
Abstract
Background
While large genome-wide association studies have identified nearly one thousand loci associated with variation in blood pressure, rare variant identification is still a challenge. In family-based cohorts, genome-wide linkage scans have been successful in identifying rare genetic variants for blood pressure. This study aims to identify low frequency and rare genetic variants within previously reported linkage regions on chromosomes 1 and 19 in African American families from the Trans-Omics for Precision Medicine (TOPMed) program. Genetic association analyses weighted by linkage evidence were completed with whole genome sequencing data within and across TOPMed ancestral groups consisting of 60,388 individuals of European, African, East Asian, Hispanic, and Samoan ancestries.
Results
Associations of low frequency and rare variants in RCN3 and multiple other genes were observed for blood pressure traits in TOPMed samples. The association of low frequency and rare coding variants in RCN3 was further replicated in UK Biobank samples (N = 403,522), and reached genome-wide significance for diastolic blood pressure (p = 2.01 × 10− 7).
Conclusions
Low frequency and rare variants in RCN3 contributes blood pressure variation. This study demonstrates that focusing association analyses in linkage regions greatly reduces multiple-testing burden and improves power to identify novel rare variants associated with blood pressure traits.
Similar content being viewed by others

Background
Compared to European Americans (EA), African Americans (AA) consistently have higher blood pressure (BP) levels with earlier onset of hypertension [1]. The excess risks from elevated blood pressure directly affect the life expectancy of AA, which is considerably lower than that of EA. Compared to their EA counterparts, AA men are twice as likely to have a stroke, with earlier onset, or develop stroke-related disabilities [2]. Despite these alarming statistics, there are few genetic studies focusing on BP traits in AA with relatively smaller sample sizes than in European-ancestry studies [3,4,5]. We propose that leveraging linkage evidence from family-based studies can expedite the discovery of rare variants using WGS data.
Previous studies have shown that linkage evidence could facilitate the discovery of low frequency and rare variants associated with BP or other traits [6,7,8,9,10]. The same approach could be applied to family-based studies with AA. A linkage analysis using 4394 AA in 1802 families from the Family Blood Pressure Program (FBPP) identified several linkage peaks on chromosomes 1, 17, and 19 (maximum logarithm of the odds [LOD] > 3) for BP traits [11]. Wang et al. have examined the 1q31 region using exome array data and have detected multiple genes and rare variants contributing to pulse pressure (PP) variation [11]. Because exome array data is limited to exonic regions with mostly coding variants, regulatory non-coding variants as well as very rare variants (minor allele frequency [MAF] < 0.001) cannot be studied with high confidence. These two challenges could be overcome with the Trans-Omics for Precision Medicine (TOPMed) whole genome sequencing (WGS) project, which surveys the whole genome and provides a target coverage of 30x on average [12]. A large number of AA families from FBPP have been whole-genome sequenced as part of TOPMed. To date, most of the large BP genetic studies have focused on samples with European ancestry, and the discovery in African ancestry falls far behind [3, 5]. TOPMed contains one of the largest samples of WGS data in AA, which makes it a suitable dataset to study rare variants found in individuals of African ancestry. In this study, we use the linkage evidence observed from AAs to guide association analysis in multiple ancestral population samples.
Results
Linkage analysis of AA families with TOPMed WGS data
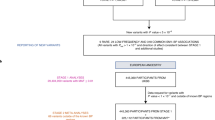
The overall analysis workflow is illustrated in Fig. 1. After conducting linkage analysis on chromosomes 1, 17, and 19 using TOPMed Freeze 6a WGS data, the linkage peaks on chromosomes 1 and 19 from Wang et al. [11] remained but the peak on chromosome 17 was no longer significant (Fig. 2). There were 2 significant linkage peaks for PP on chromosome 1q31 (maximum LOD = 3.28) and chromosome 19q13.33 (MLOD = 3.06). Two additional regions with maximum LOD > 1.87 were followed up on chromosomes 1 and 19: 1q42 for DBP (maximum LOD = 2.41) and 19q13.11 for PP (maximum LOD = 1.87). These four genomic regions on chromosomes 1 and 19 were followed-up for association analysis.

Overview of analysis workflow. Abbreviations: MLOD (maximum LOD score), LODj (family-specific LOD score for family j), QC (quality control), PCs (principal components), R-INT (rescaled inverse normal transformation), AA (African American), EA (European American), EAS (Eastern Asian/Asian American), HA (Hispanic American)

Linkage analysis with HyperGEN and GENOA subjects in TOPMed Freeze 6a release. Abbreviations: SBP (systolic blood pressure); DBP (diastolic blood pressure); PP (pulse pressure); cM (centimorgan); LOD (logarithm of the odds); MLOD (maximum LOD score)
Discovery association analyses with TOPMed WGS data
Discovery gene-based association analyses were completed for variant set 1, 2, and 3 in each of the four linkage regions and a Bonferroni correction adjusting for the number of genes tested was applied in each region to establish four discovery significance thresholds. Any genes with at least one p-value in any trait or any variant group passing the corresponding region’s discovery significance threshold were followed up with additional analyses (Table 1). There were four genes from 1q31, seven genes from 1q42, four genes from 19q13.11, and 12 genes from 19q13.33 that passed the corresponding thresholds. Of these 18 genes, variant set 1 (low frequency coding variants) of RCN3, reticulocalbin 3, showed the strongest association evidence for DBP in TOPMed trans-ancestry samples (burden p = 1.36 × 10− 5; beta = − 0.051, Tables 2 and 3). One variant (rs146159696) overlapped between coding and non-coding variant sets as it is both a missense and intronic variant for different transcripts. This variant also has the most significant p-value in the single variant association analysis (DBP p = 1 × 10− 4). The association direction of these coding variants in EA, AA and HA ancestries were consistent and neither EAS nor Samoan cohorts carried these variants. These 18 genes were carried forward for replication analysis in UKB and gene expression association analysis with GTEx.
Replication association analyses of unrelated samples in TOPMed-imputed UK Biobank
Independent replication analysis was performed using the UKB TOPMed-imputed genotype data and baseline phenotype data. The two variant sets described in the Methods section were analyzed using GENESIS [13]. The top gene from the UKB replication analysis was also RCN3. Coding variants of RCN3 were nominally associated with all three BP traits in the two gene-based association tests for Europeans and Africans, with the lowest p-value being burden p = 5.90 × 10− 5 for SBP, which also significant after Bonferroni correction for multiple comparisons (18 genes × 2 independent traits × 3 variant sets × 3 ethnic populations × 2 statistical tests).
Meta-analyses of TOPMed and UK Biobank
Finally, trans-ancestry meta-analysis and ancestry-specific meta-analyses for European and African ancestries were conducted for RCN3 in all variant sets (Tables 4) using TOPMed and UKB data. In the trans-ancestry meta-analysis, gene-based association test of variant set 1 (low frequency coding variants) in RCN3 reached genome-wide significance for DBP (burden p = 2.01 × 10− 7), which was also significant after adjusting for multiple testing (547 genes from Table 1 × 2 independent traits × 3 variant sets × 2 statistical tests). Among all individuals of European ancestry, RCN3 variant set 1 was also significant (burden p = 3.88 × 10− 6) with DBP after adjusting for multiple tests (547 genes × 2 independent traits × 3 variant sets × 2 statistical tests). Among all individuals of African ancestry, we also observed suggestive evidence for DBP (burden p = 3.16 × 10− 5). Finally, when coding and noncoding variants are combined (set 3), the association evidence of RCN3 gene remained, although the p-values were slightly inflated (Table 4). The variants in this set were further examined in single SNP association analysis (Table 5). Ancestry-specific single SNP association results are shown for SNPs that were observed in both TOPMed and UKB. For RCN3, there were seven low frequency and rare coding variants selected using linkage evidence in African-American families in HyperGEN and GENOA. Of those seven variants, three can be found in TOPMed EA (rs142564622, rs34218348, and rs146159696), all of which were also observed in the UKB European data plus an additional variant (rs770319784).
Gene expression association analysis
Tissue-specific gene expression association analyses were completed for 18 genes of interest using GTEx v7 WGS data (N = 635) and cis-eQTL gene expression data in 48 tissues (including 2 cell lines). The availability of gene expression data varies by tissue along with varying sample size on a tissue-by-tissue basis. For RCN3, gene expression gene-based tests were completed for coding variants only, noncoding variants only, and the aggregated set. P-values from SKAT and burden tests are illustrated on a heat map (Fig. 3). Although none of the associations passed the Bonferroni correction (p = 0.05/(48×2) = 5.2 × 10− 4), the heat map shows that RCN3 variants were nominally associated with gene expression in multiple tissues of the artery, brain, and thyroid, which have shown to be relevant to BP regulation [14, 15].

Heat map of p-values from GTEx tissue-specific gene expression association analysis. Gene expression-association analyses were conducted in EPACTS using variable threshold burden test (BurdenVT) and Sequence Kernel Association Test (SKAT)
Discussion
This study showed that leveraging linkage evidence from family-based studies could effectively and efficiently detect rare variants associated with complex BP traits. This approach successfully identified rare variants associated with BP traits without conducting computationally intensive sliding window-based association analysis across the whole genome and running a large number of tests. Therefore, our approach can be considered as complementary to genome-wide based approaches, which may miss the rare variants or genes identified in this study. Though the variants included for analysis were initially selected from AA families, association evidence for the genes can be observed and replicated well in independent multi-ancestry samples, including African ancestry samples (Tables 1 and 4), demonstrating the robustness of using linkage evidence to guide association analysis of low frequency and rare variants. Across multiple ancestries, we observed evidence of allelic heterogeneity as the top genes in ancestry-specific analyses included low frequency and rare variants that are more common or specific to their corresponding ancestries.
Meanwhile, it is also challenging to study rare variants in trans-ancestry samples as many rare variants are ancestry-specific due to their rarity. Because the sample sizes for non-European cohorts are often much smaller, the statistical power is reduced and replication may be challenging. For example, low frequency coding variants of VSIG10L demonstrated suggestive association evidence for DBP in the Samoan Adiposity Study (burden p = 9.24 × 10− 6; beta = 0.521), but not in any other ancestry. The significant gene-based test was mostly driven by a single variant rs141732375 (p = 9.82 × 10− 5; beta = 7.01). Due to data availability, replication in other Samoan cohorts was not feasible at the time of the analysis.
The significant gene after correcting for multiple testing identified from this study was RCN3. The association of the RCN3 coding variants in samples of African ancestry has p-values of 0.01, 4.89 × 10− 4 and 3.16 × 10− 5 for PP, SBP and DBP, respectively (Table 4), despite the relatively small sample size. This association evidence is consistent with the linkage evidence. Similar association is also present in samples of European ancestry with larger sample size. It is encouraging to observe that rare coding variants in RCN3 are associated with both AA and EA in UKB replication analysis (Table 2), suggesting the association evidence is not a false positive.
However, the association evidence for non-coding variants (variant set 2) was less consistent because TOPMed cohorts did not show any association evidence for BP traits, but RCN3 non-coding variants in UKB European samples showed significant association evidence in SKAT for PP (p = 2.97 × 10− 4) and SBP (p = 4.67 × 10–5) after adjusting for multiple comparisons (Table 3). In the single SNP association analysis (Table 5), there were seven low frequency or rare coding variants identified from HyperGEN and GENOA using the approach described in the Methods section. Among European ancestry samples, three out of seven SNPs (rs142564622, rs34218348, and rs146159696) were observed in both TOPMed EA and UKB European, and one SNP (rs770319784) was only observed in UKB European. The four SNPs observed in UKB European were also observed in UKB African. For the four SNPs observed in both TOPMed and UKB, the directions of effect in DBP was completely consistent for rs142564622 and rs770319784 and nearly consistent for rs34218348 and rs146159696.
The p-values in the discovery stage might be inflated because linkage analysis and variant selection for association analysis were performed within the same pedigrees. Our previous simulation study suggested such inflation is minimal [8]. However, to be conservative, we used the Bonferroni-corrected p-value threshold in the UKB replication data (p = 7.72 × 10− 5) after correcting for 2 independent BP traits, 2 statistical tests, 3 variant sets, and 3 UKB populations (European, African, Asian), and 18 genes. For the trans-ethnic TOPMed and UKB meta-analysis, a p-value threshold of 7.62 × 10− 6 was used to declare significance after adjusting for 547 genes, 2 independent traits, 3 variant sets and 2 statistical methods. Thus, the association evidence of RCN3 with DBP and SBP reported in this study is significant in both UKB replication data as well as combined TOPMed and UKB trans-ethnic data.
There is some genetic evidence supporting the association between RCN3 and BP traits. UKB GWAS by Neale et al. [16] found two genome-wide significant SNPs associated with hypertension: rs61760904 (missense; non-Finnish European allele frequency = 0.007; OR = 1.2; p-value = 1.8E × 10− 9; CADD > 23.4) and rs73046792 (3′ UTR variant; non-Finnish European allele frequency = 0.15; OR = 0.96; p-value = 3.6 × 10− 8; CADD > 0.89). One SNP downstream of RCN3, rs189349094, is associated with systolic blood pressure [17] and linked to RCN3 through GeneHancer [18]. None of these previously reported SNPs overlap with SNPs selected by linkage evidence, suggesting the variants we identified in RCN3 are novel.
One pattern observed in the gene-based association analysis was that the strongest association evidence did not come from PP, the trait with the linkage signal. One possible explanation is that when the directions of effect are the same for SBP and DBP, the effect size for PP is reduced because PP is the difference of SBP and DBP; thus, canceling the association of PP.
There are a number of known imputation challenges for rare variants, particularly for non-European individuals, in the UKB data imputed using the Haplotype Reference Consortium [19]. Therefore, it was necessary to re-impute these regions using the multi-ancestry TOPMed reference panel. Unpublished results from our group and recent TOPMed publications have shown that the TOPMed reference panel can successfully impute rare variants found in populations of African ancestry [12, 20, 21]. With the TOPMed imputation, we were able to examine UKB samples with European, African, and Asian ancestries.
Common genetic variants discovered from GWAS face a challenge of pinpointing causal genes and therefore are difficult to interpret. On the other hand, rare variants may contribute to a trait’s “missing heritability” but are extremely difficult to uncover and to replicate due to insufficient statistical power for currently available samples with WGS data, such as TOPMed. The primary goal of this study is to search for rare variants using the TOPMed WGS data with an approach that is not widely used in WGS association analysis. However, our study demonstrates that this approach can be successful in identifying rare variants and is complementary to purely population-based approaches. The association of the coding variants identified in RCN3 gene is replicable and present across multiple ancestries, although the original linkage evidence was identified from AA families. Additionally, these coding variants are more interpretable; however, further functional studies are needed to understand the mechanisms underlying how these variants contribute to BP variation.
There are some limitations of our study. The major limitation is the wide range of study designs and phenotype collection procedures in the studies included. While adjustments were included in analyses for study and data collection centers, it was difficult to control for the study design differences, which may reduce statistical power.
Conclusions
This study examined low frequency and rare variants under linkage peaks on chromosomes 1 and 19 that were detected in AA families. By focusing on linkage regions and following up with gene-based and single SNP association analyses, multiple genes were found to be associated with BP traits. In particular, low frequency and rare coding variants from RCN3 were significantly associated with DBP in trans-ancestry samples. While our finding is supported by genetic evidence, additional analyses are warranted to examine the underlying biological mechanisms. This study demonstrates that leveraging linkage evidence in WGS expedites the process of identifying functional rare variants associated with complex traits. Individually, these rare variants might only explain a small portion of heritability in the population level, but they could facilitate our understanding of the genetic determinants of hypertension in diverse populations. Additionally, functional rare variants identified from this type of study could further facilitate the identification of disease targets.
Methods
Study population
The discovery analysis included all TOPMed Freeze 8 samples with the harmonized BP phenotype at the time of analysis, which consisted of 18 TOPMed studies (32 ancestry- and study-specific cohorts). These 18 studies (N = 60,388) included 26,590 EA, 15,284 AA, 3826 East Asians or Asian Americans (EAS), 13,419 Hispanic Americans (HA), and 1269 Samoans from the following studies: Genetics of Cardiometabolic Health in the Amish (Amish; EA), Atherosclerosis Risk in Communities Study from the Venous Thromboembolism (VTE) project (ARIC; EA and AA), Mount Sinai BioMe Biobank (BioMe; EA, AA, EAS, and HA), Coronary Artery Risk Development in Young Adults (CARDIA; EA and AA), Cleveland Family Study (CFS; EA and AA), Cardiovascular Health Study (CHS; EA and AA), Framingham Heart Study (FHS; EA), Genetic Epidemiology Network of Salt Sensitivity (GenSalt; EAS), Genetic Studies of Atherosclerosis Risk (GeneSTAR; EA and AA), Hispanic Community Health Study – Study of Latinos (HCHS_SOL; HA), Hypertension Genetic Epidemiology Network and Genetic Epidemiology Network of Arteriopathy (HyperGEN_GENOA; AA), GENOA from the African American Coronary Artery Calcification project (part of HyperGEN_GENOA; AA), Jackson Heart Study (JHS; AA), Multi-Ethnic Study of Atherosclerosis (MESA; EA, AA, EAS, and HA), MESA Family Study from the African American Coronary Artery Calcification project (MESAFam; AA), San Antonio Family Studies (SAFS; HA), Samoan Adiposity Study (Samoan), Taiwanese Study of Hypertension using Rare Variants (THRV; EAS), and the Women’s Health Initiative (WHI; EA, AA, EAS, and HA). These studies vary in design: BioMe, CARDIA, CHS, HCHS_SOL, and the Samoan study are primarily community-based studies; JHS and MESA are community-based studies that include a nested family-based design; Amish, CFS, FHS, GeneSTAR, GenSalt, HyperGEN_GENOA, MESAFam, SAFS, and THRV are family-based studies; and ARIC and WHI are population-based cohort studies in which case-control samples were selected for TOPMed. Descriptions of these studies and data collection procedures are included in (Additional File 1. Supplemental Materials & Methods).
The UK Biobank (UKB) version 3 GWAS data [22] were used as the replication cohort. These samples were collected from across the United Kingdom from participants between 40 to 69 years old. The UKB replication cohort included individuals of European ancestry (N = 417,634), African ancestry (N = 7297), and Asian ancestry (N = 10,215). Ethnic subgroups were clustered. Individuals with ethnic subgroup coding of 1 (White), 1001 (British), 1002 (Irish), and 1003 (any other white background) were considered as European ancestry, those with coding of 4 (Black or Black British), 4001 (Caribbean), 4002 (African), 4003 (any other black background) were considered as African ancestry, and those with coding of 3 (Asian), 3001 (Indian), 3002 (Pakistani), 3003 (Bangladeshi), 3004 (any other Asian background), and 5 (Chinese) were considered as Asian ancestry.
Genotyping and quality control (QC)
The TOPMed Informatics Research Center (IRC) and Data Coordinating Center (DCC) centrally performed sample and genotype quality control (QC). Detailed QC procedures are described in the TOPMed flagship paper [12] and TOPMed Freeze 8 website (https://topmed.nhlbi.nih.gov/topmed-whole-genome-sequencing-methods-freeze-8). The software BCFtools [23] was used to apply the following QC filters: 1) bi-allelic single nucleotide polymorphisms (SNPs) and small insertion-deletion polymorphisms (INDELs) passing all genotype filters; 2) a minimum 10x sequencing depth. The participant must not have any known identity problems (such as sex or pedigree mismatches) reported by the DCC to be included for analysis. In this study, unique participants from 18 TOPMed studies from the Freeze 8 release (GRCh38) were included, reflecting the May 30, 2019 sample annotation from the TOPMed DCC. After excluding individuals under 18 years old and those with missing BP measurements or covariates, the combined study sample contained 60,388 individuals. Principal components (PCs) and kinship matrix were both made available by the TOPMed DCC. As described in the TOPMed Flagship paper [12], the PCs were calculated using PC-AiR [13], and the kinship matrix was calculated using the pcrelate function in the GENESIS R package [24]. This approach estimates kinship coefficients and identical-by-descent (IBD) sharing probabilities conditional on ancestry. A fourth-degree sparse kinship matrix provided by TOPMed was used as the covariance matrix in the linear mixed model for optimal computational efficiency. The TOPMed DCC has determined that the top 11 PCs well represent global ancestry patterns among TOPMed Freeze 8 samples. Therefore, these PCs were adjusted in the phenotype residuals and linear mixed model to account for genetic ancestry background.
The UKB data were genotyped using the Affymetrix UK Biobank Axiom array [22]. Principal components were calculated by UKB with genotype data within each ancestry to account for population structure (http://www.ukbiobank.ac.uk/wp-content/uploads/2014/04/UKBiobank_genotyping_QC_documentation-web.pdf). Because the UKB imputed genotype data were originally imputed using the Haplotype Reference Consortium [25] reference panel, which is predominantly of European ancestry, we re-imputed Europeans, Africans, and Asians using the TOPMed reference panel. The TOPMed reference panel is a diverse reference panel including information from 97,256 deeply sequenced human genomes, and we were able to impute rare variants for non-European individuals with high confidence (r2 > 0.3). Ancestry-specific genotype imputation was conducted on the TOPMed Imputation Server (https://imputation.biodatacatalyst.nhlbi.nih.gov/). The software QCTOOL v2 (https://www.well.ox.ac.uk/~gav/qctool_v2/index.html) was used to convert the BGEN format genotype files to VCF format. The following pre-imputation quality control were done in PLINK 1.9 [26]: variants with MAF < 1%, genotyping rate < 97%, or Hardy-Weinberg Equilibrium < 1 × 10− 6 were removed. Variants were remapped from GRCh37 to GRCh38 using the TOPMed Imputation Server, and those variants that cannot be remapped were excluded. Imputed variants with a r2 > 0.3 were retained for analysis. Further analysis by increasing the threshold to r2 > 0.5 did not affect the result (Additional file 1. Supplemental Materials and Methods). Sample QC was performed for UKB by excluding outliers in heterozygosity and missing rates defined by UKB.
The SeqArray R package [27] was used to convert VCF format into GDS format to be used in the GENESIS R package [13] for association analysis. Related individuals with pairwise kinship coefficient greater than 0.0884 [28], which is the threshold for third degree relatives calculated using software KING [29], were removed from analysis, resulting in 386,813 individuals of European ancestry, 6937 individuals of African ancestry, and 9772 individuals of Asian ancestry from the UKB.
Phenotype harmonization
TOPMed phenotype data were collectively harmonized by members of the TOPMed BP Working Group. Details on TOPMed phenotype harmonization for systolic blood pressure (SBP), diastolic blood pressure (DBP), and pulse pressure (PP) were described in our previous study [7]. Covariates used in the analyses were measured at the same visit as the BP measurements.
For the UKB cohort, baseline BP and covariates (Additional file 2. Table S1) were extracted from the phenotype data. Because two SBP and DBP measurements were taken at baseline, the average of the two measurements was used to generate the phenotypes for association analyses. Individuals with missing BP data at baseline were excluded from analysis.
Transformation of phenotype data for association analyses
As each TOPMed project has a different study design and sample population, it is important to standardize the quantitative trait values by applying data transformation and rescale to restore the original measurement for genetic effects. In this study, the phenotype residuals were calculated separately by ancestry and phenotype transformation was applied to account for between-study heterogeneity. Harmonized BP phenotypes were pooled within each ancestry and BP traits were adjusted for anti-hypertensive medications use by adding 15 mmHg and 10 mmHg to raw SBP and DBP measurements, respectively [30]. The regression residuals were calculated for medication-adjusted SBP, DBP, and PP after adjusting for age, age2, sex, body mass index (BMI), field center (for multi-center studies), case-control status for stroke or venous thromboembolism (WHI only), and the top 11 PCs. Next, inverse normal transformation was applied to the ancestry-specific residuals. The inverse normal transformed residuals were re-scaled using the standard deviation (SD) of raw BP measurement, prior to medication adjustment, in each study. This results in a rescaled inverse normal transformation (R-INT) that makes the phenotype to follow a normal distribution and restores the original scale of measurement [31]. The phenotype distributions and transformations are shown in (Additional files 3, 4, 5, 6 and 7: Figs. S1-S5).
The R-INT residuals of BP phenotypes were analyzed in both gene-based and single variant association analyses. The covariates described above were adjusted for the second time in the linear mixed model. Previously, Softer et al. used TOPMed data to show that a two-stage approach to adjust for covariates can improve statistical power and reduce type I error [32]. Ancestry-specific phenotypes were pooled for the trans-ethnic analysis in TOPMed. In the UK Biobank data analysis, SBP and DBP were adjusted for anti-hypertensive medications use by adding 15 mmHg and 10 mmHg, respectively. Covariates (age, BMI, assessment center) and top 10 PCs were included in the same way as described for TOPMed data.
Overview of statistical methods
The overall analysis workflow includes 3 stages and is illustrated in Fig. 1. In the preliminary stage, we conducted linkage analysis with AA families in HyperGEN and GENOA using TOPMed WGS data. In the discovery stage, we completed gene-based and single variant association analyses using the SNPs prioritized by linkage evidence. In the final stage, we performed replication for the top genes identified from the discovery stage in the TOPMed-imputed UK Biobank data and meta-analyzed TOPMed with UK Biobank by ancestry and across ancestries.
Linkage analysis of AA families with TOPMed WGS data
We performed multi-point variance-component linkage analysis of TOPMed WGS data in HyperGEN and GENOA families to obtain the family-specific LOD scores. Study-specific BP residuals, after adjusting for anti-hypertensive medication use, were used in the linkage analysis. The genetic map for GRCh38 was obtained from the University of Washington (http://bochet.gcc.biostat.washington.edu/beagle/genetic_maps/). The set of linkage disequilibrium pruned SNPs that was used in the exome array linkage analysis by Wang et al. [11] (MAF > 0.2 and linkage disequilibrium r2 < 0.1), which consists of 813 markers for chr1, 347 markers for chr17, and 384 markers for chr19, was used again in the linkage analysis with TOPMed WGS data. Linkage region was defined as a two-LOD score drop from the linkage peak SNP, which has the highest LOD score.
The linkage regions were re-defined using WGS data due to two key reasons: 1) only 3085 out of 4394 individuals (70%) could be found in both FBPP exome array data analyzed by Wang et al. [11] and TOPMed WGS data; 2) there were several pedigree relatedness problems with the exome array data (e.g. half/step siblings were separated into different families), which resulted in inaccurate family-specific LOD scores. After correcting the pedigree errors, multi-point variance-component linkage analysis was conducted using MERLIN [33] for three BP traits (SBP, DBP, and PP) on chromosomes 1, 17, and 19 using 3149 HyperGEN and GENOA individuals in the TOPMed Freeze 6a release, the latest release at the time of analysis. For HyperGEN and GENOA, the individuals are identical for Freeze 6a and Freeze 8. Chromosome 17 was excluded from further analysis due to a lack of linkage evidence.
Variant selection from HyperGEN & GENOA families in TOPMed WGS
In the preliminary stage, we performed variance component linkage analysis in African-American families and searched for linkage regions with suggestive linkage evidence. Single SNP and gene-based associations for selected variants were conducted in protein-coding genes within the linkage regions on 1q31 (chr1:188765880–202,026,147), 1q42 (chr1:232963435–240,632,149), 19q13.11 (chr19:22332449–36,438,656), and 19q13.33 (chr19:41978814–53,404,335). We examined two significant linkage peaks (maximum LOD > 3) on 1q31 and 19q13.33 and two additional regions with max LOD that are approximately 2. The analyses were limited to variants residing within protein-coding genes, as defined by GENCODE v29 [34], of each linkage region in HyperGEN and GENOA TOPMed Freeze 8 WGS data.
Next, the variants were selected using a two-step approach. Step 1, let LODj represent the LOD score for the jth family at the max LOD marker of a chromosomal region. We selected families with LODj > 0.1 after excluding parent-offspring pairs (e.g. family of two with mother-child or father-child), which are uninformative for linkage analysis. Prior simulations from our group have shown that the threshold of 0.1 for variant selection is optimal in association analysis [8]. We identified 35 families for 1q31, 18 families for 1q42, 20 families for 19q13.11 and 25 families for 19q13.33 with LODj > 0.1. SNPs or INDELs segregating at least twice in these families were selected. Step 2, let MACij be the minor allele count for family j and variant i identified from step 1. For variant i in gene x, the correlation ri between MACij and LODj was calculated. When a portion of the variants in the linkage region contribute to linkage evidence, we expect that variants contributing to linkage evidence are more likely to have ri to be positively correlated. For the variants in a gene x, their ri were fitted a mixture of two Normal distributions using the mixtools R package. Then Fisher’s Discriminant Analysis was used to identify variants in which their correlation ri is greater than the average of two component mean. Lastly, the union of variants selected by these two steps were included for association analysis. This process can be viewed as a weighting procedure of variants contributing to the observed linkage evidence.
The gene region is defined by Ensembl Variant Effect Predictor [35] as a part of the functional annotations curated by WGSA [36], which was provided by the TOPMed DCC. The variants selected for analysis were grouped into 2 sets using annotations: 1) functional coding variants that lead to an amino acid change and 2) remaining non-coding variants and synonymous variants located within the gene region and 10 kb upstream and downstream of each gene. Functional coding variants were limited to those with MAF < 5% and included splice region variant, start lost variant, stop lost/gained variant, missense variant, inframe deletions/insertions, exon loss variant (deletion of an exon), frameshift variant, initiator codon variant non-canonical start codon, and splice acceptor variant. The non-coding variants had a maximum MAF of 1% and were further examined for those with functional prediction scores [37, 38] (CADD-phred > 10, fathmmXF > 0.5). Within each coding and non-coding group, variants were aggregated by gene names. Variants located in multiple genes with overlapping positions were retained in each gene. We separately analyzed variants into two independent sets: set 1 includes coding variants with MAF < 5% and set 2 includes non-coding variants with MAF < 1%. We further combined set 1 and 2 variants (set 3) but required the set 2 variants with either CADD > 10 or fathmmXF > 0.5 [37, 38].
Discovery association analyses with TOPMed WGS data
The focus of this study was performing gene-based association analyses in all four linkage regions for the three variant sets prioritized using linkage evidence with the GENESIS [24] R package. The majority of the analyses were completed on the High Performance Computing Cluster (HPCC) at Case Western Reserve University and parts of the trans-ancestry analysis were completed in Analysis Commons [39] on the cloud computing platform DNAnexus (https://www.dnanexus.com/) for computational efficiency. Discovery samples were stratified by ancestry (AA, EA, EAS, HA, Samoan) and both ancestry-specific and pooled trans-ancestry analyses were completed for SBP, DBP, and PP. A kinship matrix was constructed for each stratum and the trans-ancestry sample using the fourth-degree sparse kinship matrix provided by the TOPMed DCC. For each trait on each stratum, a null model was fitted using linear mixed model with the transformed phenotype residuals, covariates, and kinship matrix. Next, the three collapsed variant sets described previously were used to conduct gene-based association analysis using burden (Wald) test [40] and sequence kernel association test (SKAT) [41]. Variants were weighted using the default parameters dbeta [1, 25] to give more weight to the rarer variants. Bonferroni correction for the number of genes tested in each linkage region was used as a discovery significance threshold. After identifying top associated genes, we performed single SNP based association in order to identify individual variants contributing the gene-based association evidence. Single variant association analyses were completed using linear mixed model with GENESIS [24].
Replication association analyses of unrelated samples in TOPMed-imputed UK Biobank
For the genes carried forward for replication analyses, we used the same gene collapsing groups to perform burden test and SKAT with TOPMed-imputed UK Biobank data in the GENESIS R package [24]. Single variant association analyses were only carried out for the top gene of interest, RCN3. Association analyses were performed without including a kinship matrix after removing individuals up to the 3rd degree of relatedness.
Meta-analyses of TOPMed and UK Biobank
For the gene-based analyses, meta-analyses of European cohorts, African cohorts, and trans-ethnic cohorts from TOPMed and the UK Biobank were calculated using Fisher’s combined p value method. The trans-ethnic meta-analysis of TOPMed and UKB was also performed using Fisher’s method with 8 degrees of freedom to account for three UKB ancestry-specific analyses for individuals of European, African, and Asian ancestries. The exome-wide significance threshold (p < 2.5 × 10− 6) was used to determine genome-wide significance.
Gene expression association analysis
Genotype-Tissue Expression (GTEx) expression quantitative trait loci (eQTL) gene expression matrices (GTEx V7 cis-eQTL) were downloaded from the GTEx Portal (https://www.gtexportal.org/home/datasets) and WGS data of 635 individuals were obtained from dbGaP phs000424.v7.p2. Tissue-specific gene expression association analyses were completed for genes of interest in 46 tissues and 2 cell lines. SKAT and burden test were completed in the software EPACTS [42] using both coding and non-coding variants in genes of interest identified from TOPMed (variant set 3). The residuals of the gene expression level were treated as the phenotype, after adjusting for sex, platform, PCs 1–3, and tissue-specific latent factors inferred by GTEx using the PEER method [43]. The analyzed variants were limited to variants replicated across studies, where we aggregated linkage-based selected functional coding variants and rare non-coding variants identified from HyperGEN and GENOA.
Availability of data and materials
All the TOPMed datasets generated and/or analyzed during the current study are available in the dbGaP repository and instructions for data access can be found at https://www.nhlbiwgs.org/topmed-data-access-scientific-community. The current study includes datasets: phs000956, phs001211, phs001644, phs001624, phs001612, phs000954, phs001368, phs000951, phs001218, phs001345, phs000974, phs001217, phs001395, phs001293, phs000964, phs001416, phs001215, phs000972, phs001387, phs001237. The UK Biobank data is available in the UK Biobank repository: ukbiobank.ac.uk.
Abbreviations
- AA:
-
African American
- BP:
-
Blood pressure
- BMI:
-
Body mass index
- DBP:
-
Diastolic blood pressure
- DCC:
-
Data coordinating center
- EA:
-
European American
- EAS:
-
East Asian
- FBPP:
-
Family Blood Pressure Program
- GENESIS:
-
GENetic EStimation and Inference in Structured samples
- GENOA:
-
Genetic Epidemiology Network of Arteriopathy
- GTEx:
-
Genotype-Tissue Expression
- GWAS:
-
Genome-wide association studies
- HA:
-
Hispanic American
- HyperGEN:
-
Hypertension Genetic Epidemiology Network
- LD:
-
Linkage disequilibrium
- LOD:
-
Logarithm of odds
- MAC:
-
Minor allele count
- MAF:
-
Minor allele frequency
- MLOD:
-
MLOD
- NHLBI:
-
National Heart, Lung, and Blood Institute
- PC:
-
Principal component
- PP:
-
Pulse pressure
- QC:
-
Quality control
- R-INT:
-
Rescaled inverse normal transformation
- SBP:
-
Systolic blood pressure
- SKAT:
-
Sequence kernel association test
- SNP:
-
Single nucleotide polymorphism
- TOPMed:
-
Trans-Omics for Precision Medicine
- UKB:
-
UK Biobank
- WGS:
-
Whole genome sequencing
References
Lackland DT. Racial differences in hypertension: implications for high blood pressure management. Am J Med Sci. 2014;348(2):135–8.
Howard G, Labarthe DR, Hu J, Yoon S, Howard VJ. Regional differences in African Americans' high risk for stroke: the remarkable burden of stroke for southern African Americans. Ann Epidemiol. 2007;17(9):689–96.
Franceschini N, Fox E, Zhang Z, Edwards TL, Nalls MA, Sung YJ, et al. Genome-wide association analysis of blood-pressure traits in African-ancestry individuals reveals common associated genes in African and non-African populations. Am J Hum Genet. 2013;93(3):545–54.
Zhu X, Feng T, Tayo BO, Liang J, Young JH, Franceschini N, et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am J Hum Genet. 2015;96(1):21–36.
Liang J, Le TH, Velez Edwards DR, Tayo BO, Gaulton KJ, Smith JA, et al. Correction: single-trait and multi-trait genome-wide association analyses identify novel loci for blood pressure in African-ancestry populations. PLoS Genet. 2018;14(5):e1007345.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30.
He KY, Wang H, Cade BE, Nandakumar P, Giri A, Ware EB, et al. Rare variants in fox-1 homolog a (RBFOX1) are associated with lower blood pressure. PLoS Genet. 2017;13(3):e1006678.
Liang J, Cade BE, He KY, Wang H, Lee J, Sofer T, et al. Sequencing analysis at 8p23 identifies multiple rare variants in DLC1 associated with sleep-related Oxyhemoglobin saturation level. Am J Hum Genet. 2019;105(5):1057–68.
Zhu X, Feng T, Li Y, Lu Q, Elston RC. Detecting rare variants for complex traits using family and unrelated data. Genet Epidemiol. 2010;34(2):171–87.
Feng T, Elston RC, Zhu X. Detecting rare and common variants for complex traits: sibpair and odds ratio weighted sum statistics (SPWSS, ORWSS). Genet Epidemiol. 2011;35(5):398–409.
Wang H, Nandakumar P, Tekola-Ayele F, et al. Combined linkage and association analysis identifies rare and low frequency variants for blood pressure at 1q31. Eur J Hum Genet. 2019;27(2):269-277. https://doi.org/10.1038/s41431-018-0277-1.
Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature. 2021;590(7845):290–9.
Conomos MP, Miller MB, Thornton TA. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet Epidemiol. 2015;39(4):276–93.
Danzi S, Klein I. Thyroid hormone and blood pressure regulation. Curr Hypertens Rep. 2003;5(6):513–20.
OpenStax. Anatomy & Physiology. OpenStax CNX. 2016.
Neale B. UK Biobank GWAS Round 2 [Available from: http://www.nealelab.is/uk-biobank/.
Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. 2019;104(1):65–75.
Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017;2017:bax028.
Weedon MN, Jackson L, Harrison JW, Ruth KS, Tyrrell J, Hattersley AT, et al. Very rare pathogenic genetic variants detected by SNP-chips are usually false positives: implications for direct-to-consumer genetic testing. 2019.
Quick C, Anugu P, Musani S, Weiss ST, Burchard EG, White MJ, et al. Sequencing and imputation in GWAS: cost-effective strategies to increase power and genomic coverage across diverse populations. Genet Epidemiol. 2020;44(6):537–49.
Kowalski MH, Qian H, Hou Z, Rosen JD, Tapia AL, Shan Y, et al. Use of >100,000 NHLBI trans-Omics for precision medicine (TOPMed) consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15(12):e1008500.
Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–9.
Danecek P, Bonfield JK, Liddle J, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008. https://doi.org/10.1093/gigascience/giab008.
Gogarten SM, Sofer T. Chen H, Yu C. Thornton TA, et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics: Brody JA; 2019.
McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–83.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Zheng X, Gogarten SM, Lawrence M, Stilp A, Conomos MP, Weir BS, et al. SeqArray-a storage-efficient high-performance data format for WGS variant calls. Bioinformatics. 2017;33(15):2251–7.
Hanscombe KB, Coleman JRI, Traylor M, Lewis CM. ukbtools: An R package to manage and query UK Biobank data. PLoS One. 2019;14(5):e0214311.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26(22):2867–73.
Law MR, Morris JK, Wald NJ. Use of blood pressure lowering drugs in the prevention of cardiovascular disease: meta-analysis of 147 randomised trials in the context of expectations from prospective epidemiological studies. BMJ. 2009;338:b1665.
Tang ZZ, Lin DY. Meta-analysis for discovering rare-variant associations: statistical methods and software programs. Am J Hum Genet. 2015;97(1):35–53.
Sofer T, Zheng X, Gogarten SM, Laurie CA, Grinde K, Shaffer JR, et al. A fully adjusted two-stage procedure for rank-normalization in genetic association studies. Genet Epidemiol. 2019;43(3):263–75.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101.
Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47(D1):D766–D73.
Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83(3):311–21.
Liu X, White S, Peng B, Johnson AD, Brody JA, Li AH, et al. WGSA: an annotation pipeline for human genome sequencing studies. J Med Genet. 2016;53(2):111–2.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D94.
Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, Campbell C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. 2018;34(3):511–3.
Brody JA, Morrison AC, Bis JC, O'Connell JR, Brown MR, Huffman JE, et al. Analysis commons, a team approach to discovery in a big-data environment for genetic epidemiology. Nat Genet. 2017;49(11):1560–3.
Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5(2):e1000384.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89(1):82–93.
Kang HM. Efficient and parallelizable association container toolbox (EPACTS) 2013 [Available from: http://genome.sph.umich.edu/wiki/EPACTS.
Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protoc. 2012;7(3):500–7.
Acknowledgements
Trans-Omics for Precision Medicine (TOPMed) Acknowledgements
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Amish Complex Disease Research Program (phs000956) was performed at the Broad Institute of MIT and Harvard (3R01HL121007-01S1). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Atherosclerosis Risk in Communities study (phs001211) was performed at the Baylor Human Genome Sequencing Center (3U54HG003273-12S2 and HHSN268201500015C) and the Broad Institute for MIT and Harvard (3R01HL092577-06S1). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the IPM BioMe Biobank (phs001644) was performed at the Baylor Human Genome Sequencing Center (HHSN268201600033I) and the McDonnell Genome Institute at Washington University (HHSN268201600037I). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Coronary Artery Risk Development in Young Adults (phs001612) was performed at the Baylor Human Genome Sequencing Center (HHSN268201600033I). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Cleveland Family Study (phs000954) was performed at the University of Washington Northwest Genomics Center (3R01HL098433-05S1 and HHSN268201600032I). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Cardiovascular Health Study (phs001368) was performed at the Baylor Human Genome Sequencing Center (HHSN268201600033I). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Framingham Heart Study (phs000974) was performed at the Broad Institute of MIT and Harvard (3U54HG003067-12S2). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Genetic Studies of Atherosclerosis Risk (phs001218) was performed by the Broad Institute of MIT and Harvard (HHSN268201500014C), Psomagen, Inc. (3R01HL112064-04S1), and Illumina Genomic Services (R01HL112064). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Genetic Epidemiology Network of Arteriopathy (phs001345) was performed at the Mayo Clinic Genotyping Core, the DNA Sequencing and Gene Analysis Center at the University of Washington Northwest Genomics Center (3R01HL055673-18S1), and the Broad Institute (HHSN268201500014C). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Genetic Epidemiology Network of Salt-Sensitivity (phs001217) was performed at the Baylor Human Genome Sequencing Center (HHSN268201500015C). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Hispanic Community Health Study – Study of Latinos (phs001395) was performed at the Baylor Human Genome Sequencing Center (HHSN268201600033I). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Hypertension Genetic Epidemiology Network (phs001293) was performed at the University of Washington Northwest Genomics Center (3R01HL055673-18S1). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Jackson Heart Study (phs000964) was performed at the University of Washington Northwest Genomics Center (HHSN268201100037C). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Multi-Ethnic Study of Atherosclerosis (phs001416) was performed at the Broad Institute of MIT and Harvard (3U54HG003067-13S1). Genome sequencing for “NHLBI TOPMed: Whole Genome Sequencing to Identify Causal Genetic Variants Influencing CVD Risk - San Antonio Family Studies” (phs001215) was performed at Illumina (3R01HL113323-03S1, 3R01HL113323-03S1). WGS for NHLBI TOPMed: Genome-wide Association Study of Adiposity in Samoans (phs000972) was performed at the University of Washington Northwest Genomics Center (HHSN268201100037C) and the New York Genome Center (HHSN268201500016C). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Taiwan Study of Hypertension using Rare Variants (phs001387) was performed at the Baylor Human Genome Sequencing Center (3R01HL111249-04S1 and HHSN26820150015C). WGS for NHLBI TOPMed: WGS and Related Phenotypes in the Women’s Health Initiative (phs001237) was performed at the Broad Institute of MIT and Harvard (HHSN268201500014C). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. The authors would like to acknowledge contributions from the investigators of the NHLBI TOPMed Consortium (Additional file 9; https://www.nhlbiwgs.org/topmed-banner-authorship).
Study-Specific Acknowledgements and Funding Sources
Amish: The Amish studies upon which these data are based were supported by NIH grants R01 AG18728, U01 HL072515, R01 HL088119, and R01 HL121007.
Analysis Commons: The Analysis Commons was funded by R01HL131136.
ARIC: The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the NHLBI, National Institutes of Health (NIH), Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN268201700003I, HHSN268201700004I and HHSN268201700005I). The authors thank the staff and participants of the ARIC study for their important contributions.
WGS for the Genome Sequencing Program (GSP) was funded by the National Human Genome Research Institute (NHGRI), the NHLBI, and the National Eye Institute (NEI). The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. The Centers for Common Disease Genomics (CCDG) program was supported by NHGRI and NHLBI, and WGS was performed at the Baylor College of Medicine Human Genome Sequencing Center (UM1 HG008898 and R01HL059367).
Paul S. de Vries was supported by American Heart Association grant number 18CDA34110116.
BioMe: The Mount Sinai BioMe Biobank has been supported by The Andrea and Charles Bronfman Philanthropies and in part by Federal funds from the NHLBI and NHGRI (U01HG00638001; U01HG007417; R56HG010297; R01DK110113; R01DK107786; X01HL134588). We thank all participants in the Mount Sinai Biobank. We also thank all our recruiters who have assisted and continue to assist in data collection and management and are grateful for the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai.
CARDIA: CARDIA is conducted and supported by the NHLBI in collaboration with the University of Alabama at Birmingham (HHSN268201800005I & HHSN268201800007I), Northwestern University (HHSN268201800003I), University of Minnesota (HHSN268201800006I), and Kaiser Foundation Research Institute (HHSN268201800004I).
WGS for the TOPMed program was supported by the NHLBI. WGS for the CARDIA study (phs001612) was performed at the Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I. Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1; contract HHSN268201800001I).
CFS: CFS was supported by the NHLBI (R35HL135818, R01HL113338, R01HL098433, and R01HL46380).
CHS: This research was supported by contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, 75N92021D00006, HL105756 and grants U01HL080295 and U01HL130114 from the National Heart, Lung, and Blood Institute (NHLBI), with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
FHS: Support for the FHS was provided by the NIH contract N01-HC-25195 (Boston University). We would also like to thank the FHS participants and families who participate in FHS research. In addition, we acknowledge the contributions of the research scientists and staffs of the Division of Intramural Research, NHLBI, NIH and Boston University.
GENOA: Support for the Genetic Epidemiology Network of Arteriopathy (GENOA) was provided by the National Heart, Lung and Blood Institute (U01 HL054457, U01 HL054464, U01 HL054481, R01 HL119443, and R01 HL087660) of the National Institutes of Health. DNA extraction for “NHLBI TOPMed: Genetic Epidemiology Network of Arteriopathy” (phs001345) was performed at the Mayo Clinic Genotyping Core, and WGS was performed at the DNA Sequencing and Gene Analysis Center at the University of Washington (3R01HL055673-18S1) and the Broad Institute (HHSN268201500014C). We would also like to thank the GENOA participants.
GenSalt: GenSalt was supported by research grants (U01HL072507, R01HL087263, and R01HL090682) from the NHLBI and partially supported by the National Institute of General Medical Sciences of the NIH under Award Number P20GM109036 and the Collins C. Diboll Private Foundation, New Orleans, LA.
GenSalt investigator, Tanika N. Kelly, is supported by NHLBI grant 5U01HL120393.
GeneSTAR: GeneSTAR was supported by grants from the NIH/NHLBI (U01 HL72518, HL087698, HL49762, HL58625, HL071025, HL112064), the NIH/National Institute of Nursing Research (NR0224103), and by a grant from the NIH/National Center for Research Resources (M01-RR000052) to the Johns Hopkins General Clinical Research Center.
HCHS-SOL: The authors thank the staff and participants of HCHS/SOL for their important contributions. A complete list of HCHS/SOL staff and investigators can be found at http://www.cscc.unc.edu/hchs/. The Hispanic Community Health Study/Study of Latinos is a collaborative study supported by contracts from the NHLBI to the University of North Carolina (HHSN268201300001I / N01-HC-65233), University of Miami (HHSN268201300004I / N01-HC-65234), Albert Einstein College of Medicine (HHSN268201300002I / N01-HC-65235), University of Illinois at Chicago – HHSN268201300003I / N01-HC-65236 Northwestern Univ), and San Diego State University (HHSN268201300005I / N01-HC-65237). The following Institutes/Centers/Offices have contributed to the HCHS/SOL through a transfer of funds to the NHLBI: National Institute on Minority Health and Health Disparities (NIMHD), National Institute on Deafness and Other Communication Disorders, National Institute of Dental and Craniofacial Research (NIDCR), National Institute of Diabetes and Digestive and Kidney Diseases, National Institute of Neurological Disorders and Stroke, NIH Institution-Office of Dietary Supplements. The Genetic Analysis Center at the University of Washington was supported by NHLBI and NIDCR contracts (HHSN268201300005C AM03 and MOD03). WGS for “NHLBI TOPMed: Whole Genome Sequencing and Related Phenotypes in the HCHS/SOL” (phs001395) was performed at the Baylor College of Medicine Human Genome Sequencing Center (HHSN268201600033I).
HyperGEN: We thank the HyperGEN Study participants and investigators for their significant contributions. HyperGEN is part of the NHLBI Family Blood Pressure Program; collection of the data represented here was supported by grants U01 HL054472, U01 HL054473, U01 HL054495, and U01 HL054509. The HyperGEN: Genetics of Left Ventricular Hypertrophy Study was supported by NHLBI grant R01 HL055673 with whole-genome sequencing made possible by supplement -18S1.
ARIC study investigator, Paul Stefan de Vries, was independently supported by American Heart Association grant number 18CDA34110116.
JHS: The JHS is supported and conducted in collaboration with Jackson State University (HHSN268201800013I), Tougaloo College (HHSN268201800014I), the Mississippi State Department of Health (HHSN268201800015I/HHSN26800001) and the University of Mississippi Medical Center (HHSN268201800010I, HHSN268201800011I and HHSN268201800012I) contracts from the NHLBI and the NIMHD. The authors also wish to thank the staffs and participants of the JHS. JHS study investigator, Adam P. Bress was supported by 1K01HL133468-01 from the NHLBI, Bethesda, MD.
The project described was also supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant KL2TR002490 (Laura M. Raffield).
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
MESA: WGS for the TOPMed program was supported by the NHLBI. WGS for “NHLBI TOPMed: Multi-Ethnic Study of Atherosclerosis (MESA)” (phs001416.v1.p1) was performed at the Broad Institute of MIT and Harvard (3U54HG003067-13S1). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1). Phenotype harmonization, data management, sample-identity QC, and general study coordination, were provided by the TOPMed Data Coordinating Center (3R01HL-120393-02S1). MESA and the MESA SHARe project are conducted and supported by the NHLBI in collaboration with MESA investigators. Support for MESA is provided by contracts 75N92020D00001, HHSN268201500003I, N01-HC-95159, 75N92020D00005, N01-HC-95160, 75N92020D00002, N01-HC-95161, 75N92020D00003, N01-HC-95162, 75N92020D00006, N01-HC-95163, 75N92020D00004, N01-HC-95164, 75N92020D00007, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420. Funding for SHARe genotyping was provided by NHLBI Contract N02-HL-64278. Genotyping was performed at Affymetrix (Santa Clara, California, USA) and the Broad Institute of Harvard and MIT (Boston, Massachusetts, USA) using the Affymetrix Genome-Wide Human SNP Array 6.0. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. MESA Family is conducted and supported by the NHLBI in collaboration with MESA investigators. Support is provided by grants and contracts R01HL071051, R01HL071205, R01HL071250, R01HL071251, R01HL071258, R01HL071259, and by the National Center for Research Resources, Grant UL1RR033176. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
SAFS: Collection of the San Antonio Family Study data was supported in part by National Institutes of Health (NIH) grants R01 HL045522, MH078143, MH078111 and MH083824; and whole genome sequencing of SAFS subjects was supported by U01 DK085524 and R01 HL113323. We are very grateful to the participants of the San Antonio Family Study for their continued involvement in our research programs.
Samoan Study: The Samoan study is supported by NIH/NHLBI grants, R01HL093093 and R01HL133040. WGS for the TOPMed program was supported by the NHLBI. We acknowledge the support from the Samoan Ministry of Health and Ministry of Women, Community and Social Development, local village government officials, and the generosity of our research staff, and all participants.
The Samoan Obesity, Lifestyle, and Genetic adaptations (OLaGA) Study group is comprised of the following members (also in Additional file 8):
Brandon Blobner, Department of Human Genetics, University of Pittsburgh, Pittsburgh, PA, USA.
Jenna C. Carlson, Department of Biostatistics, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA, USA.
Ranjan Deka, Department of Environmental Health, University of Cincinnati, Cincinnati, OH, USA Nicola L. Hawley, Department of Chronic Disease Epidemiology, Yale University, New Haven, CT, USA Stephen T McGarvey, Department of Epidemiology and International Health Institute, and Department of Anthropology, Brown University, Providence, RI, USA Ryan L Minster, Department of Human Genetics, University of Pittsburgh, Pittsburgh, PA, USA Take Naseri, Ministry of Health, Government of Samoa, Apia, Samoa Muagututi‘a Sefuiva Reupena, Lutia i Puava ae Mapu i Fagalele, Apia, Samoa Daniel E. Weeks, Depts. of Human Genetics and Biostatistics, University of Pittsburgh, Pittsburgh, PA, USA.
THRV: THRV is supported by the NHLBI grant (R01HL111249). THRV is a collaborative study between Washington University in St. Louis, Lundquist Institute for Biomedical Innovation at Harbor UCLA Medical Center, University of Texas in Houston, Taichung Veterans General Hospital, Taipei Veterans General Hospital, Tri-Service General Hospital, National Health Research Institutes, National Taiwan University, and Baylor University. THRV is based (substantially) on the parent SAPPHIRe study, along with additional population-based and hospital-based cohorts. SAPPHIRe was supported by NHLBI grants (U01HL54527, U01HL54498) and Taiwan funds, and the other cohorts were supported by Taiwan funds.
UK Biobank: The TOPMed-imputed UK Biobank association tests were conducted using the UK Biobank resource through the application of Aravinda Chakravarti (project # 34201).
WHI: The WHI program is funded by the NHLBI through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C. The TOPMed component of WHI was funded by NIH award X01 HL139376. The authors would like to acknowledge contributions from WHI investigators: https://www-whi-org.s3.us-west-2.amazonaws.com/wp-content/uploads/WHI-Investigator-Short-List.pdf.
WHI investigator Nora Franceschini is funded by the NIH awards R01-MD012765, R01-DK117445-01A1 and R21-HL140385.
Funding
KYH was partially supported by grant T32 HL007567 from the National Heart, Lung, and Blood Institute (NHLBI). This work was supported by HL086694 from NHLBI, HG003054 and HG011052 from the National Human Genome Research Institute. The funders do not have a role in this study.
Author information
Authors and Affiliations
Consortia
Contributions
XZ designed and led the study. KYH performed all of the analyses and wrote the manuscript. TNK led the effort to harmonize the TOPMed phenotype data. HW, JL, and BEC contributed to the study design. LZ contributed to UKB data analysis. BDM, MEM, BDM, and YPCC designed and provided the TOPMed Amish data. EB, PSDV, and ACM designed and provided the TOPMed ARIC data. EK and RJFL designed and provided the TOPMed BioMe data. MF, LH, and DLJ designed and provided the TOPMed CARDIA data. BEC and SR designed and provided the TOPMed CFS data. JAB, KMR, BMP and RT designed and provided the TOPMed CHS data. SJW, CTL, CL, LAC, RSV, and DL designed and provided the TOPMed FHS data. LCB, RRK, LRY, and RAM designed and provided the TOPMed GeneSTAR data. TNK and JH designed and provided the TOPMed GenSalt data. RCK and KEN designed and provided the TOPMed HCHS_SOL data. LFB, MRI, JAS, WZ, SLRK, PAP, DKA and DCR designed and provided the TOPMed HyperGEN_GENOA data. APP, ERF, ALF, YG, PMM, SM, WP, DS, AC and LR designed and provided the TOPMed JHS data. YDIC, SSR, XG, JIR and KDT designed provided the TOPMed MESA data. RD, JB, and JEC designed and provided the TOPMed SAFS data. TN, STM, and MSR designed and provided the TOPMed Samoan data. DCR, XG, JIR, KDT, YCC, YJH, and WHHS designed and provided the TOPMed THRV data. TLA, NF, JH, LW, BS, CK, and APP designed and provided the TOPMed WHI data. AC acquired UK biobank data and contributed to the study design. The Samoan Obesity, the Lifestyle and Genetic Adaptations Study Group designed and provided the data for the Samoan Study. The NHLBI Trans-Omics for Precision Medicine Consortium sequenced, processed, analyzed and distributed the data for analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the institutional review board (IRB) at Case Western Reserve University. Each individual cohort study was approved by the appropriate IRB in the corresponding institute and written consent from each individual was obtained in the study.
Amish: All study protocols were approved by the institutional review board at the University of Maryland Baltimore. Informed consent was obtained from each study participant.
ARIC: The ARIC study was approved by the IRBs of the University of North Carolina at Chapel Hill, Univerity of Minnesota, and Johns Hopkins University.
BioMe: The BioMe cohort was approved by the Institutional Review Board at the Icahn School of Medicine at Mount Sinai. All BioMe participants provided written, informed consent for genomic data sharing.
CARDIA: All CARDIA participants provided informed consent, and the study was approved by the Institutional Review Boards of the University of Alabama at Birmingham and the University of Texas Health Science Center at Houston.
CFS: Cleveland Family Study was approved by the Institutional Review Board (IRB) of Case Western Reserve University and Mass General Brigham (formerly Partners HealthCare). Written informed consent was obtained from all participants.
CHS: All CHS participants provided informed consent, and the study was approved by the Institutional Review Board [or ethics review committee] of University Washington.
FHS: The Framingham Heart Study was approved by the Institutional Review Board of the Boston University Medical Center. All study participants provided written informed consent.
GENOA: The Framingham Heart Study was approved by the Institutional Review Board of the Boston University Medical Center. All study participants provided written informed consent.
GenSalt: All subjects provided informed consent and the GenSalt study was approved by the Institutional Review Board (IRB) of all participating institutes in the US and China.
GeneSTAR: All participants provided written informed consent and the study was approved by the Johns Hopkins Medicine Institutional Review Board.
HCHS-SOL: This study was approved by the institutional review boards (IRBs) at each field center, where all participants gave written informed consent, and by the Non-Biomedical IRB at the University of North Carolina at Chapel Hill, to the HCHS/SOL Data Coordinating Center. All IRBs approving the study are: Non-Biomedical IRB at the University of North Carolina at Chapel Hill. Chapel Hill, NC; Einstein IRB at the Albert Einstein College of Medicine of Yeshiva University. Bronx, NY; IRB at Office for the Protection of Research Subjects (OPRS), University of Illinois at Chicago. Chicago, IL; Human Subject Research Office, University of Miami. Miami, FL; Institutional Review Board of San Diego State University. San Diego, CA.
HyperGEN: All HyperGEN participants provided informed consent, and the study was approved by the Institutional Review Board of the University of Kentucky.
JHS: The JHS study was approved by Jackson State University, Tougaloo College, and the University of Mississippi Medical Center IRBs, and all participants provided written informed consent.
MESA: All MESA participants provided written informed consent, and the study was approved by the Institutional Review Boards at The Lundquist Institute (formerly Los Angeles BioMedical Research Institute) at Harbor-UCLA Medical Center, University of Washington, Wake Forest School of Medicine, Northwestern University, University of Minnesota, Columbia University, and Johns Hopkins University.
SAFS: All SAFS participants provided informed consent, and the study was approved by Institutional Review Board at the University of Texas Rio Grande Valley.
Samoan Study: All Samoan study participants provided written informed consent, and the study was approved by the Institutional Review Board at Brown University, and the Health Research Committee of the Samoa Ministry of Health.
THRV: All THRV participants provided informed consent, and the study was approved by the Institutional Review Board at The Lundquist Institute (formerly Los Angeles BioMedical Research Institute) at Harbor-UCLA Medical Center. All THRV participants provided informed consent, and the study was approved by the Institutional Review Board at Washington University in St. Louis.
WHI: All WHI participants provided informed consent and the study was approved by the Institutional Review Board (IRB) of the Fred Hutchinson Cancer Research Center.
Consent for publication
Not applicable.
Competing interests
Karen Y. He is currently an employee of The Janssen Pharmaceutical Companies of Johnson & Johnson. This publication does not represent the views of her employer. Adam P. Bress receives research support to his institution from Novartis, Amgen, and Amarin. Bruce M. Psaty serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplemental Materials & Methods.
Additional file 2: Table S1.
Characteristics of UK Biobank European samples.
Additional file 3: Fig. S1.
TOPMed Freeze 8 phenotype distributions in African Americans.
Additional file 4: Fig. S2.
TOPMed Freeze 8 phenotype distributions in European Americans.
Additional file 5: Fig. S3.
TOPMed Freeze 8 phenotype distributions in East Asian/Asian Americans.
Additional file 6: Fig. S4.
TOPMed Freeze 8 phenotype distributions in Hispanic Americans.
Additional file 7: Fig. S5.
TOPMed Freeze 8 phenotype distributions in Samoans.
Additional file 8.
Members of the Samoan Obesity, Lifestyle and Genetic Adaptations Study (OLaGA) Group.
Additional file 9.
Members of the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, K.Y., Kelly, T.N., Wang, H. et al. Rare coding variants in RCN3 are associated with blood pressure. BMC Genomics 23, 148 (2022). https://doi.org/10.1186/s12864-022-08356-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08356-4