trypanosomatidae, Kinetoplastida), a parasite of biting midges (Ceratopogonidae, Diptera). Int J Syst Evol Microbiol. 2007;57(Pt 2):423–32.
Kostygov AY, Grybchuk-Ieremenko A, Malysheva MN, Frolov AO, Yurchenko V. Molecular revision of the genus Wallaceina. Protist. 2014;165(5):594–604.
Hamilton PT, Votýpka J, Dostalova A, Yurchenko V, Bird NH, Lukeš J, Lemaitre B, Perlman SJ. Infection dynamics and immune response in a newly described Drosophila-trypanosomatid association. mBio. 2015;6(5):e01356-01315.
Lukeš J, Kachale A, Votypka J, Butenko A, Field MC. African trypanosome strategies for conquering new hosts and territories: the end of monophyly? Trends Parasitol. 2022;38(9):724–36.
Ramirez JL. Trypanosoma cruzi genome 15 years later: What has been accomplished? Trop Med Infect Dis. 2020;5(3):129.
Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest. 2008;118(4):1301–10.
Barratt J, Kaufer A, Peters B, Craig D, Lawrence A, Roberts T, Lee R, McAuliffe G, Stark D, Ellis J. Isolation of novel trypanosomatid, Zelonia australiensis sp. nov. (Kinetoplastida: Trypanosomatidae) provides support for a Gondwanan origin of dixenous parasitism in the Leishmaniinae. PLoS Negl Trop Dis. 2017;11(1):e0005215.
Kostygov AY, Yurchenko V. Revised classification of the subfamily Leishmaniinae (Trypanosomatidae). Folia Parasitol. 2017;64:020.
Lukeš J, Tesařová M, Yurchenko V, Votýpka J. Characterization of a new cosmopolitan genus of trypanosomatid parasites, Obscuromonas gen. nov. (Blastocrithidiinae subfam. nov.). Eur J Protistol. 2021;79:125778.
Záhonová K, Kostygov A, Ševčíková T, Yurchenko V, Eliáš M. An unprecedented non-canonical nuclear genetic code with all three termination codons reassigned as sense codons. Curr Biol. 2016;26(17):2364–9.
Ishemgulova A, Butenko A, Kortišová L, Boucinha C, Grybchuk-Ieremenko A, Morelli KA, Tesařová M, Kraeva N, Grybchuk D, Pánek T, et al. Molecular mechanisms of thermal resistance of the insect trypanosomatid Crithidia thermophila. PLoS ONE. 2017;12(3):e0174165.
Morsy TA, Schnur LF, Feinsod FM, Michael SA, Saah A, Salama MM, Wahba MM. The discovery and preliminary characterization of a novel trypanosomatid parasite from Rattus norvegicus and stray dogs from Alexandria. Egypt Ann Trop Med Parasitol. 1988;82(5):437–44.
Yurchenko V, Butenko A, Kostygov AY. Genomics of Trypanosomatidae: where we stand and what needs to be done? Pathogens. 2021;10(9):1124.
Kraeva N, Butenko A, Hlaváčová J, Kostygov A, Myškova J, Grybchuk D, Leštinová T, Votýpka J, Volf P, Opperdoes F, et al. Leptomonas seymouri: adaptations to the dixenous life cycle analyzed by genome sequencing, transcriptome profiling and co-infection with Leishmania donovani. PLoS Pathog. 2015;11(8):e1005127.
El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309(5733):404–9.
Pita S, Diaz-Viraque F, Iraola G, Robello C. The Tritryps comparative repeatome: insights on repetitive element evolution in trypanosomatid pathogens. Genome Biol Evol. 2019;11(2):546–51.
Cruz-Saavedra L, Schwabl P, Schwabl P, Vallejo GA, Carranza JC, Muñoz M, Patino LH, Paniz-Mondolfi A, Llewellyn MS, Ramirez JD. Genome plasticity driven by aneuploidy and loss of heterozygosity in Trypanosoma cruzi. Microb Genom. 2022;8(6):mgen000843.
Bennett RJ, Forche A, Berman J. Rapid mechanisms for generating genome diversity: whole ploidy shifts, aneuploidy, and loss of heterozygosity. Cold Spring Harb Perspect Med. 2014;4(10):a019604.
Mannaert A, Downing T, Imamura H, Dujardin JC. Adaptive mechanisms in pathogens: universal aneuploidy in Leishmania. Trends Parasitol. 2012;28(9):370–6.
Franssen SU, Durrant C, Stark O, Moser B, Downing T, Imamura H, Dujardin JC, Sanders MJ, Mauricio I, Miles MA, et al. Global genome diversity of the Leishmania donovani complex. eLife. 2020;9:e51243.
Králová J, Grybchuk-Ieremenko A, Votýpka J, Novotný V, Kment P, Lukeš J, Yurchenko V, Kostygov AY. Insect trypanosomatids in Papua New Guinea: high endemism and diversity. Int J Parasitol. 2019;49(13–14):1075–86.
Schoener E, Uebleis SS, Cuk C, Nawratil M, Obwaller AG, Zechmeister T, Lebl K, Radrova J, Zittra C, Votypka J, et al. Trypanosomatid parasites in Austrian mosquitoes. PLoS ONE. 2018;13(4):e0196052.
Ganyukova AI, Zolotarev AV, Frolov AO. Geographical distribution and host range of monoxenous trypanosomatid Crithidia brevicula (Frolov et Malysheva, 1989) in the northern regions of Eurasia. Protistology. 2020;14:70–8.
Kostygov AY, Malysheva MN, Ganyukova AI, Razygraev AV, Drachko DO, Yurchenko V, Agasoi VV, Frolov AO. The roles of mosquitoes in the circulation of monoxenous trypanosomatids in temperate climates. Pathogens. 2022;11(11):1326.
Husnik F, Keeling PJ. The fate of obligate endosymbionts: reduction, integration, or extinction. Curr Opin Genet Dev. 2019;58–59:1–8.
Mendonça AG, Alves RJ, Pereira-Leal JB. Loss of genetic redundancy in reductive genome evolution. PLoS Comput Biol. 2011;7(2):e1001082.
Wolf YI, Koonin EV. Genome reduction as the dominant mode of evolution. BioEssays. 2013;35(9):829–37.
Peyretaillade E, El Alaoui H, Diogon M, Polonais V, Parisot N, Biron DG, Peyret P, Delbac F. Extreme reduction and compaction of microsporidian genomes. Res Microbiol. 2011;162(6):598–606.
Yuan J, Zhang X, Wang M, Sun Y, Liu C, Li S, Yu Y, Gao Y, Liu F, Zhang X, et al. Simple sequence repeats drive genome plasticity and promote adaptive evolution in penaeid shrimp. Commun Biol. 2021;4(1):186.
Fischer G, Rocha EPC, Brunet F, Vergassola M, Dujon B. Highly variable rates of genome rearrangements between hemiascomycetous yeast lineages. PloS Genet. 2006;2(3):253–61.
Wickham H. ggplot2: Elegant graphics for data analysis. New York: Springer-Verlag; 2016.
Negreira GH, de Groote R, Van Giel D, Monsieurs P, Maes I, de Muylder G, Van den Broeck F, Dujardin JC, Domagalska MA. The adaptive roles of aneuploidy and polyclonality in Leishmania in response to environmental stress. EMBO Rep. 2023:e57413.
Negreira GH, Monsieurs P, Imamura H, Maes I, Kuk N, Yagoubat A, Van den Broeck F, Sterkers Y, Dujardin JC, Domagalska MA. High throughput single-cell genome sequencing gives insights into the generation and evolution of mosaic aneuploidy in Leishmania donovani. Nucleic Acids Res. 2022;50(1):293–305.
Minning TA, Weatherly DB, Flibotte S, Tarleton RL. Widespread, focal copy number variations (CNV) and whole chromosome aneuploidies in Trypanosoma cruzi strains revealed by array comparative genomic hybridization. BMC Genom. 2011;12:139.
Davey JW, Catta-Preta CMC, James S, Forrester S, Motta MCM, Ashton PD, Mottram JC. Chromosomal assembly of the nuclear genome of the endosymbiont-bearing trypanosomatid Angomonas deanei. G3-Genes Genom Genet. 2021;11(1):jkaa018.
Albanaz ATS, Gerasimov ES, Shaw JJ, Sádlová J, Lukeš J, Volf P, Opperdoes FR, Kostygov AY, Butenko A, Yurchenko V. Genome analysis of Endotrypanum and Porcisia spp., closest phylogenetic relatives of Leishmania, highlights the role of amastins in shaping pathogenicity. Genes. 2021;12(3):444.
Mulindwa J, Ssentamu G, Matovu E, Kamanyi Marucha K, Aresta-Branco F, Helbig C, Clayton C. In vitro culture of freshly isolated Trypanosoma brucei brucei bloodstream forms results in gene copy-number changes. PLoS Negl Trop Dis. 2021;15(9):e0009738.
Cosentino RO, Brink BG, Siegel TN. Allele-specific assembly of a eukaryotic genome corrects apparent frameshifts and reveals a lack of nonsense-mediated mRNA decay. NAR Genom Bioinform. 2021;3(3):lqab082.
Clayton C. Regulation of gene expression in trypanosomatids: living with polycistronic transcription. Open Biol. 2019;9(6):190072.
Boynton PJ, Janzen T, Greig D. Modeling the contributions of chromosome segregation errors and aneuploidy to Saccharomyces hybrid sterility. Yeast. 2018;35(1):85–98.
Reis-Cunha JL, Rodrigues-Luiz GF, Valdivia HO, Baptista RP, Mendes TA, de Morais GL, Guedes R, Macedo AM, Bern C, Gilman RH, et al. Chromosomal copy number variation reveals differential levels of genomic plasticity in distinct Trypanosoma cruzi strains. BMC Genom. 2015;16(1):499.
Farrer RA, Henk DA, Garner TW, Balloux F, Woodhams DC, Fisher MC. Chromosomal copy number variation, selection and uneven rates of recombination reveal cryptic genome diversity linked to pathogenicity. PLoS Genet. 2013;9(8):e1003703.
Laffitte MN, Leprohon P, Papadopoulou B, Ouellette M. Plasticity of the Leishmania genome leading to gene copy number variations and drug resistance. F1000Re. 2016;5:2350.
Almeida LV, Coqueiro-Dos-Santos A, Rodriguez-Luiz GF, McCulloch R, Bartholomeu DC, Reis-Cunha JL. Chromosomal copy number variation analysis by next generation sequencing confirms ploidy stability in Trypanosoma brucei subspecies. Microb Genom. 2018;4(10):e000223.
Gutiérrez-Corbo C, Domínguez-Asenjo B, Martínez-Valladares M, Pérez-Pertejo Y, García-Estrada C, Balaña-Fouce R, Reguera RM. Reproduction in trypanosomatids: Past and present. Biology. 2021;10(6):471.
Dutta A, Dutreux F, Schacherer J. Loss of heterozygosity spectrum depends on ploidy level in natural yeast populations. Mol Biol Evol. 2022;39(11):msac214.
Yurchenko V, Kostygov A, Havlová J, Grybchuk-Ieremenko A, Ševčíková T, Lukeš J, Ševčík J, Votýpka J. Diversity of trypanosomatids in cockroaches and the description of Herpetomonas tarakana sp. n. J Eukaryot Microbiol. 2016;63(2):198–20.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinform. 2018;34(17):884–90.
Bushnell B, Rood J, Singer E. BBMerge - accurate paired shotgun read merging via overlap. PLoS ONE. 2017;12(10):e0185056.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2019. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Allam A, Kalnis P, Solovyev V. Karect: accurate correction of substitution, insertion and deletion errors for next-generation sequencing data. Bioinform. 2015;31(21):3421–8.
Mapleson D, Garcia Accinelli G, Kettleborough G, Wright J, Clavijo BJ. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinform. 2017;33(4):574–6.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, Yabana M, Harada M, Nagayasu E, Maruyama H, et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24(8):1384–95.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1(1):18.
Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021;38(10):4647–54.
Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. Versatile genome assembly evaluation with QUAST-LG. Bioinform. 2018;34(13):142–50.
Vasimuddin M, Misra S, Li H, Aluru S. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. Int Parall Distrib. 2019:314–324.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10:giab008.
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–5.
Shen W, Le S, Li Y, Hu F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE. 2016;11(10):e0163962.
Laetsch DR, Blaxter ML. BlobTools: Interrogation of genome assemblies [version 1; peer review: 2 approved with reservations]. F1000Res. 2017;6:1287.
Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18(4):366–8.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinform. 2009;10:421.
Maslov DA, Yurchenko VY, Jirků M, Lukeš J. Two new species of trypanosomatid parasites isolated from Heteroptera in Costa Rica. J Eukaryot Microbiol. 2010;57(2):177–88.
Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci U S A. 2020;117(17):9451–7.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
An J, Lai J, Sajjanhar A, Batra J, Wang C, Nelson CC. J-Circos: an interactive Circos plotter. Bioinformatics. 2015;31(9):1463–5.
Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44(W1):W160-165.
Pedersen BS, Quinlan AR. Mosdepth: quick coverage calculation for genomes and exomes. Bioinform. 2018;34(5):867–8.
Kokot M, Dlugosz M, Deorowicz S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics. 2017;33(17):2759–61.
Ranallo-Benavidez TR, Jaron KS, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11(1):1432.
Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, Marth GT. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinform. 2011;27(12):1691–2.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SR, Wilkie AO, McVean G, Lunter G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46(8):912–8.
Acknowledgements
Not applicable.
Funding
This work was supported by the Grant Agency of the Czech Republic (grants 23-07695S to AB and AYK [sequencing, genome assembly, annotation and analyses of Trypanosoma spp., analyses of repetitive elements, SNP, and CNV for the complete dataset] and 22-14356S to JL and VY [sequencing, genome assembly, annotation and analyses of O. modryi]), the Russian Science Foundation (grant 19–15-00054 to ESG and VY [sequencing, genome assembly, annotation and analyses of a phylogenetic relative of Leishmania, Crithidia breviculae, and analysis of somy for the complete dataset]), and the INTER-EXCELLENCE II/ INTER-ACTION program of the Czech Ministry of Education, Youth and Sports (grant LUASK22033 to VY [sequencing, genome assembly, annotation and analyses of Crithidia thermophila, Jaenimonas drosophilae, Sergeia podlipaevi, Vickermania spadyakhi, and Wallacemonas spp.]). The computational resources used in this work were funded by the EU’s Operational Program “Just Transition” CZ.10.03.01/00/22_003/0000003 LERCO to VY, European Regional Funds CZ.02.1.01/16_019/0000759 to VY, AYK, JV, and JL.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design, material preparation, and data collection. The analyses were performed by ATSA. The first draft of the manuscript was written by ATSA, AB and VY, and edited by all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Yes, from all authors.
Competing interests
AB and VY are members of the Editorial board of BMC Genomics and are Guest Editors of the collection “Omics of parasites”. All other authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Genome and transcriptome sequencing and assembly statistics. The genomes of C. fasciculata Cf-Cl, L. major Friedlin, T. brucei brucei TREU927 and V. ingenoplastis COLPROT021 are used for comparative purposes.
Additional file 2.
Blobplots showing the state of genome assemblies before and after decontamination for the following species: Borovskyia barvae (panel A), Obscuromonas modryi (B), Trypanosoma avium (C), Trypanosoma boissoni (D), Trypanosoma mega (E), Trypanosoma platydactyli (F), and Trypanosoma scelopori (G).
Additional file 3.
Repetitive content of genome assemblies. Overall statistics on the repetitive content and classification in all genomes assembled in this study and the references for comparison.
Additional file 4.
J-circos plots showing the distribution of repeats along ten longest scaffolds for each trypanosomatid species. The tracks are the following (from inside out): repeat distribution, GC skew, transcriptomic read mapping, scaffold borders and IDs. Total length of the ten largest scaffolds is shown in the center of each circle. Repeats are colorcoded: yellow - LINEs, light green - low complexity, magenta - LTRs, blue - rolling circles, dark green - satellites, orange - simple repeats, red - transposons, turquoise - unclassified repeats.
Additional file 5.
Somy levels and repeat content for 50 longest scaffolds from the genome assemblies obtained in this study.
Additional file 6.
Violin plot representation of somy for the 50 longest scaffolds for each genome assembly. Estimated scaffold somy is color-coded. Scaffold IDs and the ratio of median-of-means coverage values to the median genome overage are shown on X and Y axes, respectively. Bar plot shows the median coverage value and interquartile range.
Additional file 7.
Genome ploidy estimation based on the analysis of heterozygous k-mer pairs. Total coverage of k-mer pairs and normalized minor k-mer coverage are plotted on Y and X axes, respectively.
Additional file 8.
Examples of command lines used for the genome assembly and downstream analyses.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Albanaz, A.T.S., Carrington, M., Frolov, A.O. et al. Shining the spotlight on the neglected: new high-quality genome assemblies as a gateway to understanding the evolution of Trypanosomatidae. BMC Genomics 24, 471 (2023). https://doi.org/10.1186/s12864-023-09591-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09591-z