
Abstract
Background
Green feed diet in ruminants exerts a beneficial effect on rumen metabolism and enhances the content of milk nutraceutical quality. At present, a comprehensive analysis focused on the identification of genes, and therefore, biological processes modulated by the green feed in buffalo rumen has never been reported. We performed RNA-sequencing in the rumen of buffaloes fed a total mixed ration (TMR) + the inclusion of 30% of ryegrass green feed (treated) or TMR (control), and identified differentially expressed genes (DEGs) using EdgeR and NOISeq tools.
Results
We found 155 DEGs using EdgeR (p-values < 0.05) and 61 DEGs using NOISeq (prob ≥0.8), 30 of which are shared. The rt-qPCR validation suggested a higher reliability of EdgeR results as compared with NOISeq data, in our biological context.
Gene Ontology analysis of DEGs identified using EdgeR revealed that green feed modulates biological processes relevant for the rumen physiology and, then, health and well-being of buffaloes, such as lipid metabolism, response to the oxidative stress, immune response, and muscle structure and function. Accordingly, we found: (i) up-regulation of HSD17B13, LOC102410803 (or PSAT1) and HYKK, and down-regulation of CDO1, SELENBP1 and PEMT, encoding factors involved in energy, lipid and amino acid metabolism; (ii) enhanced expression of SIM2 and TRIM14, whose products are implicated in the immune response and defense against infections, and reduced expression of LOC112585166 (or SAAL1), ROR2, SMOC2, and S100A11, encoding pro-inflammatory factors; (iii) up-regulation of NUDT18, DNAJA4 and HSF4, whose products counteract stressful conditions, and down-regulation of LOC102396388 (or UGT1A9) and LOC102413340 (or MRP4/ABCC4), encoding detoxifying factors; (iv) increased expression of KCNK10, CACNG4, and ATP2B4, encoding proteins modulating Ca2+ homeostasis, and reduced expression of the cytoskeleton-related MYH11 and DES.
Conclusion
Although statistically unpowered, this study suggests that green feed modulates the expression of genes involved in biological processes relevant for rumen functionality and physiology, and thus, for welfare and quality production in Italian Mediterranean dairy buffaloes.
These findings, that need to be further confirmed through the validation of additional DEGs, allow to speculate a role of green feed in the production of nutraceutical molecules, whose levels might be enhanced also in milk.
Similar content being viewed by others

Introduction
In mammals, a homeostatic orchestration of several metabolic processes in the different tissues is required for the maintenance of lactation. In particular, the gastrointestinal tract is the central site of feed digestion, nutrient uptake, and has a pivotal role in the endocrinal control of ruminant metabolism [1]. Exploring the biology of the different tissues and organs of gastrointestinal system, including their gene expression profiles and metabolic pathways, may significantly increase the knowledge of the ruminant biology and lead to discovering new potential candidate genes for future increases in animal production, in terms of both quantity and quality. Several studies have demonstrated the relevance of RNA sequencing (RNA-seq) and other omics approaches for the identification of metabolic processes potentially involved in lactating dairy cows [2,3,4]. It has been reported that milk composition could be influenced by many factors such as breed, diseases, environment, parity, days in milk and many others [5]. Among these factors, the diet, whether or not mediated by the rumen metabolism, has a pivotal role since it can deeply modify the organoleptic and nutraceutical quality of both milk and dairy products. Several studies have demonstrated that pasture-based diets can positively influence milk nutrient profile improving the concentration of several beneficial compounds such as Omega-3 polyunsaturated fatty acids, vaccenic and linolenic acid, and conjugated linoleic acid (CLA), as well as reducing the levels of Omega-6 fatty acids and palmitic acid [6,7,8,9,10].
Rumen metabolic processes have a significant impact on the composition of milk nutrients. The products of the rumen fermentation, such as short-chain fatty acids (SCFAs) produced in the forestomaches, indeed, are largely absorbed across the epithelium of the rumen and of the omasum [11] and, together with other nutraceutical molecules, are extracted into the blood, metabolized by the liver and finally transported to the mammary gland [3]. Of note, SCFAs meet approximately 80% of the energy needs for these animals, and therefore, they play important roles in the maintenance of energy homeostasis of ruminants [12].
Improving the nutraceutical properties of milk is an attempt to meet consumers demand of healthy, “natural”, and sustainable dairy products. Consumers have started to ask for “natural” foods, and this could be made possible modifying milk composition directly at the farm stage through dietary intervention, without using mechanical modifications such as processing, fat separation, ultrafiltration, etc. [8, 13, 14]. When it is not possible to use pastures, the utilization of green feed could represent a low-cost opportunity. It has already been seen that the inclusion of green forage in the diet enhances the content of some components with nutraceutical properties [15], such as vaccenic and rumenic acids [16], and betaines and carnitines [17]. In nature, feed regimens have the potential to induce changes in the transcriptional program of the cells, often through the modulation of the epigenome, and this could have a significant impact on many biological processes, such as metabolism, health and development [18]. Angus beef cattle that received a grass-based diet showed changes in the rumen expression of gene networks associated with immune mechanisms, cellular development, and the biosynthesis of key molecules [19]. In buffalo species, few molecular studies have been performed so far, taking advantage of RNA-seq and other omics approaches [20, 21], and no information is available on the impact of different feed regimens on the transcriptomic landscape in these species. In a previous study [17], it has been shown that the administration of green forage in Italian Mediterranean dairy buffaloes enhanced the antioxidant and antineoplastic activity of buffalo milk. Based on this evidence, it is conceivable that the provision of green feed to Italian Mediterranean dairy buffaloes would also induce, in the rumen, changes in the expression of genes whose products participate to networks linked to the metabolism of important biomolecules.
The aim of the present study was to identify in rumen the impact of green feed diet on molecular mechanisms relevant for rumen functionality and physiology, through the analysis of ruminal transcriptomic profiles of buffaloes that received a standard total mixed ration (TMR) or a TMR + ryegrass green feed (30% of diet). The obtained results might highlight the beneficial effect of this diet on welfare-related molecular processes, and contribute to elucidate the role of green feed in the production of nutraceutical molecules, whose levels might be enhanced also in milk.
Results
Transcriptomic analysis in the ruminal wall of dairy buffaloes
RNA-seq yielded an average of 47,967,818 high-quality 150 bp reads (23,983,909 paired reads) per sample. Approximately 93% of the reads were mapped to the Bubalus bubalis reference genome sequence (version UOA_WB_1 from NCBI) and almost 84% of the reads were uniquely mapped (Additional file 1: Table S1). A total of 18,386 and 18,380 genes were expressed (Trimmed Mean of M values, TMM > 1) in ruminal wall of buffaloes that received TMR + green feed (treated group) and TMR (control group), respectively (Additional file 2: Table S2).
Green feed re-modulates the expression of several genes in rumen of dairy buffaloes
The identification of differentially expressed genes (DEGs) between treated and control groups (six animals for each condition) has been carried out using two different approaches: EdgeR and NOISeq software packages.
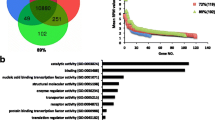
Initial analysis of DEGs performed using EdgeR with FDR-adjusted p-values < 0.05 as significance threshold failed to yield significant DEGs. Considering that the two diets were isonitrogenous and isoenergetic, and varied only for the inclusion of green forage, we expect small variations of gene expression following the change of feed regimen, that could be masked by the inter-individual variability of the animals belonging to the same group. In the light of this hypothesis, we decided to identify DEGs using nominal p-values < 0.05 as significance threshold. This analysis identified 155 DEGs (Fig. 1; Additional file 3: Table S3). The volcano plot depicts the statistical significance of the differential expression between the two groups and the difference in gene expression levels (Fig. 1a, top). Among DEGs, 71 genes showed lower expression and 84 displayed higher expression in the rumen of treated buffaloes (Fig. 1a, bottom). In Fig. 1b is shown the heatmap that illustrates, through a hierarchical clustering analysis, the significant DEGs across all the samples.

Effect of green feed on transcriptional profile in rumen of dairy buffaloes (six animals for each condition), identified using EdgeR (nominal p-values < 0.05). a Top: Volcano plot that illustrates differentially expressed genes (DEGs) in rumen of dairy buffaloes fed a TMR + green feed (Treated) versus animals fed the TMR diet (Control). The significance of the differential expression (−log10 (p-value), y-axis) is plotted versus fold change expression level (x-axis). Red and green dots depict the genes that are significantly up- and down-regulated in treated versus control animals; black dots are genes whose expression levels did not reach the statistical significance (p-value < 0.05) between the two experimental groups. Bottom: Bar graph that shows the number of up- and down-regulated genes in treated vs control animals. b Heatmap representation of significant DEGs (155) across all the samples. Blue-red gradient shows low to high expression levels for each gene. Dendrogram represents a hierarchical clustering based on Pearson correlation values
Among the genes found to be deregulated in rumen of treated versus control animals, using EdgeR software package, the top 20 up-regulated (Table 1) and down-regulated (Table 2) genes were selected in terms of the magnitude of differential expression, log2 (fold change), to identify the processes being affected by the different diet composition.
Included in the top up-regulated genes, we found LOC102407663 (or interferon alpha-inducible protein 27-like protein 2, IFI27L2), single-minded homolog 2 protein (SIM2) and interferon-induced protein 44-like (IFI44L) genes, which are associated with immune response; the lipid metabolism-related 17-beta-hydroxysteroid dehydrogenase 13 (HSD17B13) and LOC102411389 (or Vanin 1, VNN1) genes; LOC102395439 (or four and a half LIM domains protein 1, FHL1), tubulin polymerization-promoting protein family member 3 (TPPP3), and LOC102397197 (or tubulin alpha-1D chain, TUBA1D) genes, whose products are involved in cytoskeletal organization; the low-affinity IgE receptor Fc Epsilon Receptor II (FCER2) gene; the oxidative stress- and immune system-linked dual oxidase maturation factor 1 isoform X2 (DUOXA1) gene; insulin-like growth factor-binding protein 6 (IGFBP6) gene.
Among the top down-regulated genes, we identified the LOC112585166 (or serum amyloid A protein-like, SAAL1), tyrosine-protein kinase transmembrane receptor (ROR2), and SPARC-related modular calcium-binding protein 2 (SMOC2) genes, encoding pro-inflammatory factors; the LDL receptor related protein 2 (LRP2) and LOC102399021 (or cytochrome P450 1A1, CYP1A1) genes, which are related to lipid metabolism; the energy metabolism-linked LOC102389855 (or cytochrome C oxidase subunit 7C, Cox7C) gene; the LOC102399215 (or nuclear distribution protein nudE homolog 1-like, NDEL1) gene, involved in cytoskeletal organization; LOC112578513 (or multidrug resistance-associated protein 4-like (ABCC4) gene, involved in the response to oxidative stress; the extracellular matrix-associated synaptopodin-2 (SYNPO2) gene; LOC102400888 (or olfactory receptor 51E1, OR51E1) gene; the transporter of α-tocopherol (vitamin E) alpha tocopherol transfer protein (TTPA) gene; the membrane-bound glycosylated Mucin-15 (MUC15) gene.
A parallel analysis, performed using the NOISeq software package with a posterior probability (prob) ≥ 0.8 as significance threshold, identified 61 DEGs, 35 of which showed lower expression and 26 displayed higher expression in the rumen of treated buffaloes (Fig. 2a). Among DEGs, 11 of these have a prob. ≥0.9 and only 1 DEG shows a prob. ≥0.95 (Additional file 3: Table S3). The statistical significance of the differential expression between the two groups and the difference in gene expression levels are illustrated with volcano plot (Fig. 2a). The heatmap reported in Fig. 2b illustrates the significant DEGs with a prob. ≥0.8 across all the samples, through a hierarchical clustering analysis.

Effect of green feed on transcriptional profile in rumen of dairy buffaloes (six animals for each condition), identified using NOISeq software (prob ≥0.8). a Top: Volcano plot that illustrates differentially expressed genes (DEGs) in rumen of dairy buffaloes fed a TMR + green feed (Treated) versus animals fed the TMR diet (Control). The significance of the differential expression (probability, y-axis) is plotted versus fold change expression level (x-axis). Red and green dots depict the genes that are significantly up- and down-regulated in treated versus control animals; black dots are genes whose expression levels did not reach the statistical significance (prob ≥0.8) between the two experimental groups. Bottom: Bar graph that shows the number of up- and down-regulated genes in treated vs control animals. b Heatmap representation of significant DEGs (61) across all the samples. Blue-red gradient shows low to high expression levels for each gene. Dendrogram represents a hierarchical clustering based on Pearson correlation values. c Venn diagram that shows the overlap between DEGs identified with EdgeR (nominal p-values < 0.05) and NOISeq (prob ≥0.8) tools
The comparison of DEGs identified with EdgeR and NOISeq software packages highlighted 30 genes that showed a coherent differential expression in treated vs control groups using the two different approaches (Fig. 2c), 3 of which with a prob. ≥0.9 (Additional file 3: Table S3).
In Tables 3 and 4 are reported the top 10 up-regulated and down-regulated genes in terms of the magnitude of differential expression, log2 (fold change), identified in rumen of treated versus control animals using NOISeq software package.
Among the top up-regulated genes, we identified LOC102397197 (or TUBA1D), IGFBP6, LOC102411389 (or VNN1), DUOXA1, TPPP3, bone morphogenetic protein 6 (BMP6), poly(A)-specific ribonuclease PNLDC1 (PNLDC1), and laminin subunit alpha-1 (LAMA1) genes, which are also included in the list of up-regulated genes identified using EdgeR software (see above and Additional file 3: Table S3). In addition, among the top up-regulated genes identified by NOISeq analysis, we found also LOC112583793 (or WAP four-disulfide core domain protein 18-like, WFDC18) gene, encoding a proteinase inhibitor, and transcobalamin-1 (TCN1) gene, encoding a vitamin B12-binding protein.
Included in the top down-regulated genes, we found ROR2, SMOC2, LOC102400339 (or collagen alpha-1(I) chain-like, COL1A1) genes, and the teneurin-3 (TENM3) gene, encoding a transmembrane protein, all down-regulated also with EdgeR analysis (see above and Additional file 3: Table S3). Moreover, the list of the top down-regulated genes identified with NOISeq analysis includes also acyl-CoA synthetase short-chain family member 3 (ACSS3) gene, whose product has a role in energy and lipid metabolism, fraser extracellular matrix complex subunit 1 (FRAS1) gene, integrin alpha-8 (ITGA8) gene, Wilms tumor protein 1-interacting protein (WTIP) gene, encoding a factor involved in the response to hypoxia, and LOC112580184 (or glutathione S-transferase A1, GSTA1) gene, whose product has a role in the response to the oxidative stress.
The lists of all DEGs in treated group in comparison with the control one, identified using EdgeR and NOISeq software packages, and the DEGs identified with both approaches (common DEGs) are reported in Additional file 3: Table S3.
To validate the differential expression observed by RNA-seq analysis, the expression of randomly selected DEGs identified with EdgeR and NOISeq software packages was analyzed in rumen of treated and control buffaloes by reverse-transcription qPCR (rt-qPCR), using the same samples analyzed by RNA-seq. We analyzed the expression of 12 DEGs identified with EdgeR tool and 8 DEGs identified with NOISeq tool, 4 of which resulted commonly deregulated in RNA-seq experiment analyzed with both approaches.
For all 12 genes identified with EdgeR tool (nominal p-values < 0.05), the rt-qPCR assays showed an expression pattern concordant with RNA-seq results (Fig. 3). On the other hand, among randomly selected genes found to be deregulated with NOISeq analysis, differential expression of genes identified with both methods [common DEGs: selenium binding protein 1 (SELENBP1), DUOXA1, and IGFBP6 with 0.8 < prob. < 0.9; TPPP3 with 0.9 < prob. < 0.95] has been validated by rt-qPCR assay, whereas DEGs identified only with NOISeq analysis [LOC112583793 (or WFDC18) with prob. > 0.95; WTIP with 0.9 < prob. < 0.95; insulin-like growth factor-binding protein 2 (IGFBP2) and testis-specific serine kinase substrate (TSKS) with 0.8 < prob. < 0.9] showed an expression pattern conflicting with RNA-seq data. These findings suggest a higher reliability of EdgeR data analysis with respect to the NOISeq approach in our experimental conditions, and prompted us to perform subsequent analyses using DEGs selected with EdgeR approach.

Validation of the differential expression observed by RNA-seq. The expression of randomly selected DEGs [12 DEGs identified with EdgeR tool and 8 DEGs identified with NOISeq tool, 4 of which are deregulated with both approaches (common DEGs)] was analyzed by rt-qPCR in ruminal wall of dairy buffaloes fed a TMR + green feed (Treated) in comparison with animals fed the TMR diet (Control). Data were normalized to β-actin and are reported as mean fold increases ± standard error of the mean (SEM) relative to transcript levels of control group samples. Six control and six treated animals were used, and two independent qPCR experiments were performed. * p < 0.05; * p < 001; *** p < 0.001 (one-tailed Student’s t-test). §: rt-qPCR data conflicting with RNA-seq results
Gene ontology analysis of differentially expressed genes
Gene Ontology Enrichment Analysis (GOEA) was undertaken on the 155 DEGs between treated versus controls buffaloes, identified using EdgeR tool. Overall, DEGs were functionally associated with 431 significantly enriched gene ontology (GO) terms (FDR-adjusted p-value < 0.05), 195 of which were enriched in up-regulated genes and 236 were enriched in down-regulated genes (Additional file 4: Table S4).
The 35% (68) of GO terms enriched in up-regulated genes were classified as molecular function, 17% (33) as cellular component, and 48% (94) as biological process. The 38% (89) of GO terms enriched in down-regulated genes were categorized as molecular function, 22% (51) as cellular component, and 40% (96) as biological process. Selected GO terms enriched in up- and down-regulated genes are displayed in Fig. 4a and b. Among GO terms enriched in up-regulated genes, the category of molecular functions included those related to nitric oxide production, JAK-STAT pathway, NAD(P)H oxidase activity, peroxidase and oxidoreductase activity, calcium channel activity and heat shock proteins. In the category of cellular components we found GO terms related to extracellular space and Z Disc - a component of the striated muscle with a key role for the muscle contraction -, while the category of biological processes included GO terms linked to immune system (cellular response to interleukin-4, immune response, T cell homeostasis, positive regulation of interferon-gamma production, defense response to bacterium and cytokine-mediated signaling pathway and negative regulation of inflammatory response), to lipid metabolism (triglyceride metabolic process and fatty acid metabolic process) and to the response to oxidative stress. Down-regulated genes were enriched in molecular function GO terms related to methanethiol oxidase activity, to selenium homeostasis (selenium binding), to oxidoreductase activity, to S-adenosyl methionine (SAM)-dependent methyltransferase activity, and to structural constituent of muscle. In the category of cellular components, we found an enrichment in down-regulated genes in GO terms related to the external side of plasma membrane, Z disc and calcium channel complex. The category of biological processes included GO terms associated with positive regulation of fat cell differentiation, with BMP signaling pathway, with cytokine-mediated signaling pathway, with positive regulation of apoptotic signaling pathway, and with negative regulation of canonical Wnt signaling pathway.

Gene ontology enrichment analysis of differentially expressed genes. Selected GO-terms, enriched in genes up-regulated (a) and down-regulated (b) in rumen of buffaloes fed a TMR + green feed (Treated) in comparison with those fed the TMR diet (Control), are shown. GO terms were classified as molecular function (purple bars), cellular component (green bars) and biological process (blue bars). Bar graphs indicate the statistical significance of the enrichment, as -log10 (adj p-value). Vertical yellow bars indicate the cut-off level for significance (p < 0.05, adjusted by Benjamini-Hochberg correction)
In summary, the Gene Ontology analysis indicated that green feed modulated biological processes related to health and well-being in buffaloes, such as the immune response, lipid metabolism, the response to the oxidative stress and muscle structure and function.
Gene-by-gene analysis of differential expression
Further analysis of individual genes found to be differentially expressed in rumen of treated dairy buffaloes in comparison with control animals, using EdgeR with a nominal p-values < 0.05, allowed us to highlight transcriptional changes in genes involved in biological processes relevant for rumen functionality and physiology, and thus, for animal welfare and quality production.
Genes linked to energy, lipid and amino acid metabolism
Our analysis highlighted differential expression of genes encoding factors involved in energy, lipid, and amino acid metabolism (especially cysteine and methionine), in treated versus control buffaloes (Fig. 5a). We found enhanced expression of LOC102410803 (or Phosphoserine Aminotransferase 1, PSAT1), and Hydroxylysine Kinase (HYKK), and decreased levels of cysteine dioxygenase Type 1 (CDO1), SELENBP1 and phosphatidylethanolamine N-methyltransferase (PEMT) genes, encoding enzymes with a function in amino acid metabolism. Furthermore, we found up-regulation of HSD17B13, and ARFGEF family member 3 (ARFGEF3) genes, and down-regulation of LRP2, encoding factors implicated in energy and lipid metabolism.

Transcriptomic data of differential expression of genes involved in selected biological processes. Boxplots illustrate the Trimmed Mean of M values (TMM) expression values of selected genes deregulated in rumen of dairy buffaloes fed a TMR + green feed (Treated) versus animals fed the TMR diet (Control) and related to energy, lipid or amino acid metabolism (a), immune system and inflammation (b), oxidative stress and cellular response to stress (c), extracellular matrix organization (d) and muscle structure and function (e). Whiskers show the 5–95% percentiles of the confidence intervals, the horizontal lines within each box show the medians, and outliers are shown as dots. * p-value < 0.05; ** p-value < 0.01; *** p-value < 0.001
In conclusion, these findings allow to speculate that green feed has an impact on amino acid and lipid metabolism that, in turn, influence energy production.
Genes linked to immune system and inflammation
Green feed seems to modulate the expression of several genes encoding factors with a role in immune system or inflammation (Fig. 5b). Among them, we found enhanced expression of dual oxidase 1 (DUOX1), DUOXA1, IGFBP6, MTOR associated protein LST8 homolog (MLST8), transcription factor 7 (TCF7), LOC102408976 (or IFI27), LOC102407663 (or IFI27L2), tripartite motif containing 14 (TRIM14), and suppressor of cytokine signaling 2 (SOCS2) genes, which are correlated with the innate or adaptative immune response. Moreover, the expression of the receptor of immunoglobulin E FCER2, LOC102411389 (VNN1), ISL LIM homeobox 1 (ISL1), and Secreted LY6/PLAUR Domain Containing 1 (SLURP1) genes, which are linked to the regulation of inflammatory response, was increased. On the other side, we found reduced levels of the immune system-related lymphoid enhancer binding factor 1 (LEF1) and complement C1r (C1R) genes, and inflammation-related heat shock protein 90 beta family member 1 (HSP90B1), S100 calcium binding protein A11 (S100A11), LOC112585166 (or serum amyloid A protein-like, SAAL1), interleukin 6 cytokine family signal transducer (IL6ST), leukemia inhibitory factor receptor (LIFR), ROR2, and SMOC2 genes.
In summary, the large number of genes with a role in immune response and inflammation, whose expression is re-modulated upon green feed administration, allows to hypothesize a beneficial effect of green diet on the animal health.
Genes linked to oxidative stress and cellular response to stress
Upon green feed administration, changes in the expression levels of a number of factors linked to oxidative stress and cellular response to stress have been observed (Fig. 5c). Among them, we found up-regulation of nudix hydrolase 18 (NUDT18), DnaJ heat shock protein family (Hsp40) member A4 (DNAJA4) and heat shock transcription factor 4 (HSF4) genes, encoding factors that counteract oxidative and heat stress conditions, and down-regulation of LOC102396388 (or UDP glucuronosyltransferase family 1 member A9, UGT1A9), LOC102413340 (or ATP binding cassette subfamily C member 4, MRP4/ABCC4) and LOC102399021 (CYP1A1) genes, involved in detoxifying cellular response.
Our findings prompt to hypothesize that green feed modulates the response of animals to different kind of stressful stimuli.
Genes linked to extracellular matrix organization (ECM)
Green feed seems to induce a significant deregulation of genes encoding factors with a function in the organization of extracellular matrix (ECM) (Fig. 5d), a complex and dynamic non-cellular scaffold that acts as a structural support for cells, and known to regulate several functions, including shape, adhesion, and migration. We found up-regulation of collagen type IV alpha 6 chain (COL4A6) and laminin subunit alpha-1 (LAMA1) genes, and down-regulation of LOC102400339 (or COL1A1), encoding structural constituents of ECM. Moreover, in treated buffaloes, the ECM glycoprotein tenascin XB (TNXB), whose product plays a role in the expression and deposition of collagen into the ECM, was significantly up-regulated, whereas the gene encoding the integrin subunit alpha 2 (ITGA2), which connects the ECM with the cytoskeleton, and LOC102390617 (or UDP-glucose 6-dehydrogenase-like, UGDH) gene, encoding an oxidoreductase involved in the production of extracellular matrix components, were significantly down-regulated.
Our results allow to hypothesize that green feed influences the organization of extracellular matrix in the rumen of buffaloes.
Genes linked to muscle structure and function
Green feed induced differential expression of several genes encoding proteins involved in muscle structure and function (Fig. 5e). We found enhanced expression of Potassium two pore domain channel subfamily K member (KCNK10), calcium voltage-gated channel auxiliary subunit gamma 4 (CACNG4), and ATPase plasma membrane Ca2+transporting 4 (ATP2B4), which encode factors that regulate homeostasis of Ca2+. Moreover, we detected up-regulation of the cytoskeleton-related tubulin polymerization promoting protein (TPPP), LOC102397197 (or TUBA1D), LOC102395439 (or FHL1), and TPPP3 genes, and down-regulation of the desmin (DES) and myosin heavy chain 11 (MYH11) genes.
Overall, changed expression of muscle-related genes suggests an impact of green feed on the muscularity of ruminal wall.
Discussion
The present study aimed to investigate the impact of green forage on the ruminal transcriptional program through RNA-seq, which represents an excellent tool for large scale gene expression studies. Although many different bioinformatic procedures are currently available to analyze RNA-seq data, there is no consensus about which are the most reliable, and the choice of the most appropriate tool is still arduous [22]. For this reason, we approached the identification of DEGs by using two different algorithms, EdgeR and NOISeq, both considered to be robust methods [22, 23].
The EdgeR approach with a nominal p-values < 0.05 allowed the identification of 155 DEGs in rumen of treated versus control buffaloes. Conversely, the use of FDR-adjusted p-values as significance threshold did not reveal significant DEGs between treated and control groups. We hypothesized that the lack of FDR significant genes might be due to the moderate impact of green feed on gene expression, which might, in turn, be masked by the inter-individual variability of the buffaloes belonging to the same group. It is known that different feed regimens are known to induce large changes in gene expression [1]. However, the aim of our study was to understand if the solely inclusion of green forage would be responsible for differences in rumen gene expression profile, and hence we use two diets isonitrogenous and isoenergetic; therefore, we expect subtle differences between treated and control groups. In support of our hypothesis, we observed that the 155 DEGs identified with a p-value < 0.05 had a median absolute log2 fold change of 0.82. In addition, Barton and coworkers [24] showed that most differential expression analysis tools, including EdgeR, have lower performance at lower fold changes, as indicated by their True Positive Rate (TPR). We may hypothesize that inclusion of 30% of ryegrass green feed in the treated group was not great enough to have considerable impacts on the gene expression. Additional studies performed feeding the animals a diet with a higher proportion of green ryegrass, as well as the use of a greater number of animals, might improve the statistical power, thus emphasizing gene expression changes. However, it has been already observed that an inclusion higher than 30% of high-quality green feed in the diet of dairy cows fed TMR have reduced both the dry matter intake and milk yield [25], and hence would not be practically suitable for breeders.
The lack of FDR significant genes is suggestive of a limitation of our study, which appears statistically underpowered. However, rt-qPCR assay on 12 randomly selected DEGs identified using nominal p-values < 0.05, confirmed their differential gene expression observed by RNA-seq experiment and suggests a genuine impact of green feed on gene expression. On the other hand, we cannot rule out the occurrence of false positive and negative DEGs, due to the validation of only a subset of them by rt-qPCR. It is conceivable that validation of additional DEGs identified in our study should further confirm the impact of green diet on the ruminal transcriptional program.
A parallel analysis of differential expression performed using NOISeq software with a prob. ≥0.8 as significance threshold, highlighted 61 DEGs, which dropped to 11 or 1 DEGs, with prob. ≥0.9 or prob. ≥0.95, respectively. Among DEGs identified with NOISeq, only 30 genes showed a differential expression coherent with EdgeR results, 3 of which have prob. ≥0.9, and none had prob. ≥0.95. It is interesting to note that rt-qPCR assay validated the differential expression of randomly selected DEGs identified with both methods (common DEGs), while differential expression of genes highlighted exclusively with NOISeq tool, including the gene with prob. ≥0.95, was not confirmed by this assay.
In summary, in our biological context, results obtained using EdgeR software, although statistically unpowered, seems to be more reliable as compared with those obtained with NOISeq tool, and this agrees with the proposed higher performance of EdgeR in certain experimental conditions [23]. Based on these premises, we considered, for the subsequent analyses, DEGs identified with EdgeR tool, emphasizing those revealed with both approaches.
RNA-seq data, obtained using EdgeR tool, indicated that green forage impacted on the expression of several genes encoding factors implicated in the regulation of biological processes relevant for buffalo welfare, such as those related to cellular response to stress, immune system and inflammation, as well as genes encoding factors involved in rumen functionality, such as muscle structure/function, and energy, lipid, and amino acid metabolism. This evidence suggested an enhancement of rumen activity by green feed and allows to speculate a role of this diet in the production of nutraceutical molecules, whose levels might be enhanced also in milk. It was previously shown that 30% green forage increases the concentration of beneficial biomolecules, including betaine and carnitine, in milk and other dairy products in buffaloes [17, 26]. In the present study, green feed modulated the expression of genes linked to amino acid metabolism, including those related to cysteine/methionine, serine and lysine metabolism. These compounds mitigate oxidative damage in tissues by acting as precursors of anti-oxidant molecules, such as S-adenosylmethionine, hydrogen sulfide (H2S), taurine, and glutathione [27]. This aspect is relevant, since betaines, which have a high nutraceutical action, are the methyl donor in the cysteine/methionine pathway that converts methionine to cysteine. In particular, green feed was associated with decreased expression of CDO1 and SELENBP1. Of note, decreased levels of SELENBP1 has been detected also with NOISeq analysis, and both genes are included in the subset of DEGs randomly validated by rt-qPCR.
CDO1 encodes the cysteine dioxygenase enzyme that has a key role in the catabolism of cysteine and taurine biosynthesis, thus regulating cysteine intracellular levels and availability [28, 29]. SELENBP1 encodes an enzyme important for oxidation of methanethiol, a factor linked to the methionine catabolism [30]. Decreased CDO1 expression in the rat liver was associated with high levels of many amino acids and their metabolites, including betaine [28]. As noted above, green feed increases levels of betaines in buffalo milk [17]. Based on the above findings, we speculate that green feed impacts betaine levels through the regulation of genes implicated in the cysteine/methionine pathway, such as CDO1 and SELENBP1. Moreover, PEMT, whose expression is modulated by green feed, transcribes for the phosphatidylethanolamine N-methyltransferase, a rate-limiting enzyme of the biosynthetic pathway of choline, which is the precursor of betaine (trimethyl-glycine). Choline, in turn, participates in homocysteine production [31]. CDO1 over-expression is known to decrease cellular glutathione (GSH) levels by approximately 40% [32], thus CDO1 down-regulation observed upon green feeding could be responsible for enhanced antioxidant activity. Since SELENBP1 is considered an “H2S-producing enzyme”, its reduced levels might be responsible for lower production of H2S which is involved in the mitochondrial transport of carnitine/acyl-carnitine [33, 34]. Green feed induced also up-regulation of LOC102410803 (or PSAT1) and HYKK. PSAT1 encodes a phosphoserine aminotransferase involved in the biosynthesis of serine, a non-essential amino acid implicated in many biological processes and crucial for the formation of glycine, cysteine, and taurine amino acids [35]. HYKK encodes hydroxylysine kinase that is involved in the catabolism of lysine [36]. This is related to the metabolism of carnitine since trimethyl-lysine is the main precursor of carnitine [37, 38]. We might speculate that green feed influences carnitine levels [17] by interfering with lysine metabolism.
Among genes implicated in the lipid metabolism there was up-regulation of HSD17B13, which encodes the 17-beta-hydroxysteroid dehydrogenase 13 enzyme that is thought to be involved in fatty acid metabolism [39], although this remains controversial. The over-expression of HSD17B13 was associated with increased lipogenesis and triglyceride levels in the liver of mice [39]. Under-expression of HSD17B13 in mice leads to increased levels of C16 and C18:1 acyl-carnitine, which suggest a key role of HSD17B13 in β-oxidation of fatty acids through the modulation of their mitochondrial transport [40]. It is possible that the increased expression of HSD17B13 in buffaloes that received green forage stimulated the β-oxidation of fatty acids, influencing the transport of acyl-carnitine into the mitochondria. Moreover, it is interesting to note that HSD17B13 has also an anti-inflammatory role, since its deficiency triggers hepatic steatosis and inflammation in mice [40].
Green feed seems to impact also on energy metabolism. We found, indeed, with both EdgeR and NOISeq methods of analysis, up-regulation of ARFGEF3, encoding the Brefeldin A-inhibited guanine nucleotide-exchange protein 3 (BIG3), a factor that participates to the regulation of systemic glucose homeostasis, through the regulation of insulin and glucagon secretion [41, 42].
Metabolism is well known to fuel cell activities. In ruminants, for instance, amino acid metabolism is of utmost importance, since it generates several effects on animal immunity and production [43]. Our results suggest that green feed exerts a positive impact on immune system, through the modulation of the expression of genes encoding factors implicated in the immune response and inflammation. There was up-regulation of LOC102408976 (or IFI27), LOC102407663 (or IFI27L2), TRIM14, DUOXA1, DUOX1, IGFBP6, and SLURP1 genes. IFI27 and IFI27L2 genes encode two factors belonging to the family of interferon-induced proteins, which are involved in both innate immune response [44] and energy metabolism [45]. TRIM14 encodes a protein belonging to TRIM family, which are also known to promote host defense against virus infections [46]. DUOXA1 is implicated in the maturation/activation of DUOX1 that is, in turn, a NADPH oxidase known to catalyze ROS production and implicated in innate immune defense and antimicrobial function [47]. IGFBP6 is involved in the immune response, with both pro- and anti-inflammatory roles [48], and its expression has been reported to be enhanced following an increase in the fiber/starch ratio in cattle diet [49]. SLURP1 is a nicotinergic peptide known to exert an anti-inflammatory effect in the human intestinal epithelial cells [48]. Notably, up-regulation of DUOXA1, DUOX1, IGFBP6, and SLURP1 has been highlighted also with NOISeq analysis. Furthermore, we found up-regulation of SIM2 that encodes a factor involved in the anti-microbial response in the gastrointestinal tract by regulating the expression of anti-microbial peptides and defensins [50]. Overall, these data seem to suggest a beneficial role of green feed, by stimulating the immune system and protecting against pathogens.
It is known that the rumen functionality depends on the microbiota composition. The cross-talk between rumen microbiota and innate immune cells in the ruminal epithelium may contribute to the balance of immune tolerance and inflammatory response in the rumen, which is partly dependent on metabolites and, thus, diet [51, 52]. High amount of vaccenic acid, the dietary CLA precursor, were observed in milk of buffaloes receiving green feed [17]. This is of particular interest considering that CLA exerts an anti-inflammatory function in sheep ruminal epithelial cells through the inhibition of pro-inflammatory cytokines production and the regulation of cell proliferation and lipid metabolism, which are potentially related to injury repair and immune response [53]. Accordingly, we found that green feed modulated the expression of many genes with a role in the inflammation. Among the down-regulated genes, we found LOC112585166 (or SAAL1), ROR2, SMOC2, and S100A11, which are associated with pro-inflammatory activity [54,55,56,57]. Of note, down-regulation of serum amyloid A1 (SAA1) was observed in rumen papillae of dairy cow fed green forage [58], which underlines the susceptibility of its expression to the diet. On the other hand, ISL1 gene, whose expression was increased upon green feed administration, is known to negatively regulates the inflammatory response [59]. Additionally, we found up-regulation of LOC102411389 (or VNN1), encoding the enzyme pantethinase, which is involved in the production of pantothenic acid (vitamin B5) and coenzyme A (CoA). VNN1 has been linked to inflammation, stress response, lipid metabolism and energy production [60, 61]. Of note, VNN1 gene has been previously reported to be differentially expressed in ruminal epithelium of heifers fed low or high grain diet [62]. Among the above discussed inflammation-related genes, ROR2, SMOC2, and VNN1 were differentially expressed also with NOISeq analysis. Altogether, these data allow to hypothesize an anti-inflammatory role of green forage, through the modulation of genes encoding pro- and anti-inflammatory factors.
Cells are continuously exposed to several stressful conditions that could negatively impact on cell metabolism, promote cell and tissue damages and contribute to the onset of several pathologies. Among the main sources of stress there are the oxidative stress, a detrimental condition arising from imbalanced levels of reactive oxygen species (ROS) and antioxidants, and stress conditions caused by excessive heat, cold or UV light. Green feed induced changes in the expression of genes linked to oxidative stress and cellular response to stress. Particularly notable was up-regulation of genes whose products counteract stressful conditions, such as NUDT18, DNAJA4 and HSF4 genes. NUDT18 catalyzes the hydrolysis of 8-oxo-dGTP - one of the best characterized DNA damages caused by oxidative stress - to produce the monophosphate form 8-oxo-dGMP (odGMP), which is unusable for DNA synthesis, thus preserving cells from oxidation-mediated mutations [63]; DNAJA4 belongs to the family of heat shock proteins HSP40, which cooperate with HSP70 chaperones to counteract protein misfolding and aggregation in stress conditions [64]; HSF4 gene encodes a factor that modulates the expression of heat shock proteins under stressful conditions [65] and exerts a protective effect against oxidative stress [66]. These findings suggest that green feed may exert an anti-oxidant function, by positively modulating factors that counteract stressful stimuli. In addition, we found down-regulation of genes encoding factors involved in detoxifying cellular response, such as LOC102396388 (or UGT1A9), coherently deregulated with both EdgeR and NOISeq approaches, LOC102413340 (or MRP4/ABCC4), and LOC102399021 (or CYP1A1), which might arise from the healthy nature of the green feed. Noteworthy, the expression of HSF4, LOC102396388, LOC102413340, and LOC102399021 has been reported to be modulated by different diet composition [62, 67,68,69].
Extracellular matrix has a crucial role in proliferation, differentiation, adhesion and inflammation. Many reports underlined a diet-related re-modulation of extracellular matrix [70,71,72], according with our data, which suggest an impact of green forage on the expression of ECM-related genes. Among them, we found up-regulation of LAMA1 and COL4A6 and down-regulation of LOC102400339 (or COL1A1) and LOC102390617 (or UGDH). Of note, LAMA1, LOC102400339, and LOC102390617 showed similar deregulation with NOISeq analysis. COL4A6, COL1A1 and LAMA1 are three of the major structural constituents of ECM [73, 74], whereas UGDH is an enzyme involved in the production of glycosaminoglycans and proteoglycans of ECM [75]. These findings allow to speculate that green forage influence the ECM modelling and thus, the establishment of the structure and the function of the cells.
The ruminal wall has an important role in nutrient uptake and represents a barrier against pathogens and mechanical injuries [76]. Buffaloes that received green feed showed changes in the expression of many genes encoding factors associated with muscle structure and function. Of particular interest is the increased expression of KCNK10, CACNG4, and ATP2B4 genes, encoding factors that modulate the homeostasis of Ca2+ [77,78,79,80], a second messenger important for muscle contraction and differentiation, which allows to suppose that green feed might stimulate the ruminal muscular function. Furthermore, green feed modulated, with a different direction, genes encoding structural components of the cytoskeleton, such as MYH11, DES, and LOC102397197 (or TUBA1D), and factors promoting its assembling, such as TPPP, TPPP3, thus suggesting an impact of this diet on muscular structure. LOC102397197 and TPPP3 genes resulted differentially expressed also using NOISeq tool. Interestingly, DES expression was previously shown to be modulated by diet [81].
Conclusions
Although the low statistical power of our study suggests a limitation of this work and point out the need of additional investigations, we provide, for the first time, a broad overview of the different biological processes that seems to be influenced by a green feed diet in buffalo rumen epithelium. Taking advantage of a transcriptomic approach, we deepened our understanding on the response of ruminal metabolism, activity, and physiology to different diet regimens. This work adds a piece of knowledge on the potential effects of the diet on molecular processes linked to the animal welfare and production of nutraceutical molecules. This study provides clues for future research on ruminants, and novel aspects that should be considered in addition to rumen fermentation, such as immune activity and oxidative response, for the setup of feeding strategy focused to enhance animal welfare and production. Our findings will pave the way for the optimization of novel strategies based on the precision feeding system, respectful of the ethologic and physiologic needing of the animals. However, additional studies are needed to further confirm the impact of green forage on ruminal transcriptional program, possibly through the validation of additional DEGs here identified and the increase of sample size. Moreover, further omics investigations, including methylomic studies, might allow to decipher the molecular impact of green feed at multiple layers.
Materials and methods
Animals, dietary treatment, and ruminal tissue collection
All experimental procedures were performed according to the European Directive 2010/63/EU and the Italian Legislative Decree No. 26 dated 4th March 2014 and received institutional approval from the Ethical Animal Care and Use Committee of the University of Naples “Federico II” (Protocol No. 25532–2022). The study was carried out over 60 days at a commercial buffalo dairy in southern Italy, by using Italian Mediterranean dairy buffaloes (n 12; 8.2 ± 0.6 years old), for which the slaughter was already scheduled. The animals were maintained in pens with a concrete floor and were milked twice daily in the morning and afternoon and had undergone a 14-day adaptation period before beginning the trial. Buffaloes were randomly assigned to two equal groups (Control and Treated) according to parity (5.2 ± 0.4 vs 5.3 ± 0.7 in treated vs control group, respectively; P > 0.05, two-tailed Student’s t-test), and average milk production (6.0 ± 0.7 vs 6.8 ± 0.7 kg/day in treated vs control group, respectively; P > 0.05, two-tailed Student’s t-test), whose values were recorded in the 10 days before commencement of the study. The diet of control buffaloes was a total mixed ration (TMR) whilst treated buffaloes received TMR + green feed which comprised ryegrass (approximately 30% of the diet, Table 5).
The forage was just ryegrass at the re-blossoming stage cut twice a day to avoid any fermentation and immediately put in the mixing wagon, with no storage and administered to animals. The forage to concentrates ratio of control buffaloes was 56:44 and that of treated buffaloes was 69:31. The two diets were isonitrogenous and isoenergetic and differed only in the inclusion of green feed in treated buffaloes (Table 5). Animals were fed twice daily: in the morning and in the evening. Refusals were recorded and then removed. Animals were euthanized all together by penetrating captive bolt, according to the AVMA Guidelines for the Humane Slaughter of Animals, following procedures approved by the Ethical Animal Care and Use Committee of the University of Naples “Federico II”. Ruminal wall biopsies were collected, then, they were immediately washed with cold PBS, and stored at − 80 °C before RNA extraction.
Isolation of total RNA from ruminal wall
Total RNA was isolated from 100 to 200 mg of frozen ruminal wall tissue of 12 dairy buffaloes, of which 6 were fed a TMR (control group) and 6 received TMR + green feed (treated group). RNA was extracted using the EuroGold TriFast (EuroClone, Pero, Milano, Italy) reagent, according to the manufacturer’s instructions. Tissue samples were homogenized in 1 ml of TriFast reagent by using TissueLyser (Qiagen, Hilden, Germany), then, RNA samples were treated with Turbo DNA-freeTM kit (Ambion, Austin, TX, USA) to remove genomic DNA contaminant, according to the manufacturer’s instructions. The quality and the quantity of RNA samples were determined using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Samples with an RNA integrity number (RIN) greater than 7.0 were used for library construction. RNA samples were stored at − 80 °C until further processing.
RNA-seq library preparation and sequencing
RNA-seq library preparation and sequencing were carried out at Macrogen (Seoul, Korea). Briefly, the RNA-seq libraries were prepared using TruSeq Stranded Total RNA Sample Prep kit (Illumina, San Diego, CA) according to the manufacturer’s recommendations, using 1 μg of total RNA as input. Starting RNA samples and final RNA libraries were quantified by using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and quality tested by Agilent 2100 Bioanalyzer RNA Nano assay (Agilent technologies, Santa Clara, CA). Libraries were then processed with Illumina cBot for cluster generation on the flow cell, following the manufacturer’s instructions and sequenced (paired end sequencing, 150 bp, 30 M reads per sample) at the multiplexing level requested on NovaSeq6000 (Illumina, San Diego, CA). The CASAVA 1.8.2 version of the Illumina pipeline was used to process raw data for both format conversion and de-multiplexing.
RNA-seq reads processing and mapping
Bioinformatic analysis were carried out by Sequentia Biotech (Barcelona, Spain). The quality of raw RNA-seq reads was assessed with the software FastQC v0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adapters and low-quality bases were then removed using Trimmomatic v0.39 setting a minimum base quality of 25, minimum length of 35 bp and leaving the other options as default [82]. Trimmed reads were then aligned to the buffalo genome (Bubalus bubalis, version UOA_WB_1 from NCBI) with STAR aligner v2.5.2b [83] using default parameters. Read summarization to produce gene counts matrix was performed with featureCounts v2.0.0 [84] with the following parameters: -Q 30 -s 2 -p.
Identification of differentially expressed genes
Differential expression analysis was carried out using two approaches. With the first one, lowly expressed genes and those showing high variability were filtered out using the HTSFilter R package [85] using the TMM normalization [86], which selected a threshold of 41.64 TMM. Sample clustering was assessed performing a Principal Component Analysis (PCA) on the TMM normalized values after the HTSFilter step. Differentially expressed genes were detected using EdgeR [87] by nominal p-value < 0.05. With the second approach, lowly expressed genes were filtered out using NOISeq [88] setting a threshold of 1 CPM, then the ARSyN correction was applied on the log2 TMM normalized values using the same package, in order to reduce experimental noise. Finally, the NOISeq function was used to perform the statistical function assigning a probability of differential expression to each gene. Genes showing a probability value higher than or equal to 0.8, 0.9 and 0.95 were considered for further analyses.
Functional analysis
The differentially expressed genes were split into up- and down-regulated genes and then a Gene Ontology Enrichment Analysis was performed on each list using an in-house developed script based on the methods used by the AgriGOv2 algorithm [89]. Significantly Enriched Gene Ontology terms were identified applying an FDR (p-value adjusted by Benjamini-Hochberg correction) filter of 0.05 [90].
Reverse-transcription quantitative PCR (RT-qPCR) validation of selected DEGs
To validate RNA-seq results, DEGs were randomly selected and analyzed by reverse-transcription qPCR (rt-qPCR). cDNA was obtained by reverse transcription of 500 ng of total RNA using the SuperScript II First-Strand Synthesis system (Life Technologies, Carlsbad, CA, USA), following the manufacturer’s instructions. The obtained cDNA was, then, amplified by qPCR with SsoAdvance Universal SYBR Green supermix (Bio-Rad, Hercules, CA, USA) on a CFX Opus Real-Time PCR system (Bio-Rad), according to the manufacturer’s protocols. The 2-ΔΔCq method was used to determine the relative quantitative levels, and the data were normalized with respect to the expression of β-actin, which showed a similar distribution of FPKM values with no statistically significant difference between the two experimental groups (Wilcoxon Test, p = 0.96; data not shown). List of primer sequences used in this study is reported in the Additional file 5: Table S5.
Availability of data and materials
The RNA-sequencing dataset supporting the conclusions of this article is available in the GEO repository (accession number GSE207815).
Abbreviations
- ABCC4 :
-
Multidrug resistance-associated protein 4-like
- ACSS3 :
-
Acyl-CoA synthetase short-chain family member 3
- ARFGEF3 :
-
Brefeldin A-inhibited guanine nucleotide-exchange protein 3 Family Member 3
- ATP2B4 :
-
ATPase plasma membrane Ca2+ transporting 4
- BIG3:
-
Brefeldin A-inhibited guanine nucleotide-exchange protein 3
- BMP6 :
-
Bone morphogenetic protein 6
- C1R :
-
Complement component 1 subcomponent R
- CACNG4 :
-
Calcium voltage-gated channel auxiliary subunit gamma 4
- CDO1 :
-
Cysteine Dioxygenase Type 1
- CLA:
-
Conjugated linoleic acid
- COL1A1 :
-
Collagen alpha-1(I) chain
- COL4A6 :
-
Collagen type IV alpha 6 chain
- Cox7C :
-
Cytochrome C oxidase subunit 7C
- CYP1A1 :
-
Cytochrome P450 1A1
- DEGs:
-
Differentially expressed genes
- DES :
-
Desmin
- DNAJA4 :
-
DnaJ heat shock protein family member A4
- DUOX1 :
-
Dual oxidase 1
- DUOXA1 :
-
Dual oxidase maturation factor 1 isoform X2
- ECM:
-
Extracellular matrix
- FCER2 :
-
Low affinity immunoglobulin epsilon Fc receptor
- FHL1 :
-
Four and a half LIM domains 1
- FRAS1 :
-
Fraser extracellular matrix complex subunit 1
- GO:
-
Gene ontology
- GOEA:
-
Gene ontology enrichment analysis
- H2S:
-
Hydrogen sulfide
- GSTA1 :
-
Glutathione S-transferase A1
- HSD17B13 :
-
17-beta-hydroxysteroid dehydrogenase 13
- HSF4 :
-
Heat Shock Transcription Factor 4
- Hsp40:
-
Heat Shock Protein 40
- HSP90B1 :
-
Heat Shock Protein 90 Beta Family Member 1
- HYKK :
-
Hydroxylysine Kinase
- IFI27 :
-
Interferon Alpha Inducible Protein 27
- IFI27L2:
-
Interferon alpha-inducible protein 27-like protein 2
- IFI44L :
-
Interferon-induced protein 44-like
- IGFBP6 :
-
Insulin-like growth factor-binding protein 6
- IL6ST :
-
Interleukin 6 cytokine family signal transducer
- ISL1 :
-
ISL LIM homeobox 1
- ITGA2 :
-
Integrin subunit alpha 2
- ITGA8 :
-
Integrin alpha-8
- KCNK10 :
-
Potassium two pore domain channel subfamily K member
- LAMA1 :
-
Laminin Subunit Alpha-1
- LEF1 :
-
Lymphoid Enhancer Binding Factor 1
- LIFR :
-
Leukemia inhibitory factor receptor
- LRP2 :
-
Low-density lipoprotein receptor-related protein 2
- MLST8 :
-
MTOR associated protein, LST8 homolog
- MRP4/ABCC4 :
-
ATP Binding Cassette Subfamily C Member 4
- MUC15 :
-
Mucin-15
- MYH11 :
-
Myosin heavy chain 11
- NDEL1 :
-
Nuclear distribution protein nudE homolog 1-like
- NUDT18 :
-
Nudix hydrolase 18
- odGMP:
-
8-oxo-dGMP
- OR51E1 :
-
Olfactory receptor 51E1
- PEMT :
-
Phosphatidylethanolamine N-Methyltransferase
- PNLDC1 :
-
Poly(A)-specific ribonuclease PNLDC1
- PSAT1 :
-
Phosphoserine aminotransferase 1
- RNA-seq:
-
RNA sequencing
- ROR2 :
-
Tyrosine-protein kinase transmembrane receptor
- ROS:
-
Reactive oxygen species
- rt-qPCR:
-
Reverse-transcription quantitative PCR
- S100A11 :
-
S100 calcium binding protein A11
- SAA1 :
-
Serum Amyloid A1
- SAAL1 :
-
Serum amyloid A protein-like
- SAM:
-
S-adenosyl methionine
- SCFAs:
-
Short-chain fatty acids
- SELENBP1 :
-
Selenium Binding Protein 1
- SIM2 :
-
Single-minded homolog 2 protein
- SLURP1 :
-
Secreted LY6/PLAUR Domain Containing 1
- SMOC2 :
-
SPARC-related modular calcium-binding protein 2
- SOCS2 :
-
Suppressor of cytokine signaling 2
- SYNPO2 :
-
Synaptopodin-2
- TCF7 :
-
Transcription factor 7
- TCN1 :
-
Transcobalamin-1
- TENM3 :
-
Teneurin-3
- TSKS :
-
Testis-specific serine kinase substrate
- TMM:
-
Trimmed Mean of M values
- TMR:
-
Total mixed ration
- TNXB :
-
Tenascin XB
- TPPP :
-
Tubulin polymerization promoting protein
- TPPP3 :
-
Tubulin polymerization promoting protein family member 3
- TPR:
-
True Positive Rate
- TRIM14 :
-
Tripartite motif containing 14
- TTPA :
-
Alpha tocopherol transfer protein
- TUBA1D :
-
Tubulin Alpha 1d
- UGT1A9 :
-
UDP Glucuronosyltransferase Family 1 Member A9
- VNN1 :
-
Vanin 1
- WFDC18 :
-
WAP four-disulfide core domain protein 18-like
- WTIP :
-
Wilms tumor protein 1-interacting protein
References
Connor EE, Li RW, Baldwin RL, Li C. Gene expression in the digestive tissues of ruminants and their relationships with feeding and digestive processes. Animal. 2010;4(7):993–1007.
Li S, Wang Q, Lin X, Jin X, Liu L, Wang C, et al. The use of “omics” in lactation research in dairy cows. Int J Mol Sci. 2017;18(5):983.
Sun HZ, Zhou M, Wang O, Chen Y, Liu JX, Guan LL. Multi-omics reveals functional genomic and metabolic mechanisms of milk production and quality in dairy cows. Bioinformatics. 2020;36(8):2530–7.
Wu JJ, Zhu S, Gu F, Valencak TG, Liu JX, Sun HZ. Cross-tissue single-cell transcriptomic landscape reveals the key cell subtypes and their potential roles in the nutrient absorption and metabolism in dairy cattle. J Adv Res. 2022;37:1–18.
Gross JJ. Limiting factors for milk production in dairy cows: perspectives from physiology and nutrition. J Anim Sci. 2022;100(3):skac044.
Marín MP, Meléndez PG, Aranda P, Ríos C. Conjugated linoleic acid content and fatty acids profile of milk from grazing dairy cows in southern Chile fed varying amounts of concentrate. J Appl Anim Res. 2018;46(1):150–4.
Morales-Almaraz E, Soldado A, Gonzalez A, Martinez-Fernandez A, Dominguez-Vara I, de la Roza-Delgado B, et al. Improving the fatty acid profile of dairy cow milk by combining grazing with feeding of total mixed ration. J Dairy Res. 2010;77(2):225–30.
O'Callaghan TF, Hennessy D, McAuliffe S, Kilcawley KN, O'Donovan M, Dillon P, et al. Effect of pasture versus indoor feeding systems on raw milk composition and quality over an entire lactation. J Dairy Sci. 2016;99(12):9424–40.
Pegolo S, Stocco G, Mele M, Schiavon S, Bittante G, Cecchinato A. Factors affecting variations in the detailed fatty acid profile of Mediterranean buffalo milk determined by 2-dimensional gas chromatography. J Dairy Sci. 2017;100(4):2564–76.
Zhang H, Ao C, Khas E, Dan N. Effects of isonitrogenous and isocaloric total mixed ration composed of forages with different quality on milk fatty acid composition and gene expression of mammary lipogenic enzymes in mid-lactating dairy cows. Anim Sci J. 2019;90(2):214–21.
Flint HJ, Bayer EA. Plant cell wall breakdown by anaerobic microorganisms from the mammalian digestive tract. Ann N Y Acad Sci. 2008;1125:280–8.
Baaske L, Gabel G, Dengler F. Ruminal epithelium: a checkpoint for cattle health. J Dairy Res. 2020;87(3):322–9.
Santos JEP. Feeding for Milk Composition. Proceedings of the VI International Congress on Bovine Medicine; Spanish Association of Specialists in Bovine Medicine (ANEMBE): Santiago de Compostela, Spain; 2002.
Sutton JD. Altering Milk composition by feeding. J Dairy Sci. 1989;72:2801–14.
Chaudry AS. Forage based animal production systems and sustainability, an invited keynote. Rev Bras Zootec. 2008;37:78–84.
Elgersma A, Soegaard K, Jensen SK. Fatty acids, alpha-tocopherol, beta-carotene, and lutein contents in forage legumes, forbs, and a grass-clover mixture. J Agric Food Chem. 2013;61(49):11913–20.
Salzano A, Neglia G, D'Onofrio N, Balestrieri ML, Limone A, Cotticelli A, et al. Green feed increases antioxidant and antineoplastic activity of buffalo milk: a globally significant livestock. Food Chem. 2021;344:128669.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13(2):97–109.
Li Y, Carrillo JA, Ding Y, He Y, Zhao C, Zan L, et al. Ruminal transcriptomic analysis of grass-fed and grain-fed Angus beef cattle. PLoS One. 2015;10(6):e0116437.
Arora R, Sharma A, Sharma U, Girdhar Y, Kaur M, Kapoor P, et al. Buffalo milk transcriptome: a comparative analysis of early, mid and late lactation. Sci Rep. 2019;9(1):5993.
Deng T, Liang A, Liang S, Ma X, Lu X, Duan A, et al. Integrative analysis of transcriptome and GWAS data to identify the hub genes associated with Milk yield trait in Buffalo. Front Genet. 2019;10:36.
Stupnikov A, McInerney CE, Savage KI, McIntosh SA, Emmert-Streib F, Kennedy R, et al. Robustness of differential gene expression analysis of RNA-seq. Comput Struct Biotechnol J. 2021;19:3470–81.
Corchete LA, Rojas EA, Alonso-Lopez D, De Las RJ, Gutierrez NC, Burguillo FJ. Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Sci Rep. 2020;10(1):19737.
Schurch NJ, Schofield P, Gierlinski M, Cole C, Sherstnev A, Singh V, et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA. 2016;22(6):839–51.
Pastorini M, Pomies N, Repetto JL, Mendoza A, Cajarville C. Productive performance and digestive response of dairy cows fed different diets combining a total mixed ration and fresh forage. J Dairy Sci. 2019;102(5):4118–30.
Salzano A, Licitra F, D'Onofrio N, Balestrieri ML, Limone A, Campanile G, et al. Short communication: space allocation in intensive Mediterranean buffalo production influences the profile of functional biomolecules in milk and dairy products. J Dairy Sci. 2019;102(9):7717–22.
Bin P, Huang R, Zhou X. Oxidation resistance of the sulfur amino acids: methionine and cysteine. Biomed Res Int. 2017;2017:9584932.
Stipanuk MH, Jurkowska H, Roman HB, Niewiadomski J, Hirschberger LL. Insights into taurine synthesis and function based on studies with cysteine dioxygenase (CDO1) knockout mice. Adv Exp Med Biol. 2015;803:29–39.
Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis. 2011;34(1):17–32.
Pol A, Renkema GH, Tangerman A, Winkel EG, Engelke UF, de Brouwer APM, et al. Mutations in SELENBP1, encoding a novel human methanethiol oxidase, cause extraoral halitosis. Nat Genet. 2018;50(1):120–9.
Noga AA, Stead LM, Zhao Y, Brosnan ME, Brosnan JT, Vance DE. Plasma homocysteine is regulated by phospholipid methylation. J Biol Chem. 2003;278(8):5952–5.
Wang Y, Li J, Matye D, Zhang Y, Dennis K, Ding WX, et al. Bile acids regulate cysteine catabolism and glutathione regeneration to modulate hepatic sensitivity to oxidative injury. JCI Insight. 2018;3(8):e99676.
Console L, Scalise M, Mazza T, Pochini L, Galluccio M, Giangregorio N, et al. Indiveri C: carnitine traffic in cells. Link with Cancer. Front Cell Dev Biol. 2020;8:583850.
Giangregorio N, Tonazzi A, Console L, Lorusso I, De Palma A, Indiveri C. The mitochondrial carnitine/acylcarnitine carrier is regulated by hydrogen sulfide via interaction with C136 and C155. Biochim Biophys Acta. 2016;1860(1 Pt A):20–7.
Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino acid. J Biol Chem. 2012;287(24):19786–91.
Veiga-da-Cunha M, Hadi F, Balligand T, Stroobant V, Van Schaftingen E. Molecular identification of hydroxylysine kinase and of ammoniophospholyases acting on 5-phosphohydroxy-L-lysine and phosphoethanolamine. J Biol Chem. 2012;287(10):7246–55.
Servillo L, D'Onofrio N, Giovane A, Casale R, Cautela D, Castaldo D, et al. Ruminant meat and milk contain delta-valerobetaine, another precursor of trimethylamine N-oxide (TMAO) like gamma-butyrobetaine. Food Chem. 2018;260:193–9.
Servillo L, D'Onofrio N, Neglia G, Casale R, Cautela D, Marrelli M, et al. Carnitine precursors and short-chain Acylcarnitines in water Buffalo Milk. J Agric Food Chem. 2018;66(30):8142–9.
Su W, Mao Z, Liu Y, Zhang X, Zhang W, Gustafsson JA, et al. Role of HSD17B13 in the liver physiology and pathophysiology. Mol Cell Endocrinol. 2019;489:119–25.
Adam M, Heikela H, Sobolewski C, Portius D, Maki-Jouppila J, Mehmood A, et al. Hydroxysteroid (17beta) dehydrogenase 13 deficiency triggers hepatic steatosis and inflammation in mice. FASEB J. 2018;32(6):3434–47.
Li H, Liu T, Lim J, Gounko NV, Hong W, Han W. Increased biogenesis of glucagon-containing secretory granules and glucagon secretion in BIG3-knockout mice. Mol Metab. 2015;4(3):246–52.
Li H, Wei S, Cheng K, Gounko NV, Ericksen RE, Xu A, et al. BIG3 inhibits insulin granule biogenesis and insulin secretion. EMBO Rep. 2014;15(6):714–22.
Lobley GE. Control of the metabolic fate of amino acids in ruminants: a review. J Anim Sci. 1992;70(10):3264–75.
Cheriyath V, Leaman DW, Borden EC. Emerging roles of FAM14 family members (G1P3/ISG 6-16 and ISG12/IFI27) in innate immunity and cancer. J Interf Cytokine Res. 2011;31(1):173–81.
Jin W, Jin W, Pan D. Ifi27 is indispensable for mitochondrial function and browning in adipocytes. Biochem Biophys Res Commun. 2018;501(1):273–9.
Giraldo MI, Hage A, van Tol S, Rajsbaum R. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr Clin Microbiol Rep. 2020;7(4):101–14.
Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol. 2008;15:164–87.
Liso A, Venuto S, Coda ARD, Giallongo C, Palumbo GA, Tibullo D. IGFBP-6: at the crossroads of immunity, tissue repair and fibrosis. Int J Mol Sci. 2022;23(8):4358.
Ma L, Zhao M, Zhao LS, Xu JC, Loor JJ, Bu DP. Effects of dietary neutral detergent fiber and starch ratio on rumen epithelial cell morphological structure and gene expression in dairy cows. J Dairy Sci. 2017;100(5):3705–12.
Chen KJ, Lizaso A, Lee YH. SIM2 maintains innate host defense of the small intestine. Am J Physiol Gastrointest Liver Physiol. 2014;307(11):G1044–56.
Minuti A, Palladino A, Khan MJ, Alqarni S, Agrawal A, Piccioli-Capelli F, et al. Abundance of ruminal bacteria, epithelial gene expression, and systemic biomarkers of metabolism and inflammation are altered during the peripartal period in dairy cows. J Dairy Sci. 2015;98(12):8940–51.
Shen H, Chen Z, Shen Z, Lu Z. Maintaining stability of the rumen ecosystem is associated with changes of microbial composition and epithelial TLR signaling. Microbiologyopen. 2017;6(3):e00436.
Yang C, Lan W, Ye S, Zhu B, Fu Z. Transcriptomic analyses reveal the protective immune regulation of conjugated linoleic acids in sheep ruminal epithelial cells. Front Physiol. 2020;11:588082.
Sato T, Fujii R, Konomi K, Yagishita N, Aratani S, Araya N, et al. Overexpression of SPACIA1/SAAL1, a newly identified gene that is involved in synoviocyte proliferation, accelerates the progression of synovitis in mice and humans. Arthritis Rheum. 2011;63(12):3833–42.
Navratilova A, Becvar V, Baloun J, Damgaard D, Nielsen CH, Veigl D, et al. S100A11 (calgizzarin) is released via NETosis in rheumatoid arthritis (RA) and stimulates IL-6 and TNF secretion by neutrophils. Sci Rep. 2021;11(1):6063.
Yuting Y, Lifeng F, Qiwei H. Secreted modular calcium-binding protein 2 promotes high fat diet (HFD)-induced hepatic steatosis through enhancing lipid deposition, fibrosis and inflammation via targeting TGF-beta1. Biochem Biophys Res Commun. 2019;509(1):48–55.
Zhang CJ, Zhu N, Liu Z, Shi Z, Long J, Zu XY, et al. Wnt5a/Ror2 pathway contributes to the regulation of cholesterol homeostasis and inflammatory response in atherosclerosis. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(2):158547.
Wang B, Wang D, Wu X, Cai J, Liu M, Huang X, et al. Effects of dietary physical or nutritional factors on morphology of rumen papillae and transcriptome changes in lactating dairy cows based on three different forage-based diets. BMC Genomics. 2017;18(1):353.
Xiang Q, Liao Y, Chao H, Huang W, Liu J, Chen H, et al. ISL1 overexpression enhances the survival of transplanted human mesenchymal stem cells in a murine myocardial infarction model. Stem Cell Res Ther. 2018;9(1):51.
Bartucci R, Salvati A, Olinga P, Boersma YL. Vanin 1: its physiological function and role in diseases. Int J Mol Sci. 2019;20(16):3891.
Schalkwijk J, Jansen P. Chemical biology tools to study pantetheinases of the vanin family. Biochem Soc Trans. 2014;42(4):1052–5.
Zhao K, Chen YH, Penner GB, Oba M, Guan LL. Transcriptome analysis of ruminal epithelia revealed potential regulatory mechanisms involved in host adaptation to gradual high fermentable dietary transition in beef cattle. BMC Genomics. 2017;18(1):976.
Markkanen E. Not breathing is not an option: how to deal with oxidative DNA damage. DNA Repair (Amst). 2017;59:82–105.
Kaida A, Iwakuma T. Regulation of p53 and Cancer signaling by heat shock protein 40/J-domain protein family members. Int J Mol Sci. 2021;22(24):13527.
Akerfelt M, Trouillet D, Mezger V, Sistonen L. Heat shock factors at a crossroad between stress and development. Ann N Y Acad Sci. 2007;1113:15–27.
Syafruddin SE, Ling S, Low TY, Mohtar MA. More than meets the eye: revisiting the roles of heat shock factor 4 in health and diseases. Biomolecules. 2021;11(4):523.
Ayemele AG, Tilahun M, Lingling S, Elsaadawy SA, Guo Z, Zhao G, et al. Oxidative stress in dairy cows: insights into the mechanistic mode of actions and mitigating strategies. Antioxidants (Basel). 2021;10(12):1918.
Ogata T, Makino H, Ishizuka N, Iwamoto E, Masaki T, Kizaki K, et al. Long-term high-grain diet alters ruminal pH, fermentation, and epithelial transcriptomes, leading to restored mitochondrial oxidative phosphorylation in Japanese black cattle. Sci Rep. 2020;10(1):6381.
Wu J, Yang D, Gong H, Qi Y, Sun H, Liu Y, et al. Multiple omics analysis reveals that high fiber diets promote gluconeogenesis and inhibit glycolysis in muscle. BMC Genomics. 2020;21(1):660.
Dionissopoulos L, Steele MA, AlZahal O, McBride BW. Adaptation to high grain diets proceeds through minimal immune system stimulation and differences in extracellular matrix protein expression in a model of subacute ruminal acidosis in non-lactating dairy cows. Am J Anim Vet Sci. 2012;7(2):84–91.
Kang L, Ayala JE, Lee-Young RS, Zhang Z, James FD, Neufer PD, et al. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes. 2011;60(2):416–26.
Tam CS, Chaudhuri R, Hutchison AT, Samocha-Bonet D, Heilbronn LK. Skeletal muscle extracellular matrix remodeling after short-term overfeeding in healthy humans. Metabolism. 2017;67:26–30.
Abe H, Matsubara T, Iehara N, Nagai K, Takahashi T, Arai H, et al. Type IV collagen is transcriptionally regulated by Smad1 under advanced glycation end product (AGE) stimulation. J Biol Chem. 2004;279(14):14201–6.
Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27.
Egger S, Chaikuad A, Kavanagh KL, Oppermann U, Nidetzky B. UDP-glucose dehydrogenase: structure and function of a potential drug target. Biochem Soc Trans. 2010;38(5):1378–85.
Aschenbach JR, Zebeli Q, Patra AK, Greco G, Amasheh S, Penner GB. Symposium review: the importance of the ruminal epithelial barrier for a healthy and productive cow. J Dairy Sci. 2019;102(2):1866–82.
Afzali AM, Ruck T, Herrmann AM, Iking J, Sommer C, Kleinschnitz C, et al. The potassium channels TASK2 and TREK1 regulate functional differentiation of murine skeletal muscle cells. Am J Physiol Cell Physiol. 2016;311(4):C583–95.
Chen RS, Deng TC, Garcia T, Sellers ZM, Best PM. Calcium channel gamma subunits: a functionally diverse protein family. Cell Biochem Biophys. 2007;47(2):178–86.
Du L, Chang T, An B, Liang M, Duan X, Cai W, et al. Transcriptome profiling analysis of muscle tissue reveals potential candidate genes affecting water holding capacity in Chinese Simmental beef cattle. Sci Rep. 2021;11(1):11897.
Kious BM, Baker CV, Bronner-Fraser M, Knecht AK. Identification and characterization of a calcium channel gamma subunit expressed in differentiating neurons and myoblasts. Dev Biol. 2002;243(2):249–59.
Kim YH, Toji N, Kizaki K, Kushibiki S, Ichijo T, Sato S. Effects of dietary forage and calf starter on ruminal pH and transcriptomic adaptation of the rumen epithelium in Holstein calves during the weaning transition. Physiol Genomics. 2016;48(11):803–9.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30.
Rau A, Gallopin M, Celeux G, Jaffrezic F. Data-based filtering for replicated high-throughput transcriptome sequencing experiments. Bioinformatics. 2013;29(17):2146–52.
Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Tarazona S, Furio-Tari P, Turra D, Pietro AD, Nueda MJ, Ferrer A, et al. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/bioc package. Nucleic Acids Res. 2015;43(21):e140.
Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, et al. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;45(W1):W122–9.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125(1–2):279–84.
Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412.
Acknowledgements
The authors are grateful to M. D’Esposito, prematurely passed away, for giving relevant advice in the conception of the study, M. R. Matarazzo and N. A. Cacciola for useful discussion and suggestions, and M. J. D’Occhio for critical reading of the manuscript.
Funding
This research was financially supported by Italian Ministry for Economic Development (grant number F/200016/01–03/X45) through a research project entitled “CAPSULE - Ottimizzazione delle teCniche di Allevamento e dei Processi produttivi del Settore lattiero-caseario bUfalino e del vino per la produzione di aLimEnti funzionali, and Agritech National Research Center and European Union Next-GenerationEU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR) - MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.4 - D.D. 1032 17/06/2022, CN00000022). This manuscript reflects only the authors’ views and opinions, neither the European Union nor the European Commission can be considered responsible for them.
Author information
Authors and Affiliations
Contributions
GC conceived the study; FDR, SF, AS and GC planned and designed the experiments with the help of NDO and MLB; GN managed the animals and organized the collection of the samples; SF and AS performed the experiments; RAC and SF performed bioinformatics analysis; GC, FDR, AS, and SF wrote the manuscript; NDO, MLB, GN and RAC gave conceptual advice and edited the manuscript; all of the authors have reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was carried out in accordance with the ARRIVE guidelines for reporting animal research [91]. All experimental procedures were performed according to the European Directive 2010/63/EU and the Italian Legislative Decree No. 26 dated 4th March 2014 and received institutional approval from the Ethical Animal Care and Use Committee of the University of Naples “Federico II” (Protocol No. 25532–2022).
The cooperating commercial buffalo dairy in southern Italy provided informed consent for the use of animals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Summary of the mapping information for each sample. CTRL: Control buffaloes; TREAT: Treated buffaloes.
Additional file 2: Table S2.
List of genes expressed in rumen of treated and control dairy buffaloes.
Additional file 3: Table S3.
List of DEGs identified with EdgeR and NOISeq analysis in treated buffaloes in comparison with control animals.
Additional file 4: Table S4.
List of GO terms significantly enriched in genes up-regulated and down-regulated in treated buffaloes in comparison with control animals.
Additional file 5: Table S5.
List of primers used in rt-qPCR experiments.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Salzano, A., Fioriniello, S., D’Onofrio, N. et al. Transcriptomic profiles of the ruminal wall in Italian Mediterranean dairy buffaloes fed green forage. BMC Genomics 24, 133 (2023). https://doi.org/10.1186/s12864-023-09215-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09215-6