
Abstract
Background
Many studies on long chain non-coding RNAs (lncRNAs) are published in recent years. But the roles of lncRNAs in aortic dissection (AD) are still unclear and should be further examined. The present work focused on determining the molecular mechanisms underlying lncRNAs regulation in aortic dissection on the basis of the lncRNA-miRNA-mRNA competing endogenous RNA (ceRNA) network.
Methods
This study collected the lncRNAs (GSE52093), mRNAs (GSE52093) and miRNAs (GSE92427) expression data within human tissue samples with aortic dissection group and normal group based on Gene Expression Omnibus (GEO) database.
Results
This study identified three differentially expressed lncRNAs (DELs), 19 differentially expressed miRNAs (DEmiRs) and 1046 differentially expressed mRNAs (DEGs) identified regarding aortic dissection. Furthermore, we constructed a lncRNA-miRNA-mRNA network through three lncRNAs (including two with up-regulation and one with down-regulation), five miRNAs (five with up-regulation), as well as 211 mRNAs (including 103 with up-regulation and 108 with down-regulation). Simultaneously, we conducted functional enrichment and pathway analyses on genes within the as-constructed ceRNA network. According to our PPI/ceRNA network and functional enrichment analysis results, four critical genes were found (E2F2, IGF1R, BDNF and PPP2R1B). In addition, E2F2 level was possibly modulated via lncRNA FAM87A-hsa-miR-31-5p/hsa-miR-7-5p or lncRNA C9orf106-hsa-miR-7-5p. The expression of IGF1R may be regulated by lncRNA FAM87A-hsa-miR-16-5p/hsa-miR-7-5p or lncRNA C9orf106-hsa-miR-7-5p.
Conclusion
In conclusion, the ceRNA interaction axis we identified is a potentially critical target for treating AD. Our results shed more lights on the possible pathogenic mechanism in AD using a lncRNA-associated ceRNA network.
Similar content being viewed by others

Introduction
Aortic dissection (AD) is a severe aortic disorder involving destruction of aortic wall medial layer, which can induce separation between intima and adventitia, and track blood within the dissection plate in the medial layer, thereby inducing the formation of true and false lumens in the aortic wall. Although the research on aortic dissection has gradually increased and many innovations have been made in research programs, the treatment of thoracic aortic dissection is still extremely challenging [1, 2]. Aortic dissections are more common in the non-whites population of elderly men [3], and the incidence rate increases sharply over the age of 50. People between the ages of 50 and 70 are at the highest risk, and increasing age was the important variable associated with AD long-term mortality [4]. In the meanwhile, because it is often misdiagnosed when it appears, it is difficult to assess the exact incidence [5]. Although studies have demonstrated that the diagnostic techniques and treatments for thoracic AD cases are improving, the mortality and morbidity are high, so early diagnosis and treatment are very necessary [6].
The current research always draws their attention to the technical means and pathogenesis of aortic dissection. More and more recent studies have suggested that non-coding RNAs (ncRNAs), including long noncoding RNAs (lncRNAs) and microRNAs (miRNAs), exert decisive roles in the development of cardiovascular diseases (CVDs) [7, 8]. lncRNAs are the ncRNAs that are over 200 bp in length, which can be used as an important type of regulatory molecule in the human genome to perform its biological functions in various ways. Many studies have shown that IncRNA can also be used to be the competitive endogenous RNA (ceRNA) as the miRNA sponges and participate in regulating the expression of target genes [9, 10]. miRNAs shows negative regulation on protein-coding genes through combining with complementary sequences [11]. As a result, the lncRNA-miRNA-mRNA interaction has a vital effect on CVD development [12, 13].
Ren and colleagues revealed that miR-193b-3p and lncRNA H19 had certain effect on vascular smooth muscle cell (VSMC) migration and proliferation, which facilitated to generate new thoughts in AD management [14]. Recently, Zhang et al. carried out a study in order to prove lncRNA XIST’s effect on AD pathogenic mechanism and identify its corresponding pathway, with the findings showing that lncRNA XIST regulated smooth muscle cell proliferation and apoptosis through the sponge of miR-17 and the regulation of the subsequent downstream PTEN to affect the development of aortic dissection in mice [15]. Simultaneously, Zhao and colleagues suggested that lncRNA CDKN2B-AS1 regulated STAT3 level through suppressing miR-320d to regulate human VSMC apoptosis and proliferation [16]. To sum up, lncRNA-miRNA-mRNA regulatory network exerts a vital part in AD genesis and progression while the lncRNA targets, roles and underlying mechanisms within different tissues and cells of AD have not been reported. At the same time, there are few reports about the ceRNA regulation mechanism of lncRNA-miRNA related to AD and the interaction between ncRNAs.
In this study, lncRNAs together with the corresponding mechanisms of action within human tissue from AD cases were explored by means of bioinformatic analysis. Firstly, this study applied Gene Expression Omnibus (GEO) database for obtaining the AD-related lncRNA, mRNA and miRNA expression data. Thereafter, we discovered differentially expressed genes (DEGs), differentially expressed miRNAs (DEmiRs), and differentially expressed lncRNAs (DELs) using RStudio. Cytoscape 3.7.2 was utilized to construct a lncRNA-miRNA-mRNA network, followed by the construction of a PPI network.
The present study aims to further screen the key lncRNA-miRNA-mRNA ceRNA axis within AD with microarray data collected from public databases and bioinformatics methods. Besides, our results shed more lights on the AD molecular mechanism, thus providing a novel direction for targeted therapy of AD. To further examine the key gene regulatory axis, we established a lncRNA-miRNA-mRNA regulatory subnetwork.
Materials and methods
Microarray data and data processing
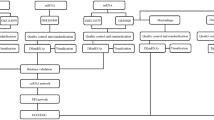
Two datasets were obtained by setting the screening criteria for the species type as “Homo sapiens”, from GEO database of National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/geo/), with such keywords being searched as “Aortic dissection” (AD). Then select the study type as " Expression profiling by array", so that we get the series matrix files and platform file of non-coding RNA. as a result, a total of two datasets were included in this study, namely GSE52093 and GSE92427. As for lncRNA and mRNA expression data (GSE52093 dataset), relative to normal aortic tissue in control group, the experimental group collected the ascending aorta of patients with acute Stanford type A aortic dissection to identify differentially expressed genes. The microRNA microarray is used for expression profiling analysis from subjects of 24 plasma samples (including acute aortic dissection and healthy subjects). Each dataset contains aorta dissected group and aorta normal group. Simultaneously, we applied GEO database for downloading series matrix files and expressive data. The data flow mechanism diagram is displayed in (Figure 1).

Flow diagram of data processing. The difference expression of two datasets (miRNA dataset, lncRNA dataset and mRNA dataset) was analyzed, and then the intersection was selected; Construction of CeRNA network, protein-protein interaction network and functional enrichment analysis, and finally determine the key genes
DEGs analysis
DEGs were analyzed by R package “Linear Models for Microarray Data (limma)” function for datasets and online analysis software SPSSAU. SPSSAU (https://spssau.com/index.html) is an online data analysis and visualization software. We inputted the expression matrix data of two datasets into SPSSAU and got the difference between aorta dissected group and aorta normal group of two datasets. In addition, we also obtained the median, extremum and outliers between each sample.
For each dataset, the sva R package was used to remove batch effects and the GeneSoring GX software package was adopted to conduct the quantile normalization for preprocessing, and the annotation information of lncRNA, miRNA and mRNA was downloaded by series matrix files of GSE52093 and GSE92427 datasets. The selection criteria for GSE52093 dataset including log|FoldChange|>1 and adjustp<0.1 were regarded as threshold values. The selection criteria for GSE92427 dataset containing log|FoldChange|>1 and adjustp<0.05 were regarded as threshold values. There existed statistical significance for the selection of this threshold, and those genes that were up- and down-regulated can also be selected for performing the subsequent analysis. This indicates statistical significance. R software package was also adopted as a heat map and volcano map drawing of DELs, DEmiRs and DEmRNAs. Subsequently, these DELs, DEmiRs and DEmRNAs between two databases were classified as the up-regulation or down-regulation group. These data would be used in the following ceRNA network construction and protein interaction network construction.
Prediction of miRNA-mRNA and lncRNA-miRNA pairs
Interaction relationships between lncRNAs and miRNAs were predicted with R package and miRcode database. By using the R language program to output all the predicted results of DElncRNAs, and intersecting the output results with DEmiRs, the interaction pair of lncRNAs and miRNAs can be obtained. Cytoscape (version 3.7.2) was utilized for visualizing forecast results.
Similarly, we use the R package to predict the miRNAs in the lncRNA-miRNA interaction pair, and intersect the prediction result with the DEmRNAs. As a result, the miRNA-mRNA interaction pair can be obtained. The difference is that we use three databases when predicting mRNAs, and we extract the results with 2 occurrences in the three databases as our prediction result.
Interactions between lncRNAs and miRNAs were analyzed on the basis of lncRNA target prediction databases shown below:
miRcode (http://www.mircode.org/).
Interactions between miRNAs and mRNAs were analyzed in line with 3 miRNA target prediction databases:
-
miRTarBase (https://maayanlab.cloud/Harmonizome/resource/MiRTarBase),
-
TargetScan (http://www.targetscan.org/vert_72/),
-
miRDB (http://mirdb.org/).
-
Perl (version 5.32.0) was adopted to analyze data.
Establishment of the ceRNA network of lncRNA-miRNA-mRNA
This study established a ceRNA network with related DELs by using commonly interactive miRNAs with DELs and DEmRNAs. Cytoscape (version 3.7.2) was later utilized to visualize the ceRNA network of lncRNA/miRNA/mRNA.
PPI network analysis and key gene identification
The plug-in CytoHubba in the Cytoscape software is a visualization software that obtains the dense relationship through the degree, closeness centrality and betweenness centrality algorithms. Those hub genes in ceRNA network were identified by CytoHubba.
The present study adopted the Search Tool for the Retrieval of interacting Genes/Proteins (STRING; version 11.0) for retrieving protein interactions between DEGs identified in GSE52093. After the points without interaction were hidden, the data were imported into Cytoscape (version 3.7.2) for visualizing the protein–protein interaction (PPI) network. CytoHubba was used to identifiy the hub genes of PPI network.
The key genes were determined by taking the intersection of hub genes in the ceRNA network and PPI network. Then, we obtained the key genes for follow-up analysis.
Functional enrichment (DEGs) and differential expression analysis (key genes)
Gene ontology (GO) functional annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were done for analyzing DEGs by using R language software package (Bioconductor and pathview) and DAVID online tool (version 6.8). P<0.05 indicated that the pathways or GO biological process terms were significantly enriched.
The present work adopted GraphPad Prism 8 (version 8.0.1) to visualize key DEGs in different datasets.
Results
Differential expression analysis
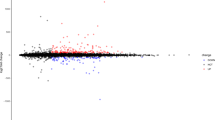
In this study, we imported the corrected expression matrix data into the SPSSAU online software, and the resulting box plot was shown in (Figure 2A and B). According to the cutoff criteria of GSE52093 (log|FoldChange|>1, adjustp<0.1) and the cutoff criteria of GSE92427 (log|FoldChange|>1, adjustp<0.05), volcano map and heat map for differential expressed lncRNAs, miRNAs and mRNAs were obtained (Figure 2C, D, E and F). The GSE52093 dataset included lncRNA and mRNA data, in which three DELs (two with up-regulation and one with down-regulation) and 1046 DEmRNAs (540 with up-regulation and 506 wtih down-regulation) were discovered through the comparison of gene expression data between AD and control samples. Altogether 19 DEmiRs of GSE92427 dataset (15 upregulated and four downregulated) were identified from plasma samples including acute aortic dissection and healthy subjects. The most significant differentially expressed genes were shown in (Table 1).

Differentially expressed analysis of LncRNAs, miRNAs and mRNAs. (A) Box plot of GSE52093, lncRNAs and mRNAs;(B) Box plot of GSE92427, miRNAs;(C) Volcano map of GSE52093, lncRNAs and mRNAs;(D) Volcano map of GSE92427, miRNAs;(E) heatmap analysis of GSE52093, lncRNAs and mRNAs;(F) heatmap analysis of GSE52093, miRNAs. Differentially expressed LncRNA and mRNA molecules were screened under the cut-off criteria log|FoldChange| > 1 and adjustp < 0.1, Differentially expressed miRNA molecules were screened under the cut-off criteria log|FoldChange| > 1 and adjustp < 0.05
Forecast of miRNA-mRNA and lncRNA-miRNA pairs
Interactive relationships between DELs and miRNAs were predicted with lncRNA mircode and R language script. We acquired the common miRNAs through the intersection of estimated DELs with DemiRs, while interactions of LncRNAs with miRNAs were predicted by LncBase Predicted v.2. LncRNA FAM87A can bind to hsa-miR-338-3p and hsa-miR-193b-3p. LncRNA UCA1 can bind to hsa-miR-1279 and hsa-miR-455-5p (Figure 3A). Several DemiRs binding sites that can bind to lncRNA were presented in (Figure 3B).

The analysis view of lncRNA, miRNA, mRNA target prediction and ceRNA network. (A) The blue quadrilateral represented lncRNA, the red oval represented miRNA that could interact with lncRNA;(B) lncRNA and predicted miRNA, miRNA and predicted mRNA binding site;(C) lncRNA-miRNA-mRNA ceRNA network, The quadrilateral: lncRNA, the triangle: miRNA, and the oval: mRNA. Red: up-regulation, blue: down-regulation
Compared with lncRNA prediction, the interaction relationships between DemiRs of binding DELs and DEGs were predicted with three databases form miRTarBase, TargetScan and miRDB. The screening conditions through the R language software package were set as follows. If the intersection was greater than 2, the DEGs interacting with DemiRs of binding DELs were exported. For instance, hsa-miR-107 can bind to SNGG (Synuclein Gamma) and CC2D1B (Coiled-Coil and C2 Domain Containing 1B). In addition, hsa-miR-16-5p can bind to LARP1 (La Ribonucleoprotein 1, Translational Regulator) and KCTD8 (Potassium Channel Tetramerization Domain Containing 8) (Table 2). MicroT-CDS was employed for predicting the associations of miRNAs with mRNAs. Several DEGs binding sites that can bind to DemiRs were illuminated in (Figure 3B).
lncRNA-miRNA-mRNA ceRNA network construction
Cytoscape (version 3.7.2) was employed to visualize the interaction between three DELs (two with up-regulation and one with down-regulation), five DemiRs (five with up-regulation) and 211 DEGs (103 with up-regulation and 108 with down-regulation). Therefore, the lncRNA-miRNA-mRNA ceRNA network was built, where LncRNA C9orf106 can be linked by one DEmiR and 51 DEGs, LncRNA UCA1 can be linked by one DEmiR and 42 DEGs and LncRNA FAM87A can be linked by three DEmiR and 185 DEGs (Figure 3C). It includes LncRNA C9orf106-hsa-miR-7-5p-MYLK (Myosin Light Chain Kinase)/TJAP1 (Tight Junction Associated Protein 1); LncRNA FAM87A- hsa-miR-16-5p-MYC (MYC Proto-Oncogene, BHLH Transcription Factor)/H2AFX (H2A.X Variant Histone), LncRNA UCA1- hsa-miR-107-CDCA4(Cell Division Cycle Associated 4)/PDK4(Pyruvate Dehydrogenase Kinase 4) and LncRNA FAM87A-hsa-miR-17-3p-LRP8 (LDL Receptor Related Protein 8)/TFRC (Transferrin Receptor).
PPI network analysis and key gene prediction
The top 30 DEGs of ceRNA network acquired based on degree, closeness centrality and betweenness centrality algorithms using cytoHubba were visualized in (Figure 4A). When uploading the DEGs identified in ceRNA network to STRING website, 395 interaction relationships were found to be present in 211 DEGs incorporated in constructing a PPI network. In the as-constructed network, E2F2(E2F Transcription Factor 2) could interact with CCNE1(Cyclin E1). IGF1R (Insulin Like Growth Factor 1 Receptor) could interact with BNDF (Brain Derived Neurotrophic Factor). BDNF could interact with IGF1R and PPP1R9B (Protein Phosphatase 1 Regulatory Subunit 9B) (Figure 4B). The top 30 DEGs of PPI network obtained by cytoHubba were visualized in (Figure 4C). There were four key gens (E2F2, IGF1R, BDNF and PPP1R9B) which were determined by Venn diagram of ceRNA network and PPI network (Figure 4D).

PPI network and key genes analysis. (A) Relationship network diagram of hub genes from ceRNA network;(B) PPI network. Red: up-regulated, Blue: down-regulated;(C) Relationship network diagram of hub genes from PPI network;(D) Venn diagram of key genes
Functional enrichment and differential expression analysis
In the present study, the DEGs used to analyze GO/KEGG were imported into DAVID website and R language script. The results of DAVID showed the functions and pathways involved in DEGs: GO (such as plasma membrane and protein binding) and KEGG (like pathways in cancer and PI3K-Akt signaling pathway) (Table 3). The results of R language software showed the functions and pathways involved in DEGs: GO (such as nuclear receptor activity and steroid binding) and KEGG (such as proteoglycans in cancer and PI3K-Akt signaling pathway) (Figure 5).

GO/KEGG analysis of DEGs. (A) Bubble chart of GO. Each bubble represented a function;(B) Bubble chart of KEGG. Each bubble represents a pathway;(C) Histogram of GO;(D) Histogram of KEGG
To accurately explore the differential expression of key genes in the three datasets, four key genes were visualized in (Figure 6). In the GSE52093 datasets, E2F2 and PPP1R9B were up-regulated while IGF1R and BDNF were down-regulated, further revealing the underlying mechanisms for key genes.

Difference Analysis Box Plot of key genes. (A) Expression of E2F2 between normal aorta and aortic dissection; (B) Expression of IGF1R between normal aorta and aortic dissection; (C) Expression of BDNF between normal aorta and aortic dissection; (D) Expression of PPP2R1B between normal aorta and aortic dissection. Green represented normal aortic samples, and purple represented aortic dissection samples
Discussion
In normal circumstances, the 3 layers of aortic wall (e.g. intima, media, and adventitia) should maintain the complete structures and physiological functions for maintaining the aortic wall stability and managing the great influence resulting from blood flow [17]. The AD pathogenic mechanisms are vascular inflammation, matrix metalloproteinases (MMPs) activity and change in vascular smooth muscle cells (VSMCs) phenotype [18]. Endothelial cell (EC) injury promotes the occurrence of vascular inflammation modulated via immune response. Afterwards, it will activate MMP, the extracellular matrix (ECM) degrading enzyme [19, 20]. Meanwhile, AD is also associated with broad alterations of VSMC phenotype and vascular wall at molecular level [21, 22].
LncRNAs, with over 200 nucleotides in length, constitute the transcript family without protein coding functions [23]. They are suggested to play roles of ceRNAs for miRNAs; in other words, lncRNAs may play roles of miRNA “decoys” for regulating gene levels [12, 24]. As suggetsed by the theory, lncRNAs act as natural sponge for the competition adsorption of certain miRNAs and reduction of miRNA binding to corresponding target genes, thus resulting in alterations of miRNA target gene expression [25]. However, it remains unclear about whether the abnormal lncRNAs play roles of ceRNA for certain miRNAs and have certain influence on arterial wall by the indirect regulation of target mRNA expression in the process of AD occurrence.
Recently, lncRNAs have been discovered to be related to CVD genesis and progression. Recent study revealed a pathogenic H19 induce aneurysm by inflammatory pathway related to the formation of AAA, and this has offered a novel treatment for AAA [26]. Meanwhile, Kumar and colleagues investigated the effects of ncRNAs together with the target genes from the perspective of their functions within AAA. They also discussed those animal models adopted to mechanically understand AAA and the possible effects of miRNAs and lncRNAs as diagnostic biomarker and therapeutic targets [27]. Several lncRNAs are suggested to have important functions in aorta-related disease pathogenesis, such as AD [28]. Few lncRNAs show obvious spatiotemporal expression and specificity in the process of tissue growth and differentiation; As a result, they were the favorable diagnostic biomarkers for AD.
E2F belongs to the transcriptional factor family, which functions in controlling G(1)/S transition. In addition, certain E2F components have been recently suggested to regulate functions in addition to cell cycle, like apoptosis induction [29]. Fujita et al. found that E2F5 was induced by fetal bovine serum in VSMC, and E2F5 inducibility has unique function in VSMC, which is related to the feedback modulation for some cell activities in cell proliferation [30]. Nevertheless, Angiotensin II (ANG II) increased E2F3 protein expression, rather than E2F-5 and E2F-1, but it did not increase the mRNA expression. The above alterations are related to the hyperplastic or hypertrophic responses to diverse stimuli or growth factors of VSMCs. Several studies suggested that modulation of E2F had certain effect on treating hypertension, atherosclerosis, restenosis following vascular damage and bypass graft failure [31, 32]. In precent study, E2F2 expression was possibly modulated via lncRNA FAM87A-hsa-miR-31-5p/hsa-miR-7-5p in human aortic dissection tissue. Recent study investigated the implication overexpression miR-7-5p, finding that miR-7-5p agomir transfection markedly suppressed mineralization of pulmonary artery smooth muscle cell matrix under hypoxic conditions [33].
It is still unclear about the AD pathogenesis, but more and more studies have suggested the important functions of ncRNAs within AD. In this study, the underlying molecular mechanisms of AD have been investigated based on the lncRNA-miRNA-mRNA ceRNA regulatory network. lncRNAs, mRNAs and miRNAs expression data are obtained from human tissues of dissection and normal groups. Wang et al. discovered the abnormally expressed miRNAs and lncRNAs within AD samples and identified the role of lncRNA OIP5-AS1 in exacerbating injuries to all the three layer of aortic wall in AD genesis and development by increasing TUB expression through the sponge of miR-143-3p [34]. As discovered by another study, lncRNA PVT1 level increased, yet miR-27b-3p level decreased within AD tissue. They reduced the lncRNA PVT1 level to suppress the migration, phenotype switch and viability of human aortic smooth muscle cells treated with growth factor-BB (PDGF-BB) through targeting miR-27b-3p [35]. For understanding the pathophysiological and biological mechanisms related to AD genesis and development, circRNAs, miRNAs, and lncRNAs with abnormal expression should be discovered and verified in relevant human AD samples or relevant animal models. Extraction of total RNA from critical types of cells, like immune cells, ECs, and SMCs, can shed more lights on alterations specific to cell type in the process of AD progression. When the differential expression is verified, it is necessary to test those mechanisms of ncRNAs in regulating AD progression in vitro and in vivo at molecular level.
Certain limitations should be noted in the present work. At first, although it has been shown that E2F2, IGF1R, and BDNF can cause arterial-related diseases, there is no research to prove the relationship between aortic dissection and these target gene. Secondly, the way of action has been predicted based on the measured RNA network, however, which has not been confirmed (dual luciferase reporter gene analysis, gene overexpression or gene knockout). Although several related genes have been screened out in the present study for the first time, further in vitro clinical research and in vivo experiments should be carried out to confirm its expression and functional mechanism in terms of AD.
Currently, little research is conducted to explore the lncRNA mechanism in AD. The present work has its certain strengths. For instance, it is the first study to construct the lncRNA-miRNA-mRNA network based on GEO database. Nonetheless, our findings were just obtained from bioinformatics analysis. Therefore, it is of necessity to conduct a thorough study for verifying the potential effects of those 7 axes within AD. In summary, we confirmed that the ceRNA networks, including the regulated networks, lncRNA FAM87A-hsa-miR-31-5p/hsa-miR-7-5p-E2F2, lncRNA C9orf106-hsa-miR-7-5p-IGF1R, and lncRNA UCA1-hsa-miR-107-BDNF, might be associated with the pathogenesis of and development of AD. Moreover, our study shed novel lights on the AD pathogenic mechanism. Nevertheless, ceRNA networks and their associations with AD should be validated.
Conclusions
In summary, the ceRNA interaction axis we identified is a potentially critical target for treating AD. According to the difference significance, PPI interaction correlation and regulation relationship, E2F2 axis (LncFAM87A-hsa-miR-7-5p/hsa-miR-31-5p-E2F2) may play a key role in the treatment of AD. Our results shed more lights on the possible pathogenic mechanism in AD using a lncRNA-associated ceRNA network.
Availability of data and materials
The datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database (Accession Number: GSE52093, GSE92427).
Abbreviations
- AD:
-
aortic dissection
- LncRNA:
-
Long non-coding RNA
- GO:
-
Gene ontology
- hsa:
-
Homo sapiens
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- mRNA:
-
Messenger RNA
References
Nienaber CA, Rousseau H, Eggebrecht H, Kische S, Fattori R, Rehders TC, Kundt G, Scheinert D, Czerny M, Kleinfeldt T, Zipfel B, Labrousse L, Ince H, INSTEAD Trial Randomized comparison of strategies for type B aortic dissection: the INvestigation of STEnt grafts in aortic dissection (INSTEAD) trial. Circulation 2009, 120(25):2519–2528, https://doi.org/10.1161/CIRCULATIONAHA.109.886408.
Saremi F, Hassani C, Lin LM, Lee C, Wilcox AG, Fleischman F, et al. Image predictors of treatment outcome after thoracic aortic dissection repair. Radiographics. 2018;38(7):1949–72. https://doi.org/10.1148/rg.2018180025.
Harris D, Klyushnenkova E, Kalsi R, Garrido D, Bhardwaj A, Rabin J, et al. Non-white race is an independent risk factor for hospitalization for aortic dissection. Ethn Dis. 2016;26(3):363–8. https://doi.org/10.18865/ed.26.3.363.
Olsson C, Thelin S, Stahle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation. 2006;114(24):2611–8. https://doi.org/10.1161/CIRCULATIONAHA.106.630400.
Salmasi MY, Al-Saadi N, Hartley P, Jarral OA, Raja S, Hussein M, et al. The risk of misdiagnosis in acute thoracic aortic dissection: a review of current guidelines. Heart. 2020;106(12):885–91. https://doi.org/10.1136/heartjnl-2019-316322.
Pape LA, Awais M, Woznicki EM, Suzuki T, Trimarchi S, Evangelista A, et al. Presentation, diagnosis, and outcomes of acute aortic dissection: 17-year trends from the international registry of acute aortic dissection. J Am Coll Cardiol. 2015;66(4):350–8. https://doi.org/10.1016/j.jacc.2015.05.029.
Liu CY, Zhang YH, Li RB, Zhou LY, An T, Zhang RC, et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat Commun. 2018;9(1):29. https://doi.org/10.1038/s41467-017-02280-y.
Lorenzen JM, Thum T. Long noncoding RNAs in kidney and cardiovascular diseases. Nat Rev Nephrol. 2016;12(6):360–73. https://doi.org/10.1038/nrneph.2016.51.
Kong X, Duan Y, Sang Y, Li Y, Zhang H, Liang Y, et al. LncRNA-CDC6 promotes breast cancer progression and function as ceRNA to target CDC6 by sponging microRNA-215. J Cell Physiol. 2019;234(6):9105–17. https://doi.org/10.1002/jcp.27587.
Paraskevopoulou MD, Hatzigeorgiou AG. Analyzing MiRNA-LncRNA interactions. Methods Mol Biol. 2016;1402:271–86. https://doi.org/10.1007/978-1-4939-3378-5_21.
Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79(1):351–79. https://doi.org/10.1146/annurev-biochem-060308-103103.
Huang Y. The novel regulatory role of lncRNA-miRNA-mRNA axis in cardiovascular diseases. J Cell Mol Med. 2018;22(12):5768–75. https://doi.org/10.1111/jcmm.13866.
Li FP, Lin DQ, Gao LY. LncRNA TUG1 promotes proliferation of vascular smooth muscle cell and atherosclerosis through regulating miRNA-21/PTEN axis. Eur Rev Med Pharmacol Sci. 2018;22(21):7439–47. https://doi.org/10.26355/eurrev_201811_16284.
Ren M, Wang T, Wei X, Wang Y, Ouyang C, Xie Y, Ye X, Han Z: LncRNA H19 regulates smooth muscle cell functions and participates in the development of aortic dissection through sponging miR-193b-3p. Biosci Rep 2021, 41(1), 41, 1, https://doi.org/10.1042/BSR20202298.
Zhang X, Wu H, Mai C, Qi Y. Long noncoding RNA XIST/miR-17/PTEN Axis modulates the proliferation and apoptosis of vascular smooth muscle cells to affect Stanford type a aortic dissection. J Cardiovasc Pharmacol. 2020;76(1):53–62. https://doi.org/10.1097/FJC.0000000000000835.
Zhao X, Cheng S, Li S, Li J, Bai X, Xi J. CDKN2B-AS1 aggravates the pathogenesis of human thoracic aortic dissection by sponge to miR-320d. J Cardiovasc Pharmacol. 2020;76(5):592–601. https://doi.org/10.1097/FJC.0000000000000907.
Kozun M, Kobielarz M, Chwilkowska A, Pezowicz C. The impact of development of atherosclerosis on delamination resistance of the thoracic aortic wall. J Mech Behav Biomed Mater. 2018;79:292–300. https://doi.org/10.1016/j.jmbbm.2018.01.009.
Silaschi M, Byrne J, Wendler O. Aortic dissection: medical, interventional and surgical management. Heart. 2017;103(1):78–87. https://doi.org/10.1136/heartjnl-2015-308284.
Luo F, Zhou XL, Li JJ, Hui RT. Inflammatory response is associated with aortic dissection. Ageing Res Rev. 2009;8(1):31–5. https://doi.org/10.1016/j.arr.2008.08.001.
Kurihara T, Shimizu-Hirota R, Shimoda M, Adachi T, Shimizu H, Weiss SJ, et al. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation. 2012;126(25):3070–80. https://doi.org/10.1161/CIRCULATIONAHA.112.097097.
Yang K, Ren J, Li X, Wang Z, Xue L, Cui S, et al. Prevention of aortic dissection and aneurysm via an ALDH2-mediated switch in vascular smooth muscle cell phenotype. Eur Heart J. 2020;41(26):2442–53. https://doi.org/10.1093/eurheartj/ehaa352.
Clement M, Chappell J, Raffort J, Lareyre F, Vandestienne M, Taylor AL, et al. Vascular smooth muscle cell plasticity and autophagy in dissecting aortic aneurysms. Arterioscler Thromb Vasc Biol. 2019;39(6):1149–59. https://doi.org/10.1161/ATVBAHA.118.311727.
Wang K, Liu CY, Zhou LY, Wang JX, Wang M, Zhao B, et al. APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nat Commun. 2015;6(1):6779. https://doi.org/10.1038/ncomms7779.
Botti G, Marra L, Malzone MG, Anniciello A, Botti C, Franco R, et al. LncRNA HOTAIR as prognostic circulating marker and potential therapeutic target in patients with tumor diseases. Curr Drug Targets. 2017;18(1):27–34. https://doi.org/10.2174/1389450117666151209122950.
Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73(13):2491–509. https://doi.org/10.1007/s00018-016-2174-5.
Sun Y, Zhong L, He X, Wang S, Lai Y, Wu W, et al. LncRNA H19 promotes vascular inflammation and abdominal aortic aneurysm formation by functioning as a competing endogenous RNA. J Mol Cell Cardiol. 2019;131:66–81. https://doi.org/10.1016/j.yjmcc.2019.04.004.
Kumar S, Boon RA, Maegdefessel L, Dimmeler S, Jo H. Role of noncoding RNAs in the pathogenesis of abdominal aortic aneurysm. Circ Res. 2019;124(4):619–30. https://doi.org/10.1161/CIRCRESAHA.118.312438.
Cheng M, Yang Y, Xin H, Li M, Zong T, He X, et al. Non-coding RNAs in aortic dissection: from biomarkers to therapeutic targets. J Cell Mol Med. 2020;24(20):11622–37. https://doi.org/10.1111/jcmm.15802.
Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature. 2000;407(6804):645–8. https://doi.org/10.1038/35036614.
Fujita N, Furukawa Y, Itabashi N, Okada K, Saito T, Ishibashi S. Differences in E2F subunit expression in quiescent and proliferating vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2002;283(1):H204–12. https://doi.org/10.1152/ajpheart.00545.2001.
Mann MJ, Whittemore AD, Donaldson MC, Belkin M, Conte MS, Polak JF, et al. Ex-vivo gene therapy of human vascular bypass grafts with E2F decoy: the PREVENT single-Centre, randomised, controlled trial. Lancet. 1999;354(9189):1493–8. https://doi.org/10.1016/S0140-6736(99)09405-2.
Morishita R, Gibbons GH, Horiuchi M, Ellison KE, Nakama M, Zhang L, et al. A gene therapy strategy using a transcription factor decoy of the E2F binding site inhibits smooth muscle proliferation in vivo. Proc Natl Acad Sci U S A. 1995;92(13):5855–9. https://doi.org/10.1073/pnas.92.13.5855.
Ma C, Gu R, Wang X, He S, Bai J, Zhang L, et al. circRNA CDR1as promotes pulmonary artery smooth muscle cell calcification by Upregulating CAMK2D and CNN3 via sponging miR-7-5p. Mol Ther Nucleic Acids. 2020;22:530–41. https://doi.org/10.1016/j.omtn.2020.09.018.
Wang P, Wang Z, Zhang M, Wu Q, Shi F. Lnc-OIP5-AS1 exacerbates aorta wall injury during the development of aortic dissection through upregulating TUB via sponging miR-143-3p. Life Sci. 2021;271:119199. https://doi.org/10.1016/j.lfs.2021.119199.
Li S, Zhao X, Cheng S, Li J, Bai X, Meng X. Downregulating long non-coding RNA PVT1 expression inhibited the viability, migration and phenotypic switch of PDGF-BB-treated human aortic smooth muscle cells via targeting miR-27b-3p. Hum Cell. 2021;34(2):335–48. https://doi.org/10.1007/s13577-020-00452-5.
Acknowledgements
Not Applicable.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities (grant number: DUT19RC(3)076), the National Natural Science Foundation of China (grant number: 81600370), and the China Postdoctoral Science Foundation (grant number: 2018M640270) for Yanshuo Han. This work was supported by the National Natural Science Foundation of China (grant: 81970402) for Jian Zhang.
Author information
Authors and Affiliations
Contributions
YSH and HZ: conceptualization. QYL, CC and SMT: investigation and validation. FXY and PPG: methodology. CB and XTS: software. YSH: supervision. HZ, JZ and YSH: writing, and original draft preparation. HZ, and YSH: writing, review and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, H., Bian, C., Tu, S. et al. Integrated analysis of lncRNA-miRNA-mRNA ceRNA network in human aortic dissection. BMC Genomics 22, 724 (2021). https://doi.org/10.1186/s12864-021-08012-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-08012-3