
Abstract
Background
GBA1 variants are the strongest genetic risk factor for Parkinson’s disease (PD). However, the pathogenicity of GBA1 variants concerning PD is still not fully understood. Additionally, the frequency of GBA1 variants varies widely across populations.
Objectives
To evaluate Oxford Nanopore sequencing as a strategy, to determine the frequency of GBA1 variants in Norwegian PD patients and controls, and to review the current literature on newly identified variants that add to pathogenicity determination.
Methods
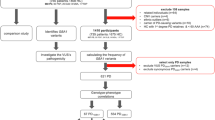
We included 462 Norwegian PD patients and 367 healthy controls. We sequenced the full-length GBA1 gene on the Oxford Nanopore GridION as an 8.9 kb amplicon. Six analysis pipelines were compared using two aligners (NGMLR, Minimap2) and three variant callers (BCFtools, Clair3, Pepper-Margin-Deepvariant). Confirmation of GBA1 variants was performed by Sanger sequencing and the pathogenicity of variants was evaluated.
Results
We found 95.8% (115/120) true-positive GBA1 variant calls, while 4.2% (5/120) variant calls were false-positive, with the NGMLR/Minimap2-BCFtools pipeline performing best. In total, 13 rare GBA1 variants were detected: two were predicted to be (likely) pathogenic and eleven were of uncertain significance. The odds of carrying one of the two common GBA1 variants, p.L483P or p.N409S, in PD patients were estimated to be 4.11 times the odds of carrying one of these variants in controls (OR = 4.11 [1.39, 12.12]).
Conclusions
In conclusion, we have demonstrated that Oxford long-read Nanopore sequencing, along with the NGMLR/Minimap2-BCFtools pipeline is an effective tool to investigate GBA1 variants. Further studies on the pathogenicity of GBA1 variants are needed to assess their effect on PD.
Similar content being viewed by others

Background
Parkinson’s disease (PD) is a multifaceted and highly complex neurodegenerative disorder. Multiple genes have been implicated in PD. Variants in GBA1 (Glucocerebrosidase A) are considered a common genetic risk factor for PD [1,2,3,4,5,6]. Biallelic (homozygous or compound heterozygous) variants in GBA1 classically cause Gaucher’s disease (GD) and an increased PD risk has been observed in patients with GD and asymptomatic carriers of heterozygous variants [7,8,9,10]. Glucocerebrosidase enzymatic activity is reduced in patients with PD who carry a GBA1 heterozygous variant compared to non-carriers, and it is even lower in GBA1 homozygotes/compound heterozygotes [10]. Common GBA1 variants in PD include p.E365K (NM_000157.4, c.1093G > A), p.T408M (NM_000157.4, c.1223 C > T), p.N409S (NM_000157.4, c.1226 A > G), and p.L483P (NM_000157.4, c.1448T > C). However, the classification of the pathogenicity of GBA1 variants and their effect on PD is still ongoing. For this reason, the reported frequencies of GBA1 variants across studies are rather inconsistent, with frequencies ranging from 1.8% up to 47% depending on the ethnicity of the samples and the GBA1 variants investigated [2, 5, 11]. In a previous study on the Norwegian population, 311 patients with PD were included and screened for the two common GBA1 variants (i.e., p.N409S and p.L483P) [12]. Seven patients (2.3%) that carried a heterozygous GBA1 variant were found: four of the patients had a p.N409S (1.3%) and three had a p.L483P (1.0%) substitution.
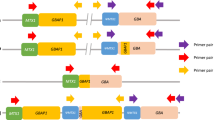
Another challenge that arises when sequencing the GBA1 gene is the nearby pseudogene GBAP1. GBAP1 shares 96% exonic sequence homology with the GBA1 coding region with the highest homology between exons 8 and 11. In this region, most pathogenic variants have been reported, usually resulting from recombination events, e.g. gene conversion, fusion, or duplication [13]. This complex regional genomic structure complicates PCR and DNA sequencing. To avoid the pseudogene, one method to analyze GBA1 is by long-read sequencing [14]. This technology provides full-length GBA1 sequencing to detect exonic and intronic variants and recombinant alleles in combination with phase information, at high multiplex capacity [15, 16].
Herein, we have comprehensively characterized GBA1 in a sample from the Norwegian population by (1) employing and evaluating Oxford Nanopore sequencing as a strategy, (2) determining the frequency of variants within the GBA1 gene in patients with PD and healthy controls by providing an update on previous reports [12, 17], and (3) reviewing current literature on newly identified variants that add to pathogenicity determination.
Results
Long-read sequencing of GBA1
After Nanopore sequencing of the long-range PCR products, we obtained a mean read length of 5.2 kb (SD = ± 2.1 kb) and a mean read quality Phred score of 14.2 (SD = ± 0.4) across all raw sequencing data. After length and quality filtering and read trimming, we obtained a mean read length of 9.0 kb (SD = ± 0.2 kb) and a mean read quality Phred score of 15.7 (SD = ± 0.5). For the filtered samples the mean coverage was 193.1X (SD = ± 187.5X), ranging between 20.6X and 1820.8X across all samples. Nevertheless, the coverage per sample was consistent over all positions (Supplementary Fig. 1).
Bioinformatic pipeline comparison
In total, 79 rare GBA1 variants (gnomAD frequency < 2%) were detected in the Nanopore sequencing analysis after filtering. Out of the 79 rare GBA1 variants, 18 variants were categorized as “pathogenic”, “likely pathogenic”, or of “uncertain significance” (Supplementary Table 1). The remaining 61 rare GBA1 variants were categorized as “benign” or “likely benign”. The number of GBA1 variants differed across all six analysis pipelines (Table 1).
BCFtools
With BCFtools, 64 rare annotated GBA1 variants were detected in 313 samples, resulting in 433 calls in total. The calls were identical with both aligners (i.e., NGMLR and Minimap2). Of these, 15 rare variants, categorized as “pathogenic”, “likely pathogenic”, or of “uncertain significance”, were found in 111 samples (i.e., 120 calls), again independent of the aligner. After Sanger sequencing, 13 variants in 110 samples (i.e., 115 calls) were validated, indicating five false-positive calls but no false-negative calls.
Clair3
For the pipeline using Clair3 as a variant caller after a preceding alignment with NGMLR, 65 rare annotated GBA1 variants were detected in 308 samples, resulting in 426 calls. Of these calls, 117 calls referred to 14 rare variants, categorized as “pathogenic”, “likely pathogenic”, or of “uncertain significance”, and were detected in 108 samples. After Sanger sequencing, 112 calls in 107 samples were validated, implying that five calls were false-positive, with an additional three calls being false-negative. When Minimap2 was used as an aligner, again 65 rare annotated GBA1 variants were detected, however, here they were detected in 311 samples (i.e., 429 calls). With this pipeline, 15 rare annotated GBA1 variants, categorized as “pathogenic”, “likely pathogenic”, or of “uncertain significance”, were found in 110 samples (i.e., 119 calls). Of these, 113 calls in 108 samples could be validated with Sanger sequencing, indicating six false-positive calls and two more false-negative calls.
Pepper-Margin-Deepvariant
Lastly, the Pepper-Margin-Deepvariant pipeline was used for variant calling. After preceding alignment with NGMLR, 453 calls of 72 rare annotated GBA1 variants were detected in 318 samples, including 123 calls of 17 rare variants, categorized as “pathogenic”, “likely pathogenic”, or of “uncertain significance”, in 114 samples. Only 108 calls in 103 samples were validated with Sanger sequencing, leading to 15 false-positive and additional seven false-negative calls. Similarly, when Minimap2 was used for the alignment, 76 rare annotated GBA1 variants were detected in 331 samples, resulting in 481 calls. Of these, 18 rare variants, categorized as “pathogenic”, “likely pathogenic”, or of “uncertain significance”, were detected in 120 samples (i.e., 130 calls). Here, 109 calls in 104 samples were Sanger validated, indicating 21 false-positive and six false-negative calls.
GBA1 variant frequencies in the Norwegian population
In total, 13/18 rare distinct variants within GBA1 were validated in 462 Norwegians with PD and 367 healthy controls (Fig. 1, Supplementary Table 2), whereas 5/18 could not be validated. Two of the 13 rare GBA1 variants were predicted to be “pathogenic” or “likely pathogenic” (p.L483P, p.S146X) and eleven GBA1 variants were of “uncertain significance” (p.G493D, p.N409S, p.T408M, p.A380T, p.R368C, p.E365K, p.D337G, p.S310G, p.R301H, p.R159W, p.R78C). The total carrier frequency of rare GBA1 variants predicted as “pathogenic”, “likely pathogenic”, or of “uncertain significance” was 17.1% (79/462) in the PD cases and 8.4% (31/367) in the controls (OR = 2.24 [1.44, 3.47]) (Table 2). The carrier frequency of known GBA1 risk variants for PD, including p.L483P, p.N409S, p.T408M, p.E365K, and p.R159W, was 15.2% in the PD cases and 7.9% in the controls (OR = 2.08 [1.32, 3.29]) (Table 2). With regard to the common and frequently investigated GBA1 risk variants for PD, the frequency of carrying either a p.L483P or p.N409S variant was 4.3% in patients with PD and 1.1% in healthy controls (OR = 4.11 [1.39, 12.12]) (Table 2).

Schematic representation of the exonic (gray boxes) and intronic (gray lines) structure of the GBA1 gene highlighting “pathogenic” (red)/“likely pathogenic” (orange)/“uncertain” (yellow) variants found in our Norwegian series
In addition, samples with a rare “pathogenic”, “likely pathogenic”, or “uncertain” GBA1 variant were further examined for possible structural variants (SVs). However, all samples tested negative for SVs.
Literature review
We performed a systematic literature review to summarize GBA1 variants detected in PD and their frequencies across different populations. In total, 100 articles on GBA1 variant frequencies across populations were included in the overview (Supplementary Table 3). Most of the studies assessed the p.L483P and p.N409S variants, with p.L483P variant frequencies ranging between 0% and 8.3% for cases with PD and between 0% and 1.4% in the general population. The highest frequency in PD patients was observed in a sample from the Japanese population [18]. For p.N409S, the frequencies ranged from 0% to 26.3% in patients with PD and from 0% to 5.96% in controls, with the highest frequency reported in a sample from the Ashkenazi Jewish population [19]. In a previous study on the Norwegian population, the frequency of p.L483P was predicted to be 0.5% in patients with PD (2/442) and 1.4% in controls (6/419) (OR = 0.31 [0.06, 1.56]) [17]. For the p.N409S variant, the frequencies reported were 0.2% in patients with PD (1/442) which was comparable to controls (1/419) (OR = 0.95 [0.06, 15.2]) [17]. In addition, the study reported GBA1 p.E365K, detected in 4.3% of patients with PD (18/442) and in 6.6% of controls (29/419) (OR = 0.57 [0.13, 1.04]), and p.T408M, found in 1.7% of patients with PD (7/442) and in 3.6% of controls (16/419) (OR = 0.41 [0.17, 1]).
Discussion
Variants in the GBA1 gene are known to affect PD risk, however, the frequency and pathogenicity of these variants are still under debate. The latter is further complicated as the frequencies of GBA1 variants vary by population. In our study, we used the pathogenicity scoring of ACMG, Varsome, ClinVar, SIFT, Polyphen2, CADD, and GERP + + and categorized the variants found into “pathogenic”, “likely pathogenic”, and variants of “uncertain significance”. Here we report 13 rare variants within GBA1 in Norwegian PD cases, with two of them predicted to be “pathogenic” or “likely pathogenic”, and eleven GBA1 variants of “uncertain significance”. In total, we found a “pathogenic” or “likely pathogenic” variant (e.g., p.L483P, p.S146X) in 1.5% of PD cases and in 0% of healthy controls. However, pathogenicity scoring with ACMG does not take the association with PD into account and consequently underestimates the frequency of risk variants in GBA1. Therefore, we further investigated known GBA1 risk variants associated with PD. In our study, 15.2% of patients with PD carried a common GBA1 risk variant (i.e., p.L483P, p.N409S, p.T408M, p.E365K, and p.R159W), compared to 7.9% of controls.
In a systematic literature review, we evaluated the frequency of variants in the GBA1 gene in patients with PD and in the general population in 100 studies (Supplementary Table 3). The frequencies of GBA1 risk variants range between 0% and 26.3% in patients with PD, highlighting the importance of investigating GBA1 variants across populations. In a previous study on the Norwegian population, two known risk variants, p.L483P and p.N409S, were investigated among others [17]. The frequency of p.L483P was predicted to be 0.5% in patients with PD and 1.4% in controls, while 0.2% of the patients with PD and controls had a p.N409S variant [17]. Therefore, the odds of carrying a p.L483P or a p.N409S variant in patients with PD are lower than in healthy controls (OR = 0.31 [0.06, 1.56], OR = 0.95 [0.06, 15.2]). In contrast to this, we found slightly higher frequencies in our Norwegian series. The p.L483P variant was found in 1.3% of Norwegian PD cases and in 0% of controls, the p.N409S variant in 2.8% of PD patients and 1.1% of controls, leading to higher odds of carrying these variants in patients with PD compared to controls (OR = 2.63 [0.85, 8.13]).
These variants are classically found in GD but are also associated with PD risk. However, some variants in GBA1 show associations with PD but do not cause GD, e.g., the GBA1 variants p.E365K and p.T408M [6]. In our Norwegian series, the p.E365K variant was found in 7.1% of cases with PD and in 2.7% of the general population (OR = 2.75 [1.33, 5.65]). The p.T408M variant was found in 2.2% of cases with PD and in 3.5% of the general population (OR = 0.6 [0.26, 1.39]). In a previous study on the Norwegian population, they reported p.E365K in 4.3% of patients with PD and in 6.6% of controls (OR = 0.57 [0.13, 1.04]), and p.T408M in 1.7% of patients with PD and in 3.6% of controls (OR = 0.41 [0.17, 1]) [17]. Several other studies have also reported higher variant frequencies of p.E365K or p.T408M in their control population compared to their patients with PD [10, 20,21,22,23,24,25,26,27], questioning the pathogenicity of these GBA1 variants and their contribution to causing PD that was previously assessed in two meta-analyses. In the summary of our systematic literature review (Table 3), we have also found on average higher frequencies of these two GBA1 variants in patients with PD compared to healthy controls. Nevertheless, a better definition of variants associated with PD is strongly needed by classifying variants into categories relevant to the disease.
Although the pathogenicity and the pathomechanism of several GBA1 variants in patients with PD are still under discussion, gene-targeted therapy might help to treat GBA1-PD. So far, there are several strategies for the treatment of GBA1-PD with ongoing clinical trials. Some of the therapeutic approaches for GBA1 include substrate reduction therapy targeting glycosylceramide synthase inhibition, the inhibition of glucocerebrosidase transportation, the development of glucocerebrosidase activators, and gene therapy targeting the replacement of mutated GBA1 with WT copies of the gene [28].
In addition to variable inclusion criteria, the sequencing method and data analysis, and the chronology when it was performed, has a major influence on the detection rate and GBA1 variant frequencies reported. Through the years, DNA sequencing technologies evolved tremendously with new sequencing techniques and especially new prediction tools improving the accuracy of variant detection. Continual refinement in these tools enables more comprehensive identification of variants and highlights the need to re-evaluate known genes as time goes by. Long-read sequencing in combination with the latest data analysis tools enabled us to determine the frequency of GBA1 variants in the Norwegian population with higher precision than before. We evaluated the consensus and accuracy of six different pipelines using two different aligners (NGMLR and Minimap2), as well as three different variant callers (BCFtools, Clair3, and the Pepper-Margin-Deepvariant pipeline). BCFtools performed best with regard to the number of true-positive, false-positive, and false-negative hits, independently from the aligner used. However, one limitation is that we could not fully evaluate pipeline sensitivity. As we only confirmed variants called by our Nanopore analysis pipelines by Sanger sequencing, we underestimate false-negative variants that were not initially called. With Oxford Nanopore technology we assessed the precision of long-read sequencing and consensus data analysis and detected 115 real GBA1 variant calls, while five variant calls were false-positives. Thus, > 95% of called variants were true-positive. Nanopore long-read sequencing is an accurate tool to detect genetic variations and with further development in flow cells and sequencing kits, the accuracy of variant detection is likely to increase. Another advantage of this technology is the capacity to multiplex samples, which decreases analysis costs of the full 8.9 kb GBA1 gene to $13 USD per sample, which is lower than other DNA sequencing methods [29]. A strength of Oxford Nanopore long-range sequencing is to specifically target the GBA1 gene without sequencing the pseudogene GBAP1, and to detect all disease-causing variants including information on phase [15, 16].
Conclusions
In conclusion, we have demonstrated that Oxford Nanopore sequencing is an efficient and scalable tool for investigating GBA1 variants. We thoroughly evaluated six different data analysis pipelines and found the pipeline consisting of an alignment with either NGMLR or Minimap2 and variant calling with BCFtools to perform best with regard to detecting variants in GBA1. With our established and validated workflow, we demonstrated that the frequency of the two common GBA1 risk variants for PD (i.e., p.L483P and p.N409S) in Norwegian patients with PD is 4.3% and higher than in the general population (1.1%). Furthermore, we reviewed current literature on GBA1 variant frequencies in PD across populations, thereby adding to pathogenicity determination. Given the importance of this gene, further functional studies on the pathogenicity of GBA1 are needed to assess their effect on PD.
Methods
Demographics
A sample of 462 Norwegian patients with PD was included in this study (Table 4). All patients were referred by general practitioners and other hospitals and have been clinically examined and observed longitudinally at the outpatient clinics of three hospitals in Central Norway. One hundred eighty (39%) of the patients were men, and 282 (61%) were women. The mean age at disease onset in the patient group was 60.3 years (SD = ± 9.7 years, range 26 to 88 years). Forty-three out of 462 patients were probands with a family history of PD. Patients with a known genetic cause of PD were not included. In the clinical assessment, patients with PD had an average score of 2.72 (SD = ± 0.86) on the Hoehn and Yahr scale and 357 patients reported a tremor, while 62 had no tremor. In addition, a group of 367 healthy Norwegian individuals (mean age 64.0 years) originating from the same geographic region and without signs of a movement disorder was included to determine the variant frequency in the general population (Table 4). Some of the patients and controls included in this study have been previously included [12]. However, in this previous report only the p.N409S and the p.L483P have been screened by PCR amplification and subsequent digestion of the PCR product with restriction enzymes and separation of resulting fragments by agarose gel electrophoresis.
Genetic analysis
Long-read Oxford Nanopore sequencing
We used blood-derived genomic DNA samples from all PD patients and controls. Informed consent was obtained from all participating individuals. We enriched for GBA1 by amplifying an 8.9 kb sequence, which covered all coding exons, the introns between them, and part of the 3’ UTR region (hg38: chr1:155232501–155,241,415), described previously [15, 16] using the LongAmp Taq PCR Kit. Subsequently, 1.3 µg of each patient-derived PCR product was barcoded with the Native 96 Barcoding Kit (EXP-NBD196) and multiplexed. The libraries were generated with the Ligation Sequencing Kit (SQK-LSK109) for long-read Nanopore sequencing on R9.4.1 flow cells (FLO-MIN106) on a GridION.
Bioinformatic analyses
Data acquisition and run monitoring was carried out with MinKNOW (version v21.05.25 and later). The integrated Guppy algorithm (version v5.0.16 and later) was used for base-calling with the super-accurate base-calling model, de-multiplexing, and FAST5 and FASTQ file generation. The base-called reads were filtered with Filtlong (v.0.2.0) (https://github.com/rrwick/Filtlong) to only include the best 50% of the reads, based on Phred quality scores (q-score) in the FASTQ files, with a minimum read length of 8 kb. Afterwards, the reads were trimmed with NanoFilt [30] (v2.8.0) and 75 bp were cropped from the front of the reads and 20 bp from the end. Subsequently, the nanopore reads were aligned against the reference sequence (hg38). We used two different aligners: NGMLR [31] (v0.2.7) and Minimap2 [32] (v2.22). Then, the alignments were sorted and indexed with SAMtools [33] using v1.9 for the NGMLR alignment, and v1.15 for the Minimap2 alignment. In addition, the coverage for each sample was calculated using SAMtools [33] (v.1.15). The processed BAM files were analyzed with three different variant callers: BCFtools [33] (v1.9), Clair3 (v0.1-r11), and the Pepper-Margin-Deepvariant pipeline [34] (Supplementary Fig. 2). Finally, the resulting VCF files containing the SNPs within GBA1 were annotated using ANNOVAR [35] (version 2020-06-11).
Sanger sequencing and structural variant detection
Sanger sequencing was performed for all individuals with a rare “pathogenic”/“likely pathogenic”/“uncertain” GBA1 variant, as previously described [36]. Individuals with a rare “pathogenic”/“likely pathogenic”/“uncertain” GBA1 variant were further examined for possible structural variants (SVs) due to the high exonic sequence homology between GBA1 and GBAP1. We used two sets of primers, as previously described [16], to detect reciprocal crossovers between the gene and pseudogene resulting in a 20.6 kb deletion or a 20.6 kb duplication using the LongAmp Taq PCR Kit. The resulting PCR products were subsequently run in a 1.5% agarose gel.
Sensitivity assessment
To assess the performance of variant calling, we stratified the detected GBA1 variants into true-positive, false-positive, and false-negative calls for each data analysis pipeline (NGMLR + BCFtools, NGMLR + Clair3, NGMLR + Pepper-Margin-Deepvariant, Minimap2 + BCFtools, Minimap2 + Clair3, Minimap2 + Pepper-Margin-Deepvariant). True-positive variants were defined as GBA1 variants detected with Nanopore sequencing that were validated with Sanger sequencing. False-positive variants are those identified with Nanopore sequencing but not confirmed with Sanger sequencing. False-negative variants were determined as those not called with a specified data analysis pipeline, but later validated with Sanger sequencing. Evaluation of each data analysis pipeline was based on the ratio of false-positive to false-negative calls.
Pathogenicity scoring
GBA1 single nucleotide variants were first filtered based on GnomAD frequency < 2%. Pathogenicity classification and scoring were assessed with American College of Medical Genetics and Genomics (ACMG) [37] criteria. This included using Varsome [38], ClinVar [39], SIFT [40], Polyphen2 [41], CADD [42], and GERP++ [43]. GBA1 variants were categorized as “pathogenic”, “likely pathogenic”, or of “uncertain significance” and further validated by Sanger sequencing. The p.E365K variant, which is a known risk factor for PD, was additionally categorized as of “uncertain significance”, despite the “likely benign” score from ACMG. Variants categorized as of “uncertain significance” without information from mutation predictors were not Sanger sequenced.
Statistical analysis
To compare the number of Norwegian PD patients with and without GBA1 variants to the frequencies of GBA1 variants in controls, odds ratios were calculated. For this, the number of patients with PD carrying a GBA1 variant was multiplied by the number of controls without a GBA1 variant and subsequently divided by the number of patients with PD without a GBA1 variant that was multiplied by the number of controls carrying a GBA1 variant.
Literature review
We performed a systematic literature review to summarize GBA1 variants detected in PD and the frequency across different populations (Supplementary Fig. 3). We searched for literature via PubMed that was published before August 4, 2022, using the search term “GBA” AND “Parkinson” AND “prevalence” OR “GBA” AND “Parkinson” AND “frequency”, while setting the species filter to “Human” and the language filter to “English”, resulting in 94 articles. These were screened based on the title, abstract, and full text, excluding all articles not directly screening for variants in the GBA1 gene in patients with PD. Of these 94 articles, 41 articles were excluded. Reasons for exclusion were reviews or comments without new data (n = 11), articles that did not perform GBA1 variant screening or examine GBA1 variant frequency in their study population (n = 22), and articles that did not include patients with PD (n = 10). Multiple reasons for exclusion were possible. In addition to the articles found via the search term, suitable articles that were referenced in this literature were also included in the overview.
Data Availability
The datasets generated and analyzed during the current study are available in the Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra) repository under the accession number: PRJNA939498 (SAMN33476944-SAMN33477772).
References
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–61.
Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012;11(11):986–98.
Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology. 2008;70(24):2277–83.
Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132(Pt 7):1783–94.
Gan-Or Z, Liong C, Alcalay RN. GBA-Associated Parkinson’s Disease and other synucleinopathies. Curr Neurol Neurosci Rep. 2018;18(8):44.
Greuel A, Trezzi JP, Glaab E, Ruppert MC, Maier F, Jager C, et al. GBA Variants in Parkinson’s Disease: clinical, metabolomic, and Multimodal Neuroimaging Phenotypes. Mov Disord. 2020;35(12):2201–10.
Riboldi GM, Di Fonzo AB, GBA. Gaucher Disease, and Parkinson’s Disease: from genetic to clinic to New Therapeutic Approaches. Cells. 2019;8(4).
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41(12):937–40.
Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. 2004;81(1):70–3.
Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138(Pt 9):2648–58.
Behl T, Kaur G, Fratila O, Buhas C, Judea-Pusta CT, Negrut N, et al. Cross-talks among GBA mutations, glucocerebrosidase, and alpha-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: a comprehensive review. Transl Neurodegener. 2021;10(1):4.
Toft M, Pielsticker L, Ross OA, Aasly JO, Farrer MJ. Glucocerebrosidase gene mutations and Parkinson disease in the norwegian population. Neurology. 2006;66(3):415–7.
Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567–83.
Ip CLC, Loose M, Tyson JR, de Cesare M, Brown BL, Jain M, et al. MinION Analysis and Reference Consortium: phase 1 data release and analysis. F1000Res. 2015;4:1075.
Leija-Salazar M, Sedlazeck FJ, Toffoli M, Mullin S, Mokretar K, Athanasopoulou M, et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol Genet Genomic Med. 2019;7(3):e564.
Toffoli M, Chen X, Sedlazeck FJ, Lee CY, Mullin S, Higgins A, et al. Comprehensive short and long read sequencing analysis for the Gaucher and Parkinson’s disease-associated GBA gene. Commun Biol. 2022;5(1):670.
Lunde KA, Chung J, Dalen I, Pedersen KF, Linder J, Domellof ME, et al. Association of glucocerebrosidase polymorphisms and mutations with dementia in incident Parkinson’s disease. Alzheimers Dement. 2018;14(10):1293–301.
Li Y, Sekine T, Funayama M, Li L, Yoshino H, Nishioka K, et al. Clinicogenetic study of GBA mutations in patients with familial Parkinson’s disease. Neurobiol Aging. 2014;35(4):935e3–8.
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2004;351(19):1972–7.
Olszewska DA, McCarthy A, Soto-Beasley AI, Walton RL, Magennis B, McLaughlin RL, et al. Association between glucocerebrosidase mutations and Parkinson’s Disease in Ireland. Front Neurol. 2020;11:527.
Kalinderi K, Bostantjopoulou S, Paisan-Ruiz C, Katsarou Z, Hardy J, Fidani L. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Greece. Neurosci Lett. 2009;452(2):87–9.
Han F, Grimes DA, Li F, Wang T, Yu Z, Song N, et al. Mutations in the glucocerebrosidase gene are common in patients with Parkinson’s disease from Eastern Canada. Int J Neurosci. 2016;126(5):415–21.
Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology. 2007;69(12):1270–7.
Graham OEE, Pitcher TL, Liau Y, Miller AL, Dalrymple-Alford JC, Anderson TJ, et al. Nanopore sequencing of the glucocerebrosidase (GBA) gene in a New Zealand Parkinson’s disease cohort. Parkinsonism Relat Disord. 2020;70:36–41.
Crosiers D, Verstraeten A, Wauters E, Engelborghs S, Peeters K, Mattheijssens M, et al. Mutations in glucocerebrosidase are a major genetic risk factor for Parkinson’s disease and increase susceptibility to dementia in a Flanders-Belgian cohort. Neurosci Lett. 2016;629:160–4.
Bras J, Paisan-Ruiz C, Guerreiro R, Ribeiro MH, Morgadinho A, Januario C, et al. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Portugal. Neurobiol Aging. 2009;30(9):1515–7.
Barkhuizen M, Anderson DG, van der Westhuizen FH, Grobler AF. A molecular analysis of the GBA gene in caucasian South Africans with Parkinson’s disease. Mol Genet Genomic Med. 2017;5(2):147–56.
Senkevich K, Rudakou U, Gan-Or Z. New therapeutic approaches to Parkinson’s disease targeting GBA, LRRK2 and parkin. Neuropharmacology. 2022;202:108822.
Yohe S, Thyagarajan B. Review of clinical next-generation sequencing. Arch Pathol Lab Med. 2017;141(11):1544–57.
De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. Bioinformatics. 2018;34(15):2666–9. NanoPack: visualizing and processing long-read sequencing data.
Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15(6):461–8.
Li H. New strategies to improve minimap2 alignment accuracy. Bioinformatics. 2021.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2).
Shafin K, Pesout T, Chang PC, Nattestad M, Kolesnikov A, Goel S, et al. Haplotype-aware variant calling with PEPPER-Margin-DeepVariant enables high accuracy in nanopore long-reads. Nat Methods. 2021;18(11):1322–32.
Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10(10):1556–66.
Crossley BM, Bai J, Glaser A, Maes R, Porter E, Killian ML, et al. Guidelines for Sanger sequencing and molecular assay monitoring. J Vet Diagn Invest. 2020;32(6):767–75.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978–80.
Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48(D1):D835–D44.
Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chap. 7:Unit7 20.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D94.
Huber CD, Kim BY, Lohmueller KE. Population genetic models of GERP scores suggest pervasive turnover of constrained sites across mammalian evolution. PLoS Genet. 2020;16(5):e1008827.
Acknowledgements
This project was supported by the Institute of Neurogenetics at the University of Lübeck. Special thanks to the families and patients who have participated in this study.
Funding
This project was supported by the Institute of Neurogenetics at the University of Lübeck. Grant support was received from the DFG RU ProtectMove (FOR2488).
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
C.G. performed the data analysis, data interpretation, and writing of the manuscript. S.S. performed the data acquisition, helped with data interpretation, and reviewed the manuscript. T.L. contributed to the data analysis, data interpretation, and reviewed the manuscript. C.M. performed the data acquisition, helped with data interpretation, and reviewed the manuscript. C.K. reviewed the manuscript. J.O.A. was responsible for the sample acquisition and reviewed the manuscript. M.J.F. reviewed the manuscript. J.T. was responsible for the conceptualization of the project, data analysis, data interpretation, and writing of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All methods were carried out in accordance with relevant guidelines and regulations. The ethics board of the University of Lübeck, Germany gave ethical approval for this work. Informed consent was obtained from all participating individuals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gabbert, C., Schaake, S., Lüth, T. et al. GBA1 in Parkinson’s disease: variant detection and pathogenicity scoring matters. BMC Genomics 24, 322 (2023). https://doi.org/10.1186/s12864-023-09417-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09417-y