
Abstract
Background
Sulfate-reducing bacteria (SRB) drive the ocean sulfur and carbon cycling. They constitute a diverse phylogenetic and physiological group and are widely distributed in anoxic marine environments. From a physiological viewpoint, SRB’s can be categorized as complete or incomplete oxidizers, meaning that they either oxidize their carbon substrate completely to CO2 or to a stoichiometric mix of CO2 and acetate. Members of Desulfofabaceae family are incomplete oxidizers, and within that family, Desulfofaba is the only genus with three isolates that are classified into three species. Previous physiological experiments revealed their capability of respiring oxygen.
Results
Here, we sequenced the genomes of three isolates in Desulfofaba genus and reported on a genomic comparison of the three species to reveal their metabolic potentials. Based on their genomic contents, they all could oxidize propionate to acetate and CO2. We confirmed their phylogenetic position as incomplete oxidizers based on dissimilatory sulfate reductase (DsrAB) phylogeny. We found the complete pathway for dissimilatory sulfate reduction, but also different key genes for nitrogen cycling, including nitrogen fixation, assimilatory nitrate/nitrite reduction, and hydroxylamine reduction to nitrous oxide. Their genomes also contain genes that allow them to cope with oxygen and oxidative stress. They have genes that encode for diverse central metabolisms for utilizing different substrates with the potential for more strains to be isolated in the future, yet their distribution is limited.
Conclusions
Results based on marker gene search and curated metagenome assembled genomes search suggest a limited environmental distribution of this genus. Our results reveal a large metabolic versatility within the Desulfofaba genus which establishes their importance in biogeochemical cycling of carbon in their respective habitats, as well as in the support of the entire microbial community through releasing easily degraded organic matters.
Similar content being viewed by others

Background
Sulfate-reducing bacteria (SRB) are widely distributed in different environments, e.g., wastewater treatment systems [1], freshwater sediments [2], and marine sediments [3]. Dissimilatory sulfate reduction to sulfide by SRB is a predominant terminal pathway of organic matter mineralization in marine sediments [4]. SRB are considered strictly anaerobic, however, it is now generally accepted that a large number of strains tolerate the exposure to oxygen for periods of some length [5,6,7]. Some members of the genus Desulfovibrio, even have high rates of aerobic respiration [6, 8]. Some of them can couple this respiratory process to ATP synthesis [9,10,11]. Recently, it has been shown that different Desulfovibrio strains were able to couple respiration with oxygen to growth [12, 13]. In addition, filamentous sulfide-oxidizing bacteria, the so-called “cable-bacteria”, are able to reduce oxygen (or nitrate) at one end of the filament and oxidize sulfide at the opposing end, thereby transporting electrons over cm-scale distances [14,15,16]. They represent the only example of a member of the Desulfobulbaceae that successfully couples the reduction of oxygen with growth, most likely by reverting the canonical sulfate reduction pathway for the oxidation of sulfide.
SRBs constitute over 23 genera, which are found both within archaeal and bacterial domains with Deltaproteobacteria being the dominant class [17]. Recently, some newly discovered bacterial phyla recovered from metagenomic data were found to be SRB [18], suggesting a high phylogenetic diversity of SRB in the environment. However, a large-scale comparison of Deltaproteobacteria genomes revealed that the genomes of cultivated Deltaproteobacteria strains are phylogenetically distant with those Deltaproteobacteria metagenome-assembled genomes (MAGs) [19]. It indicates that the genomic contents of the cultured isolates need further investigation to understand their physiological traits, yet many MAGs are recovered from metagenomic data. By now, only three species, Desulfofaba fastidiosa P2 (DSM 15249) [20], Desulfofaba gelida PSv29 (DSM 12344) [21], and Desulfofaba hansenii P1 (DSM 13527) [22] have been isolated and described within Desulfofaba, the genus within Desulfofabaceae family. The genus Desulfofaba is phylogenetically distinct to other groups of SRBs based on a single gene marker (16S rRNA or DrsA/B). All isolates incompletely oxidizing propionate to acetate and CO2 in the presence of sulfate. Interestingly, phylogenetic trees both based on 16S rRNA gene sequences and on dissimilatory sulfite reductase gene amino acid sequences show that the members of the genus Desulfofaba are closer related to complete oxidizers than to incomplete oxidizers [20,21,22].
However, single-gene based study or specific experimental studies may overlook their metabolic potential without exploring their genomic contents. To get a better overview of the metabolic potential of the genus Desulfofaba, here we present results of the analysis of the genomes of the three species. We focused on the genes that relate to the handling of oxygen and its intermediates, as physiological experiments with Desulfofaba hansenii revealed that the strain was able to respire with oxygen. We also identified genes encoding for the synthesis of polyhydroxybutyrate (PHB) in Desulfofaba hansenii and Desulfofaba gelida, which potentially could be used as electron donor when oxygen is expired in Desulfofaba hansenii. We further investigated the distribution of Desulfofaba based on the homologue search of the marker gene, as well as the collection of our MAGs recovered from various marine environments. This is of particular interest as it may provide some clues regarding the habitat adaptation of Desulfofaba genus to understand their distribution in the environment.
Results and discussion
Phylogeny and distribution of Desulfofaba genus
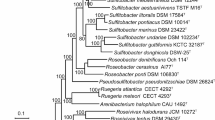
Desulfofaba genus consists of three species: Desulfofaba fastidiosa P2 (DSM 15249) [20], Desulfofaba gelida PSv29 (DSM 12344) [21], and Desulfofaba hansenii P1 (DSM 13527) [22]. A set of 120 marker genes identified using GTDB-tk and another set of 37 ribosomal protein encoding marker genes identified using Phylosift supported that Desulfofaba is a monophyletic group which is distinct from other families in the order Desulfobacterales (Figs. 1 and S1). A phylogenetic tree based on 16S rRNA gene sequences shows that the genus Desulfofaba is most closely related to the genus Desulfoluna (Fig. S2). The three species of the genus Desulfofaba were isolated from different environments, including the interior of an eelgrass root, polar surface sediments, and the methane-sulfate transition zone 1.5 m below the sediment surface [20]. Related 16S rRNA gene sequences recovered from different geographic locations (Fig. S2) suggests a wide distribution of the genus. However, when we searched 16S rRNA gene sequences against publicly available metagenomes, we could not find samples with high sequence homology (> 95%) to 16S rRNA genes. Additionally, we searched Desulfofaba genus from 194 Desulfobacterales MAGs on phylogenetic trees which were built with protein encoding marker genes extracted using GTDB-tk or Phylosift. Those 194 MAGs were part of our genome collection of over 6000 MAGs. The metagenomes were recovered from various environments, including coastal sediments in the Bohai Sea, cold seep sediments in the South China Sea, and hydrothermal vent sediment in the Southwest Indian Ocean. The search showed that none of them were closely affiliated with members of genus Desulfofaba, suggesting that members of the genus Desulfofaba are either rare in the environment or difficult to amplify by our current approach.

A maximum likelihood phylogenetic tree of 22 genomes including the 3 Desulfofaba genomes. The phylogeny is based on 37 concatenated ribosomal protein encoding genes identified using PhyloSift. Desulfofaba genomes formed a monophyletic group which is distinct from other families in the order Desulfobacterales. Acidobacteria were set as the outgroup
Average amino acid identity (AAI) revealed that the genus Desulfofaba is distinct from other described taxa. These three genomes shared at least 60.1% AAI with each other, and at most 57.5% AAI with genomes of other taxa (Fig. S3).
Overview of three Desulfofaba genomes
The overall genomic information of the three draft genomes were summarized in Table 1, and the assignment of genes into COG functional categories demonstrating the general function of three genomes is shown in Table 2. The assembled draft genomes are 99.3, 98.1, and 99.3% complete with less than 2.1% contamination based on CheckM [23] for Desulfofaba fastidiosa, Desulfofaba gelida, and Desulfofaba hansenii, respectively.
Complex organic matter degradation
These genomes include genes encoding for diverse carbohydrate-active enzymes (CAZYmes) and peptidases (Fig. S4) with the potential for degradation of complex carbohydrates and detrital proteins into simple sugars and amino acids. Most of the enzymes are assigned with intra-cellular function based on predictions of the cellular localization of the proteins. They also have genes encoding for proteins that are capable of degrading long-chain fatty acids through beta oxidation (Fig. 2), which is consistent with experimental data for Desulfofaba gelida [21]. As an incomplete oxidizer [20,21,22], they are capable of oxidizing propionate to acetate with the methylmalonyl-CoA pathway, which shares several steps with the tricarboxylic acid (TCA) cycle (Fig. 2). The acetate is excreted and may serve as a substrate for adjacent microbes in the environment.

Overview of the metabolic potential of the Desulfofaba genus based on the annotated genomes. Desulfofaba genomes have genes encoding for diverse central metabolic pathways, including glycolysis, the pentose phosphate pathway (PPP), the Wood-Ljungdahl pathway (WLP), the TCA cycle, and the reductive glycine pathway. Desulfofaba genomes have genes involved in nitrogen, sulfur, hydrogen, selenium, and arsenic cycling. Different types of cytochrome oxidases genes were annotated in Desulfofaba genomes
Central metabolism and PHB synthesis
Genes encoding central metabolic pathways, including glycolysis, the pentose phosphate pathway (PPP), the Wood-Ljungdahl pathway (WLP), the TCA cycle, and the reductive glycine pathway were identified (Fig. 2, Supplementary Dataset). This suggests a high degree of metabolic versatility of central metabolism in Desulfofaba genus, which enables them to utilize different types of substrates or be active under different environmental conditions.
All three Desulfofaba genomes have a pathway for PHB synthesis from acetyl-CoA [24, 25]. Genes encoding for acetoacetyl-CoA reductase (PhaB), reducing acetoacetyl-CoA to 3-hydroxybutyryl-CoA, were not annotated in any of these three genomes. However, genes encoding for 3-hydroxybutyryl-CoA dehydrogenase, enoyl-CoA hydratase, and 3-hydroxybutyryl-CoA dehydratase, which could reduce acetoacetyl-CoA through 3-hydroxybutanoyl-CoA and crotonoyl-CoA, were annotated in Desulfofaba genomes (Fig. 2).
Sulfur and nitrogen metabolism
The three species within Desulfofaba genus are incomplete oxidizers, i.e., oxidizing propionate incompletely to acetate and CO2 [20,21,22]. The phylogenetic tree of dissimilatory sulfite reductase (DsrAB) genes showed that the three species, together with other identified incomplete oxidizers, formed a monophyletic group (Fig. 3). This phylogenetic position contradicts with the previous study, which showed a closer phylogeny with complete oxidizers based on 16S rRNA gene or DSR sequencing [22]. According to their genetic inventory, they have the potential to reduce polysulfide to sulfide using a sulfhydrogenase and to assimilate thiosulfate into cysteine.

A maximum likelihood phylogenetic tree of genes encoding for alpha and beta subunits of dissimilatory sulfite reductase (DsrAB). DsrAB in Desulfofaba genomes belong to reductive-type and are closely related to sequences in known incomplete oxidizer genomes
The three genomes also encode several key pathways for nitrogen cycling (Fig. 2) such as nitrogenase genes involved in the fixation of nitrogen. Also, genes encoding the assimilation of ammonia into glutamine were found. On the dissimilatory side, Desulfofaba gelida encodes genes for nitrate/nitrite assimilation. Hydroxylamine is an intermediate in two important microbial processes of the nitrogen cycle: nitrification [26] and anaerobic ammonium oxidation [27]. The genomes of both Desulfofaba gelida and Desulfofaba hansenii encode enzymes to oxidize hydroxylamine (NH2OH) to nitric oxide (NO) via hydroxylamine dehydrogenase (HAO) [28, 29] or reduce NH2OH to ammonia (NH3) via hydroxylamine reductase (HCP). All genomes encode enzymes to further reduce NO to nitrous oxide (N2O) via anaerobic nitric oxide reductase. N2O is a key byproduct during denitrification and is a potent greenhouse gas and ozone destroying agent. This indicates that Desulfofaba may have important implications for Earth’s climate [30].
Hydrogen metabolism and energy conservation
Hydrogen plays a central role in the energy metabolism of sulfate reducers, which can either use H2 as an energy source or produce H2 during fermentation [31]. The genomes of three species of the genus Desulfofaba encode [NiFe] group 1b hydrogenase, which may transfer H2-liberated electrons through cytochromes to terminal reductase when sulfate, fumarate, nitrate, and metals serve as terminal electron acceptors [32]. The presence of other types of [NiFe] and [FeFe] hydrogenase genes, including cytoplasmic and transmembrane types for hydrogen metabolism, is species-specific (Fig. 2). The phylogenetic position of Desulfofaba hydrogenases is close to hydrogenases of other known sulfate reducers (Figs. S5 and S6). Besides genes encoding for respiratory complex I and II in the electron transport chain, they also encode the membrane-bound Rnf complex that can couple the electron transfer from reduced ferredoxin (Fd2−) to NAD+ with the translocation of proton (H+) for energy conservation [33]. The three species encode F-type ATPase to generate ATP (Fig. 2).
Selenium and arsenate metabolism
The genomes of the three Desulfofaba species have genes encoding for proteins involved in the mobilization of organic selenium. Those proteins catalyze the reaction of selenide with ATP to form selenophosphate via selenide water dikinase (SelD) [34] and incorporate selenium into selenocysteinyl-tRNA (Sec) with L-seryl-tRNA (Ser) selenium transferase (SelA) [35] and selenomethionyl-tRNA (Met) with methionyl-tRNA synthetase (MetG) [36], which are further utilized for protein biosynthesis.
Arsenate and arsenite are toxic to organisms by blocking general cell metabolism [37] and are the two dominant forms of inorganic arsenic in marine environments [38]. Desulfofaba has genes of the arsenic detoxification system (Fig. 2). They can reduce arsenate to arsenite via arsenate reductase (ArsC) through thioredoxin [39]. Even though arsenite is more toxic than arsenate, arsenite could be extruded from the cell by an arsenite transporter (ArsAB) or transformed to methyl arsonate, a less toxic form [40], by arsenite methyltransferase (AS3MT).
Oxygen Consumption and Defense against ROS
Most sulfate reducers are obligate anaerobes, yet some species can tolerate oxygen and have developed different strategies to cope with its presence in the environment [41]. We identified genes encoding two types of membrane-bound oxygen reductases: cytochrome bd-I ubiquinol oxidase and cytochrome cbb3-type terminal oxidase (Fig. 2). The first evidence for a membrane-bound oxygen reductase, a canonical bd quinol oxidase, in SRB was reported in Desulfovibrio gigas [11]. The genes of both identified terminal oxidases in Desulfofaba are of a high-affinity-type, and they are usually considered as important terminal oxidases under low oxygen conditions [42]. Our physiological experiments showed that Desulfofaba hansenii was able to reduce oxygen and that oxygen reduction is most likely linked to the oxidation of PHB storage compounds that are present in the cell (Supplementary material). Many sulfate reducers can reduce O2, probably as a protective mechanism, without sustainable aerobic growth [43]. Growth with energy derived from oxidative phosphorylation linked to oxygen reduction was observed in different Desulfovibrio strains [12, 13]. Based on their genomic outfit, it is possible that Desulfofaba sp. produces energy during aerobic respiration. However, we did not observe aerobic respiration linked to growth in Desulfofaba hansenii. Both Desulfofaba hansenii, isolated from Zostera marina roots, and Desulfofaba gelida, isolated from surface sediments, could encounter oxygen in their respective habitats and thus, the ability to cope with oxygen could be an important survival strategy.
The genomes that we studied contain a number of genes that enable them to deal with oxidative stress, as is the case for many other sulfate reducers [44]. These include genes to detoxify reactive oxygen species (ROS) and repair damaged DNA, as well as genes that trigger a behavioral response to the presence of O2 and ROS. ROS, including superoxide, hydrogen peroxide and hydroxyl radical, are formed during oxygen reduction in the oxygen reduction systems and during non-specific reactions of oxygen with reduced substrates, e.g., transition metals and radical species [45]. The toxicity of O2 in cells is mainly due to cellular damages caused by ROS, such as the oxidation of thiols and the release of metallic centers from proteins leading to the increase of free metals in cytosol. The increased free metals, mainly iron, cause DNA damage through fenton-type reactions that produce ROS [44]. The removal of ROS is important for cells to deal with oxidative stress. We found genes encoding nickel-containing superoxide dismutase that eliminates superoxide radicals through disproportionation into H2O2 and O2 [46] and superoxide reductase that converts toxic O2− into less toxic H2O2 [47]. These genes have been found in other SRB, e.g., in Desulfovibrio genus [48, 49]. In addition, genes encoding enzymes that can decompose H2O2 [47], such as catalase, thiol peroxidase, cytochrome c peroxidase, peroxiredoxin, and rubrerythrin, were found. Catalase is a common enzyme in aerobic organisms that catalyzes the detoxification of H2O2 and has been found in many sulfate reducers both the bacterial and archaeal domains e.g., Desulfovibrio gigas [50] and Archaeoglobus fulgidus [51],
The investigated genomes encode several damage repair systems: (1) thioredoxin, thioredoxin reductase, and glutaredoxin for disulphide bonds reductions; (2) methionine sulfoxide reductase (MsrA/MsrB) for oxidized methionines reduction; and (3) NifU like protein for the Fe-S clusters respiration or biosynthesis (Supplementary Dataset) [44]. The enzymes involved in the damage repair system are metal-free. In contrast, the damage in the detoxification system (superoxide reductase and rubrerythrin) contributes to an increase of Fe2+, leading to the formation of ROS, which requires more repair. Therefore, the system for damage repair is more important than the detoxification system in cells when oxidative conditions are more severe. For example, the expression gene that encodes enzymes of damage repair systems were highly upregulated in Desulfovibrio vulgaris after oxidative stress [52]. Apart from mechanisms dealing with oxidative stress, studied genomes encode methyl-accepting chemotaxis proteins (MCP), which are involved in behavioral strategies to avoid and thereby protect cells from contact with oxygen [10, 43, 53, 54].
Comparative genomics
The three studied genomes share 1557 and 1537 homologues which is at least 1/3 of their genome content based on two algorithms with BDBH and OMCL options (Fig. 4). These homologues have key functions for cell maintenance processes. There are still large portions of species-specific proteins, over 1/3 of proteins in each genome that do not have homologues in the other two species, that may be attributed to the different isolation sources of these three species. The number of proteins in Desulfofaba fastidiosa shared with the other two species were much lower than number of genes shared between Desulfofaba hansenii and Desulfofaba gelida. This potential syntrophic process of anaerobic oxidation of methane coupled to sulfate reduction in the sulfate-methane transition zone may increase the chance for lateral gene transfer, which further contributes to the smaller genome size in Desulfofaba fastidiosa [55]. Genes related with transcription, signal transduction, and secondary metabolite synthesis tend to lose during genome reduction in symbiotic genomes [56]. Moreover, the numbers of transport genes are positively correlated with Desulfofaba genome sizes (Table 2), which is consistent with the universal relationship with genome size [56]. It further suggests that the sulfate-methane transition zone in marine sediment, where Desulfofaba fastidiosa was isolated from, is more different from the other two environments, e.g., seagrass roots and surface sediment.

Shared protein contents among three Desulfofaba genomes based on two different algorithms: a bidirectional best-hits (BDBH) and b orthoMCL algorithm (OMCL). Over 1500 proteins were shared between the three species, and over 1/3 of protein sequences in each genome were species-specific
Conclusions
Strains belonging to the Desulfofaba genus were isolated from different environments. Genomic content of this genus showed a high degree of versatility of central metabolic pathways. The diverse central metabolism indicates that isolates have the potential to utilize a wide range of substrates. The presence of different genes involved in sulfur and nitrogen metabolism suggest that they may play a role in various aspects of sulfur and nitrogen cycling. A 16S rRNA gene homologues-based search in publicly available metagenomes and our collection of MAGs from various marine environments indicates a limited environmental distribution of this genus. This, however, does not exclude members of Desulfofaba genus from having a significant role in biogeochemical cycling in their respective habitats. Their ability to respire oxygen and the presence of genes for ROS damage defense allows them to inhabit environments with regular oxygen intrusion, such as the roots of Zostera marina plants from which Desulfofaba hansenii was isolated from. The incomplete oxidation of propionate to acetate provides easily utilized electron donors to other microbes which benefits the entire microbial community.
Methods
Bacteria cultivation and DNA extraction
Desulfofaba fastidiosa P2 (DSM 15249) [20], Desulfofaba gelida PSv29 (DSM 12344) [21], and Desulfofaba hansenii P1 (DSM 13527) [22] were grown as described in the literature [20,21,22]. The cells were harvested in the late exponential phase. DNA was extracted from the cell pellet using the PowerLyser® PowerSoil® DNA extraction kit (MoBio, Carlsbad, CA, USA) according to the manufacturer’s protocol.
Genome sequencing, assembly, annotation, and homologues search
DNA was sequenced on a MiSeq platform. The sequencing data were treated as previously described [57]. Briefly, the sequencing library was trimmed with Trimmomatic-0.36 [58] with the following trimming parameters: CROP:290 HEADCROP:25 SLIDINGWINDOW:4:20. Read quality before and after trimming was assessed by FastQC version 0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were assembled using SPAdes 3.10.1 [59]. Contigs shorter than 1,000 bp were removed after assembling. The quality of the assembled genomes was estimated using CheckM v1.1.3 [23]. The draft genome was annotated using the standard operation procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP v.4) supported by the JGI (Berkeley, CA; USA) [60]. The predicted protein sequences from MGAP were further annotated using KofamScan v.1.3.0 with the e-value cut-off of 1e-5 and the highest bit-score larger than the pre-set threshold for each gene [61], and further characterized using KAAS (KEGG Automatic Annotation Server) web server [62] using the ‘Complete or Draft Genome’ setting with parameters: GHOSTX, custom genome dataset, and BBH assignment method.
Key metabolic genes were searched using custom databases. Briefly, peptidases were identified using DIAMOND BLASTP v0.9.31.132 [63] to search against MEROPS Peptidase Protein Sequences (Downloaded on 24th, March, 2022) [64] with the settings: -e 1e-10 –subject-cover 80 –id 50 [65]. Carbohydrate active enzymes (CAZYmes) were identified using the dbCAN v2 standalone tool (CAZYDB.09242021, dbCAN-HMMdb-V10, and dbCAN-fam-aln-V9 databases) [66] with default thresholds. The localization of identified peptidases and CAZYmes was determined using the command-line version of Psort v3.0 using the option –negative for the genomes.
Genes encoding for dissimilatory sulfite reductase (DsrAB) and hydrogenase were further identified using DIAMOND BLSATP v0.9.31.132 [63] to search against different custom databases with the thresholds: -e 1e-10 –subject-cover 70 –id 50; -e 1e-10 –subject-cover 50 –id 30; and -e 1e-10 –subject-cover 50 –id 40 for DsrAB and hydrogenase sequences, respectively. The identified sequences were confirmed with the annotation from MGAP and KO assignment. The identified hydrogenase sequences were further compared with the annotation based on the assigned KO number and the web-based hydrogenase classifier (26th March, 2022) [32].
Homologues between different genomes were searched by GET_HOMOLOGUES [67, 68] with BDBH and OMCL options.
Phylogenetic analysis
A set of 120 marker genes was extracted from the three genomes and reference genomes using the Genome Taxonomy Database (GTDB)-Tk v1.7.0 (release 202) [69]. Another set of 37 single-copy, protein-coding housekeeping genes was extracted using Phylosift v1.0.1 [70]. The two sets of marker genes were separately concatenated, and aligned using MAFFT v7.450 [71] with the setting –maxiterate 1000 –localpair, trimmed using trimAl v1.2rev59 [72] with the -gappyout option, and manually checked. The two refined alignments were used to generate two maximum-likelihood trees using RAxML v8.2.4 [73] with the parameters: raxmlHPC-PTHREADS-AVX -m GTRGAMMA -N autoMRE -p 12345—× 12345. Amino acid identity (AAI) of the three genomes and reference genomes was estimated using CompareM (v0.1.2) AAI workflow (‘comparem aai_wf’, https://github.com/dparks1134/CompareM).
16S rRNA sequences were identified using Barrnap v0.9 (https://github.com/tseemann/barrnap) with the default settings, aligned, and manually curated in ARB [74] with the SILVA SSURef NR99 database (release 138). The alignment was exported to generate a maximum-likelihood tree using IQ-TREE v1.6.12 [75] with the settings: -m MFP -bb 1000 -bnni -alrt 1000.
The identified DsrA and DsrB sequences were separately aligned with reference sequences using MAFFT v7.450 with the settings: –maxiterate 1000 –globalpair –anysymbol, trimmed using trimAl v1.2rev59 [72] with the -gappyout option, manually checked, and concatenated. The maximum-likelihood tree was generated using RAxML v8.2.4 [73] with the parameters: raxmlHPC-PTHREADS-AVX -m GTRGAMMA -N autoMRE -p 12345—× 12345.
The final identified hydrogenase sequences with selected references for different types of hydrogenases [76] were aligned using ClustalW v2.1 [77], and the Neighbor-Joining tree was generated using MEGA X [78] under p-distance model with 1,000 bootstrap.
Availability of data and materials
The genome sequence and annotation can be found in IMG/JGI under the IMG Genome ID: 2928859439, 2740891818, and 2929304470.
Abbreviations
- SRB:
-
Sulfate-reducing bacteria
- Dsr:
-
Dissimilatory sulfite reductase
- MAGs:
-
Metagenome-assembled genomes
- PHB:
-
Polyhydroxybutyrate
- AAI:
-
Average amino acid identity
- CAZYmes:
-
Carbohydrate-active enzymes
- TCA:
-
Tricarboxylic acid
- PPP:
-
Pentose phosphate pathway
- WLP:
-
Wood-Ljungdahl pathway (WLP)
- NH2OH:
-
Hydroxylamine
- NO:
-
Nitric oxide
- HAO:
-
Hydroxylamine dehydrogenase
- NH3 :
-
Ammonia
- HCP:
-
Hydroxylamine reductase
- N2O:
-
Nitrous oxide
- Fd2 − :
-
Ferredoxin
- H+ :
-
Proton
- ROS:
-
Reactive oxygen species
- MCP:
-
Methyl-accepting chemotaxis proteins
- MGAP:
-
Microbial Genome Annotation Pipeline
- KAAS:
-
KEGG Automatic Annotation Server
- GTDB:
-
Genome Taxonomy Database
References
Okabe S, Itoh T, Satoh H, Watanabe Y. Analyses of spatial distributions of sulfate-reducing bacteria and their activity in aerobic wastewater biofilms. Appl Environ Microbiol. 1999;65:5107–16.
Jørgensen BB. The sulfur cycle of freshwater sediments: Role of thiosulfate. Limnol Oceanogr. 1990;35:1329–42.
Jørgensen BB. Mineralization of organic matter in the sea bed—the role of sulphate reduction. Nature. 1982;296:643–5.
Jørgensen BB, Findlay AJ, Pellerin A. The biogeochemical sulfur cycle of marine sediments. Front Microbiol. 2019;10:849.
Cypionka H, Widdel F, Pfennig N. Survival of sulfate-reducing bacteria after oxygen stress, and growth in sulfate-free oxygen-sulfide gradients. FEMS Microbiol Ecol. 1985;1:39–45.
Dannenberg S, Kroder M, Dilling W, Cypionka H. Oxidation of H2, organic compounds and inorganic sulfur compounds coupled to reduction of O2 or nitrate by sulfate-reducing bacteria. Arch Microbiol. 1992;158:93–9.
Marschall C, Frenzel P, Cypionka H. Influence of oxygen on sulfate reduction and growth of sulfate-reducing bacteria. Arch Microbiol. 1993;159:168–73.
van Niel EW, Gottschal JC. Oxygen Consumption by Desulfovibrio Strains with and without Polyglucose. Appl Environ Microbiol. 1998;64:1034–9.
Johnson MS, Zhulin IB, Gapuzan ME, Taylor BL. Oxygen-dependent growth of the obligate anaerobe Desulfovibrio vulgaris Hildenborough. J Bacteriol. 1997;179:5598–601.
Krekeler D, Teske A, Cypionka H. Strategies of sulfate-reducing bacteria to escape oxygen stress in a cyanobacterial mat. FEMS Microbiol Ecol. 1998;25:89–96.
Lemos RS, Gomes CM, Santana M, LeGall J, Xavier AV, Teixeira M. The, “strict” anaerobe Desulfovibrio gigas contains a membrane-bound oxygen-reducing respiratory chain. FEBS Lett. 2001;496:40–3.
Lefèvre CT, Howse PA, Schmidt ML, Sabaty M, Menguy N, Luther GW 3rd, et al. Growth of magnetotactic sulfate-reducing bacteria in oxygen concentration gradient medium. Environ Microbiol Rep. 2016;8:1003–15.
Schoeffler M, Gaudin A-L, Ramel F, Valette O, Denis Y, Hania WB, et al. Growth of an anaerobic sulfate-reducing bacterium sustained by oxygen respiratory energy conservation after O2 -driven experimental evolution. Environ Microbiol. 2019;21:360–73.
Nielsen LP, Risgaard-Petersen N, Fossing H, Christensen PB, Sayama M. Electric currents couple spatially separated biogeochemical processes in marine sediment. Nature. 2010;463:1071–4.
Pfeffer C, Larsen S, Song J, Dong M, Besenbacher F, Meyer RL, et al. Filamentous bacteria transport electrons over centimetre distances. Nature. 2012;491:218–21.
Marzocchi U, Trojan D, Larsen S, Meyer RL, Revsbech NP, Schramm A, et al. Electric coupling between distant nitrate reduction and sulfide oxidation in marine sediment. ISME J. 2014;8:1682–90.
Muyzer G, Stams AJM. The ecology and biotechnology of sulphate-reducing bacteria. Nat Rev Microbiol. 2008;6:441–54.
Gong X, Rio AR del, Xu L, Langwig M, Su L, Sun M, et al. New globally distributed bacteria with high proportions of novel protein families involved in sulfur and nitrogen cycling. Research Square. 2022.
Langwig MV, De Anda V, Dombrowski N, Seitz KW, Rambo IM, Greening C, et al. Large-scale protein level comparison of Deltaproteobacteria reveals cohesive metabolic groups. ISME J. 2022;16:307–20.
Abildgaard L, Ramsing NB, Finster K. Characterization of the marine propionate-degrading, sulfate-reducing bacterium Desulfofaba fastidiosa sp. nov. and reclassification of Desulfomusa hansenii as Desulfofaba hansenii comb. nov. Int J Syst Evol Microbiol. 2004;54 Pt 2:393–9.
Knoblauch C, Sahm K, Jørgensen BB. Psychrophilic sulfate-reducing bacteria isolated from permanently cold arctic marine sediments: description of Desulfofrigus oceanense gen. nov., sp. nov., Desulfofrigus fragile sp. nov., Desulfofaba gelida gen. nov., sp. nov., Desulfotalea psychrophila gen. nov., sp. nov. and Desulfotalea arctica sp. nov. Int J Syst Bacteriol. 1999;49 Pt 4:1631–43.
Finster K, Thomsen TR, Ramsing NB. Desulfomusa hansenii gen. nov., sp. nov., a novel marine propionate-degrading, sulfate-reducing bacterium isolated from Zostera marina roots. Int J Syst Evol Microbiol. 2001;51 Pt 6:2055–61.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Moskowitz GJ, Merrick JM. Metabolism of poly-β-hydroxybutyrate. II. Enzymic synthesis of D-(-)-β-hydroxybutyryl coenzyme A by an enoyl hydrase from Rhodospirillum rubrum. Biochemistry. 1969;8:2748–55.
Verlinden RAJ, Hill DJ, Kenward MA, Williams CD, Radecka I. Bacterial synthesis of biodegradable polyhydroxyalkanoates. J Appl Microbiol. 2007;102:1437–49.
Korth F, Kock A, Arévalo-Martínez DL, Bange HW. Hydroxylamine as a potential indicator of nitrification in the open ocean. Geophys Res Lett. 2019;46:2158–66.
Oshiki M, Ali M, Shinyako-Hata K, Satoh H, Okabe S. Hydroxylamine-dependent anaerobic ammonium oxidation (anammox) by “Candidatus Brocadia sinica.” Environ Microbiol. 2016;18:3133–43.
Kuypers MMM, Marchant HK, Kartal B. The microbial nitrogen-cycling network. Nat Rev Microbiol. 2018;16:263–76.
Caranto JD, Lancaster KM. Nitric oxide is an obligate bacterial nitrification intermediate produced by hydroxylamine oxidoreductase. Proc Natl Acad Sci U S A. 2017;114:8217–22.
Battaglia G, Joos F. Marine N2O Emissions From Nitrification and Denitrification Constrained by Modern Observations and Projected in Multimillennial Global Warming Simulations. Global Biogeochem Cycles. 2017;32:92–121.
Plugge CM, Zhang W, Scholten JCM, Stams AJM. Metabolic flexibility of sulfate-reducing bacteria. Front Microbiol. 2011;2:81.
Søndergaard D, Pedersen CNS, Greening C. HydDB: A web tool for hydrogenase classification and analysis. Sci Rep. 2016;6:34212.
Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front Microbiol. 2011;2:69.
Leinfelder W, Forchhammer K, Veprek B, Zehelein E, Böck A. In vitro synthesis of selenocysteinyl-tRNA(UCA) from seryl-tRNA(UCA): involvement and characterization of the selD gene product. Proc Natl Acad Sci U S A. 1990;87:543–7.
Forchhammer K, Leinfelder W, Boesmiller K, Veprek B, Böck A. Selenocysteine synthase from Escherichia coli. Nucleotide sequence of the gene (selA) and purification of the protein. J Biol Chem. 1991;266:6318–23.
Dardel F, Fayat G, Blanquet S. Molecular cloning and primary structure of the Escherichia coli methionyl-tRNA synthetase gene. J Bacteriol. 1984;160:1115–22.
Mukhopadhyay R, Rosen BP, Phung LT, Silver S. Microbial arsenic: from geocycles to genes and enzymes. FEMS Microbiol Rev. 2002;26:311–25.
Neff JM. Ecotoxicology of arsenic in the marine environment. Environ Toxicol Chem. 1997;16:917–27.
Messens J, Hayburn G, Desmyter A, Laus G, Wyns L. The essential catalytic redox couple in arsenate reductase from Staphylococcus aureus. Biochemistry. 1999;38:16857–65.
Ben Fekih I, Zhang C, Li YP, Zhao Y, Alwathnani HA, Saquib Q, et al. Distribution of Arsenic Resistance Genes in Prokaryotes. Front Microbiol. 2018;9:2473.
Dolla A, Fournier M, Dermoun Z. Oxygen defense in sulfate-reducing bacteria. J Biotechnol. 2006;126:87–100.
Morris RL, Schmidt TM. Shallow breathing: bacterial life at low O(2). Nat Rev Microbiol. 2013;11:205–12.
Cypionka H. Oxygen respiration by desulfovibrio species. Annu Rev Microbiol. 2000;54:827–48.
Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IAC. A Post-Genomic View of the Ecophysiology, Catabolism and Biotechnological Relevance of Sulphate-Reducing Prokaryotes. Adv Microb Physiol. 2015;66:55–321.
Imlay JA. Pathways of oxidative damage. Annu Rev Microbiol. 2003;57:395–418.
Shearer J. Insight into the structure and mechanism of nickel-containing superoxide dismutase derived from peptide-based mimics. Acc Chem Res. 2014;47:2332–41.
Jenney FE Jr, Verhagen MF, Cui X, Adams MW. Anaerobic microbes: oxygen detoxification without superoxide dismutase. Science. 1999;286:306–9.
Pinto AF, Rodrigues JV, Teixeira M. Reductive elimination of superoxide: Structure and mechanism of superoxide reductases. Biochim Biophys Acta. 2010;1804:285–97.
Sheng Y, Abreu IA, Cabelli DE, Maroney MJ, Miller A-F, Teixeira M, et al. Superoxide dismutases and superoxide reductases. Chem Rev. 2014;114:3854–918.
Morais-Silva FO, Rezende AM, Pimentel C, Santos CI, Clemente C, Varela-Raposo A, et al. Genome sequence of the model sulfate reducer Desulfovibrio gigas: a comparative analysis within the Desulfovibrio genus. Microbiologyopen. 2014;3:513–30.
Kengen SW, Bikker FJ, Hagen WR, de Vos WM, van der Oost J. Characterization of a catalase-peroxidase from the hyperthermophilic archaeon Archaeoglobus fulgidus. Extremophiles. 2001;5:323–32.
Mukhopadhyay A, Redding AM, Joachimiak MP, Arkin AP, Borglin SE, Dehal PS, et al. Cell-wide responses to low-oxygen exposure in Desulfovibrio vulgaris Hildenborough. J Bacteriol. 2007;189:5996–6010.
Eschemann A, Kühl M, Cypionka H. Aerotaxis in Desulfovibrio. Environ Microbiol. 1999;1:489–94.
Fu R, Wall JD, Voordouw G. DcrA, a c-type heme-containing methyl-accepting protein from Desulfovibrio vulgaris Hildenborough, senses the oxygen concentration or redox potential of the environment. J Bacteriol. 1994;176:344–50.
Dagan T, Martin W. Ancestral genome sizes specify the minimum rate of lateral gene transfer during prokaryote evolution. Proc Natl Acad Sci U S A. 2007;104:870–5.
Konstantinidis KT, Tiedje JM. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc Natl Acad Sci U S A. 2004;101:3160–5.
Gong X, Skrivergaard S, Korsgaard BS, Schreiber L, Marshall IPG, Finster K, et al. High quality draft genome sequence of Janthinobacterium psychrotolerans sp. nov., isolated from a frozen freshwater pond. Stand Genomic Sci. 2017;12:8.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Huntemann M, Ivanova NN, Mavromatis K, Tripp HJ, Paez-Espino D, Palaniappan K, et al. The standard operating procedure of the DOE-JGI Microbial Genome Annotation Pipeline (MGAP vol 4). Stand Genomic Sci. 2015;10:86.
Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35 Web Server issue:W182–5.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Rawlings ND, Barrett AJ, Finn R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2016;44:D343–50.
Zhou Z, Tran PQ, Kieft K, Anantharaman K. Genome diversification in globally distributed novel marine Proteobacteria is linked to environmental adaptation. ISME J. 2020;14:2060–77.
Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:W95-101.
Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 2013;79:7696–701.
Vinuesa P, Contreras-Moreira B. Robust Identification of Orthologues and Paralogues for Microbial Pan-Genomics Using GET_HOMOLOGUES: A Case Study of pIncA/C Plasmids. In: Mengoni A, Galardini M, Fondi M, editors. Bacterial Pangenomics: Methods and Protocols. New York, NY: Springer New York; 2015. p. 203–32.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019. https://doi.org/10.1093/bioinformatics/btz848.
Darling AE, Jospin G, Lowe E, Matsen FA 4th, Bik HM, Eisen JA. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2014;2: e243.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Greening C, Biswas A, Carere CR, Jackson CJ, Taylor MC, Stott MB, et al. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J. 2016;10:761–77.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 2018;35:1547–9.
Acknowledgements
We thank Anne B. Stentebjerg for excellent technical assistance.
Funding
This work was supported by the National Key Research and Development Program of China (grant numbers 2018YFA0605800 and 2020YFA0608301), the National Natural Science Foundation of China (grant numbers 42006134, 42106153 and 91951202), the Pilot National Laboratory for Marine Science and Technology (Qingdao) (grant number LSKJ202203901), and Shandong University Foundation for Future Scholar Plan.
Author information
Authors and Affiliations
Contributions
P.G., L.Q., K.F., and X.G. designed the study. X.Z. X.H. performed phylogenetic analysis. A.M. and X.G. assembled the genome. X.Z and X.G., performed the metabolic analysis. L.H. performed the physiological experiment. P.G., L.Q., K.F., and X.G. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
A maximum likelihood phylogenetic tree of xx genomes including the 3 Desulfofaba genomes. The phylogeny is based on 120 concatenated ribosomal protein encoding genes identified using GTDB-tk. Acidobacteria were set as the outgroup.
Additional file 2: Figure S2.
Maximum likelihood phylogenetic tree of 16S rRNA gene.
Additional file 3: Figure S3.
Hierarchical clustering heatmap using pheatmap package in R based on average amino acids identity (AAI) for each genome pair.
Additional file 4: Figure S4.
Carbohydrate-active enzymes (CAZyme) and peptidase encoded by Desulfofaba genus. (a) CAZymes include carbohydrate esterase (CE), glycoside hydrolase (GH), and polysaccharide lyase (PL). (b) Peptidases are classified by family as aspartic (A), cysteine (C), unassigned inhibitors (I), metallo (M), asparagine (N), serine (S), threonine (T), and unknown (U) by the MEROPS database. Sizes of the circle denote the number of gene copies in the genome. The number on top of the circle represents the number of sequences identified with potential secretion signal using PSORTb v3.0.
Additional file 5: Figure S5.
Maximum likelihood phylogenetic tree of NiFe hydrogenases. Bootstrap values ≥ 75 are shown in circles.
Additional file 6: Figure S6.
Maximum likelihood phylogenetic tree of FeFe hydrogenases. Bootstrap values ≥ 75 are shown in circles.
Additional file 7: Figure S7.
Schematic drawing of the reaction chambers used in this study. Type I chamber (a) was used at oxygen concentrations between 0 and 36 μM; type II chamber (b) was used up to 140 μM.
Additional file 8: Figure S8.
Oxygen respiration under different conditions. (a) Oxygen consumption rates under different initial oxygen concentrations. The oxygen consumption rates increased with increasing oxygen concentrations up to about 40 μM and decreased slowly to the lowest rates at 140 μM oxygen. Filled circles represent rates obtained in type I chamber, while open circles represent rates obtained in type II chamber. (b) Oxygen consumption started immediately after the oxygenated medium was injected (final concentration 36 μM) into the culture. The highest rates were measured in the beginning of the monitoring period. (c) The effect of formate on the rate of oxygen consumption. The experiment was initiated by injection of oxygenation medium (final concentration 25 μM of oxygen). The immediate consumption rate of oxygen was 15 nmol O2 min-1 mg protein-1. After addition of formate (final concentration 20 mM) the oxygen consumption rate increased to 22 nmol O2 min-1 mg protein-1.
Additional file 9: Figure S9.
A Lineweaver-Burk plot constructed from the first eleven measurements shown in Fig. S8a. The X and Y intercepts are used to calculate Km and Vmax.
Additional file 10: Figure S10.
Cells of D. hansenii before (a) and after (b) exposure to 60 μM oxygen for 20 h. The cells were stained with Nile blue, which binds to polyhydroxyalkanoates. The red color represents areas in the cells, which were stained by Nile blue, indicating that the amount of polyhydroxyalcanoates decreased after exposure to oxygen.
Additional file 11.
Detailed annotation of the three Desulfofaba genomes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, P., Zhang, X., Huang, X. et al. Genomic insight of sulfate reducing bacterial genus Desulfofaba reveals their metabolic versatility in biogeochemical cycling. BMC Genomics 24, 209 (2023). https://doi.org/10.1186/s12864-023-09297-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09297-2