
Abstract
Background
Zebrafish is a popular animal model used for high-throughput screening of chemical hazards, however, investigations of transcriptomic mechanisms of toxicity are still needed. Here, our goal was to identify genes and biological pathways that Aryl Hydrocarbon Receptor 2 (AHR2) Activators and flame retardant chemicals (FRCs) alter in developing zebrafish. Taking advantage of a compendium of phenotypically-anchored RNA sequencing data collected from 48-h post fertilization (hpf) zebrafish, we inferred a co-expression network that grouped genes based on their transcriptional response.
Results
Genes responding to the FRCs and AHR2 Activators localized to distinct regions of the network, with FRCs inducing a broader response related to neurobehavior. AHR2 Activators centered in one region related to chemical stress responses. We also discovered several highly co-expressed genes in this module, including cyp1a, and we subsequently show that these genes are definitively within the AHR2 signaling pathway. Systematic removal of the two chemical types from the data, and analysis of network changes identified neurogenesis associated with FRCs, and regulation of vascular development associated with both chemical classes. We also identified highly connected genes responding specifically to each class that are potential biomarkers of exposure.
Conclusions
Overall, we created the first zebrafish chemical-specific gene co-expression network illuminating how chemicals alter the transcriptome relative to each other. In addition to our conclusions regarding FRCs and AHR2 Activators, our network can be leveraged by other studies investigating chemical mechanisms of toxicity.
Similar content being viewed by others

Background
With advancements in technology and medical science, various types of chemicals (xenobiotics, drugs, etc) are being applied to both the natural environment and human body. However, the majority of these chemicals are yet to be evaluated for their potential to cause adverse health effects. High-throughput (HTP) in vitro assays are popular methods used to estimate chemical toxicity and identify underlying molecular events [1]. Despite their application to determine whether a chemical class may be toxic, we still lack adequate knowledge of the mechanisms of toxicity of many chemicals [2] preventing us from connecting assay measurements to phenotypes. Animal models can be a more translatable way of revealing chemical hazard potential to humans, with both metabolism and integrated tissue systems. However, high cost and low throughput are often barriers to testing across a large chemical space. These barriers have been serious deficiencies in advancing chemical risk assessment, and thus a HTP and accurate prediction of which chemicals may be toxic is an essential next step in the evaluation of chemical safety [3].
A popular HTP animal model, zebrafish (D. rerio), by virtue of its rapid development [4] and high physiological and genetic similarity to humans [5], is being leveraged to identify chemical hazards [6] and to determine the molecular signaling events that precede adverse phenotypic outcomes [7]. One of the main -omics techniques that is applied to zebrafish is transcriptomics, which uses RNA sequencing analysis to provide an unbiased snapshot of gene expression changes associated with a particular chemical exposure [8]. Numerous transcriptomic studies in both developing and adult zebrafish leave no doubt that diverse chemical exposures often result in diverse gene expression changes [9,10,11]. Such studies have led to the discovery of transcripts important for the manifestation of higher level toxicity effects [12, 13] and to a large relational database of chemical-zebrafish transcriptome responses.
Much of this transcriptomic data collected from developing zebrafish has examined toxicants from a variety of different classes. Polycyclic aromatic hydrocarbons (PAHs), are a large class of ubiquitous environmental pollutants, and human exposure has been linked to cardiovascular disease, respiratory problems, carcinogenicity, and developmental deficits [14, 15]. The molecular initiating event of many PAHs is the aryl hydrocarbon receptor (AHR), and PAHs have been shown to bind all three orthologs of AHR in zebrafish, with AHR2 being the predominant receptor required for toxicity [16]. 2,3,7,8-Tetrachlorodibenzodioxin (TCDD) is a halogenated aromatic hydrocarbon and the best-characterized and most potent AHR2 ligand. It is widely used as a canonical xenobiotic ligand to study responses downstream of AHR activation [17]. Many studies have focused on the roles of the cytochrome P450 (CYP) metabolizing enzymes in PAH toxicity [18, 19], while other molecular signaling events downstream of the AHR remain largely unknown. Flame-retardant chemicals (FRCs) are a diverse class of chemicals including polybrominated diphenyl ethers (PBDEs) and organophosphate flame retardants (OPFRs), commonly applied in an additive manner to manufactured materials such as furniture, clothing, and electronics, and often found to leach into surrounding environments and human bodies [20, 21]. FRCs have been associated with neurodevelopmental effects [22], altered reproductive and thyroid function [23], and impacts on the immune and endocrine systems [24]. While many research groups have conducted transcriptomic studies in developing zebrafish exposed to PAHs and TCDD, only a limited number of FRC whole-genome expression studies have been published [25,26,27,28].
The standard use of transcriptomic data, and the approach used in many of the studies above, is to compare gene expression levels between a control and a chemical treatment. However, with a transcriptomic database of sufficient size [29], it is possible to perform a meta-analysis of sequencing data and construct a network of genes based on co-expression values of each gene pair [30, 31]. Co-expressed genes show a similar pattern of either direct or inverse co-expression across multiple conditions or biological replicates. Network analysis can be used to reveal important chemical targets in a biological system [32,33,34], and to indicate processes that are critical for or distinctive between responses to certain classes of chemicals [35, 36]. Network approaches also have a distinct advantage over standard control/condition comparisons in that data from several different studies, even those conducted under variable conditions, can be collected and integrated to produce a model formed from a compendium of many datasets. Network analysis can also highlight genes, pathways, and processes that may change their expression in a significant but subtle manner (less than the 2-fold cutoff normally applied to control/condition comparisons), expanding our ability to identify processes related to chemical class or phenotypic response. A whole transcriptome approach is thus far more efficient and informative to detect subtle but potentially important shifts in gene expression patterns that describe interactions between processes impacted by chemical exposure [37]. Such patterns would be missed by smaller scale targeted expression studies for specific biomarkers. Based on the large amount of transcriptomic data collected for zebrafish exposed to a variety of different chemicals, we are now able to apply these network approaches to zebrafish response to chemicals.
While phenotypic outcomes of many chemicals have been studied in zebrafish [6, 38], we still do not know many of the transcriptomic pathways and processes that are induced by these chemicals early in response. Additionally, we do not know to what degree different classes of chemicals may overlap in their transcriptomic response compared to their potentially similar phenotypic responses. To fill this gap in knowledge, the primary goal of our study was to utilize network analyses to discover both common and distinctive biological pathways that polycyclic aromatic hydrocarbons (PAHs), 2,3,7,8-Tetrachlorodibenzodioxin (TCDD), and several flame retardant chemicals (FRCs), alter in developing zebrafish. These chemicals were selected in part because transcriptomic data reflecting a similar experiment design was available. However, these treatments not only encompassed chemicals with unknown and likely distinct modes of action, but also included those with the same or similar modes of action, allowing us to compare the individually altered transcriptomes in a single network. Networks were inferred using a random forest method [31] applied to the compendium of both new and previously published zebrafish transcriptomic data. We identified specific portions of this network that represented tightly co-expressed genes enriched for certain pathways and responding to certain chemical types. The network approach used here enabled discovery of novel genes in the AHR2 signaling pathway as well as several genes and pathways associated with FRC exposure. The inference of this gene co-expression network for zebrafish not only provides novel insight into the transcriptional responses to chemical exposure, but can serve as a resource for other studies focused on transcriptomic coordination, predictive toxicology, and identification of chemical-specific biomarkers and processes of interest in this model organism.
Results
Characterization of full dataset
In this study, we sought to identify molecular signatures and biological pathways following exposures to a diverse group of chemicals. This was done using a large array of transcriptomic data combined with the first chemical-focused gene co-expression network inferred using zebrafish. Transcriptomic data from 48-h post fertilization (hpf) zebrafish included several previously published studies [7, 39,40,41] in addition to new unpublished datasets. Details on these datasets including the exposure concentrations and the number of differentially expressed genes (DEGs) are included in Supplementary Table S1. To gain an overview of transcriptomic patterns from the 33 unique chemical treatments, we first used Ward’s method of hierarchical clustering of the 10,346 genes that responded significantly to at least a subset of chemicals. We observed that the treatments naturally clustered into six groups based on log2FC values (Fig. 1). Clusters 1 (orange) and 2 (red) represented all the flame retardant chemicals (FRCs) examined and were also more similar to each other than to other clusters in the dataset, showing that the overall transcriptomic response to FRCs is distinct from PAH and TCDD responses. In addition to differences in transcriptomic response based on chemical type, we also found that when we compared our clustering to our lab’s previous studies investigating the morphology and behavior effects caused by FRC exposure [6, 41], there were differences between transcriptomic response and developmental toxicity phenotype. For example, IPP exposure caused both morphology and behavior malformations at 120 hpf and yet, it clustered here with the relatively benign FRCs. While several PAHs in Cluster 3 (black) (9-MA, 3-NF, 2-MN, 1,5-DMN, Carbazole, and Anthracene) were previously determined to cause modest or no phenotypic responses in developing zebrafish, other chemicals in this cluster including 4 h-CPdefP, 7,12-B [a] AQ, and TCDD are known to cause overt developmental toxicity [7, 38, 42]. These observations demonstrated that transcriptional response and phenotypic outcome are often not strongly correlated. Clusters 4 and 5 consisted of the remaining PAHs in this study. We noted that within all the PAHs and TCDD, the chemicals known to predominantly activate AHR2 (“AHR Activators”, see Supplementary Table S1) did not group together. The hierarchical clustering analysis presented here indicates that chemical type is the strongest driver of overall transcriptomic response, rather than known mechanism of action or magnitude of developmental toxicity within the chemical classes.

Heatmap of 48-hpf zebrafish transcriptomic response to ten Flame Retardant Chemical (FRC), 22 Polycyclic Aromatic Hydrocarbon (PAH), and 1 TCDD treatment. Average log2 fold change (log2FC) values for each chemical treatment (y-axis) are shown, with yellow indicating higher expression and blue indicating lower expression compared to each chemical’s DMSO control. Both genes and treatments are grouped via hierarchical clustering with clusters of treatments (Clusters 1–6) indicated by colors on the left
Global analysis of full co-expression network
We next inferred a co-expression network using the random forest method, GENIE3 [31], and grouped genes into one of 23 different modules ranging in size from 14 to 425 genes. We chose to use GENIE3 as we have found it be highly accurate in our previous work [43], as well as in other studies that directly compare gene co-expression network methods [29]. While these previous studies were in bacterial or human systems, we used similar methods like those we already published [43] to compare GENIE3 to other gene co-expression methods using the zebrafish data in this study. Again, GENIE3 was found to be the most accurate and provided the most comprehensive network with our data (data not shown). Figure 2 shows the location of the 12 largest modules calculated based on the number of genes they each contain. Table 1 provides module information including number of genes in each module and colors for each module used in Fig. 2. We carried out functional enrichment on all modules using g:Profiler [44] and found that several modules were significantly enriched (adjusted p-value < 0.05, function is overrepresented in a module compared to the whole genome) for one or more processes (Supplementary Table S2). Table 1 shows the pathway(s) that had the highest functional enrichments for each module, and demonstrates the diversity in functional enrichment across all the modules, with some examples of the most enriched functions highlighted in Fig. 2. Many of the functions are related to disruptions to normal development such as nervous system (Modules 1 and 5) and axon development (Module 10), in addition to processes associated with ion transporter activities (Modules 2 and 6) and gene expression (Modules 4 and 14).

Gene co-expression network of 48-hpf zebrafish transcriptomic response to chemicals. Small colored circles represent zebrafish genes (nodes), and lines (edges) connecting the genes represent instances of high co-expression. Nodes are colored by the module they belong to with processes highly enriched in four example modules indicated
To visualize how the network groups genes that respond to particular classes of chemicals, we overlaid gene expression changes from response to either the FRCs or the AHR2 Activators onto the full co-expression network. Overlaying gene expression changes was done by determining how many FRCs or AHR2 Activator chemicals each gene responded to (i.e. whether it was a DEG with p-value < 0.05 and log2FC > 1, for a given chemical treatment). Genes in the network were then sized and colored by how many chemicals they responded to with larger green nodes responding to more chemical treatments and smaller brown nodes responding to fewer chemical treatments. Genes responding to the FRCs were spread across several modules (Modules 2, 7, and 10, with some being in Modules 1 and 6, Fig. 3A). Functional enrichment showed that Module 1 was strongly enriched for development processes including multicellular, neuronal, and anatomical structure development. Module 2 was enriched for transport mechanisms including metal ion, cation and calcium transporters among others, similar to Module 6, which was enriched for sodium channel complexes (Table 1). In addition, Module 7 was enriched for regulation of non-motile cilium assembly, while Module 10 was enriched for axon development. The large number of FRC-responsive genes making up these modules strongly suggests that response to the FRCs in our dataset centers around these pathways and processes. In contrast to the FRCs, genes responding to the AHR2 Activators were very tightly localized to one location in the network (Module 13, Fig. 3B). Module 13 was specifically enriched for known chemical response pathways such as xenobiotic phase I and phase II metabolism, and oxidative stress, consistent with AHR functions [45, 46]. This module was also very tightly clustered (there were a number of edges linking genes within Module 13), indicating that the genes in this module are highly co-expressed relative to each other and are strongly regulated in response to AHR2 Activator exposure. Genes within Module 13 include known zebrafish AHR2-regulated genes upon PAH exposure including, cyp1a, cyp1c1, ahr2, ahrra, ahrrb, and foxq1a [39]. Thus, our chemical subgroup-specific module analysis shows that gene responses to FRCs are more spread across the network and are associated with several different pathways and developmental processes, while AHR2 Activators induce a much more focused and tightly controlled response consisting specifically of pathways linked to xenobiotic exposure. The remaining modules did not contain large numbers of genes that responded to either the FRCs or the AHR2 Activators and were likely driven by the remaining chemicals in our dataset. Several were enriched for general housekeeping processes in 48-hpf zebrafish [4], including neuronal (Module 5) and eye (Module 8) development (Table 1).

Network response to FRCs and AHR2 Activators. A Nodes are colored and sized by the number of FRCs they respond to (defined as an FDR p-value < 0.05 when comparing chemical to DMSO control, no fold-change cutoff). Larger green nodes respond to a more chemicals, smaller brown nodes respond to fewer chemicals. General location of modules highly associated with FRCs (Modules 1, 2, 6, 7, and 10) are indicated. B Same as (A) but showing response to AHR2 Activators. Location of Module 13, highly associated with the AHR2 Activators, is indicated
Centrality analysis
While hierarchical clustering and module analysis can give broad overviews of pathway responses and modules associated with specific chemicals, networks also contain valuable gene-specific co-expression information. Centrality analysis of network genes based on betweenness or degree can reveal which genes are critical within the transcriptomic map of a biological system [34]. Genes of high degree are those with several connections to other genes and are termed “hubs”. Genes of high betweenness are those that occupy positions as links between larger clusters of genes, and are termed “bottlenecks”. Supplementary Figure S1A shows the top 20 genes from the network with the highest betweenness centrality values. The gene with the highest betweenness was apc, which codes for a regulator of the WNT signaling pathway [47] (Table 2). Bottleneck genes were also found throughout the network and in several different modules suggesting that such genes do not respond in a particularly strong way to any specific chemical or chemical class, but are associated either with general chemical response or with zebrafish housekeeping processes.
Genes of highest degree centrality (top 20) showed different location patterns within the network compared to genes of highest betweenness (Supplementary Figure S1B). Table 3 shows that the highest degree genes were primarily grouped into Module 13 (12 genes) which was enriched for AHR2 Activators, and Module 2 (7 genes), which was enriched for the FRCs. There was one additional high degree gene from the list of the top 20 that was found in Module 4. This degree centrality analysis shows that the most highly connected genes within our network are those specifically responding to AHR2 Activator or FRC exposure, in contrast to high betweenness genes which are more distributed throughout the network. The genes with the highest degree were cyp1a and sult6b, both previously shown to be highly induced by PAHs [39]. While the cyps, gstp1, and ahrra are all involved in response to xenobiotic stimulus and metabolism, the functional roles of the other high degree genes are less clear. SlincR and foxq1a appear to have roles in TCDD-induced toxicity in zebrafish; slincR was recently identified as a long noncoding RNA that is involved in the regulation of sox9b, one of the most highly depressed transcripts upon TCDD exposure [12], and foxq1a was induced by TCDD in the jaw primordium of developing zebrafish [13]. It is unknown what the specific roles of gng13b, wfikkn1, and mxd1 are as they relate to chemical exposure; however, identification of these hub genes is suggestive of their potentially important roles in mediating toxicity of some of the chemicals in our network. We also highlight three high degree Module 2 genes that might be involved in pathways associated with disruption of neurodevelopment due to FRC exposure: srgap3, involved in neurodevelopmental processes as reviewed previously [48], tfe3a, an important transcription factor [49], and cacna1da, associated with calcium ion transport in neuronal signaling [50, 51]. Other high degree genes associated with FRC exposure are presented in Table 4, and the high centrality within their respective modules are suggestive of their potentially important roles in the toxicity of FRCs included in this study. We also examined which of these high degree genes responded to specific chemical treatments and found that many of them responded only to a subset of conditions (with p-value < 0.05, and log2FC > 1) (Supplementary Table S3). Therefore, while there was some overlap between centrality analysis and more fundamental pairwise comparisons to identify genes critical to chemical response, our network approach also found several new genes that would not have been found by simply comparing individual treatments to control. We also identified two genes of high degree that were completely uncharacterized, NA_732 (Module 13) and NA_146 (Module 2) (Entrez GeneIDs: 108182865 and 100,332,468 respectively). Identification of these uncharacterized genes in our analysis alongside genes that are strongly linked to AHR2 Activator or FRC exposure suggests that they should be investigated in future analyses.
Analysis of module 13
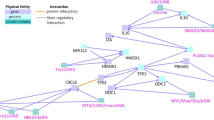
Module 13 of the network is a critical AHR2 activation module associated with xenobiotic metabolism pathways. Centered within Module 13 is cyp1a, a well-studied biomarker gene of AHR exposure that is involved in detoxification of xenobiotics. This gene has been previously shown to be highly induced by TCDD and several PAHs [39, 52], has a high degree centrality in our network, and also responds to a large number of treatments (20/33 chemical treatments induce cyp1a). We extracted a subnetwork of the second order network neighborhood for cyp1a (Fig. 4A), which is defined as any gene connected directly to cyp1a through an edge, or any gene connected to a gene that is connected directly to cyp1a. Within this subnetwork, there were 55 genes (including cyp1a), and 339 edges. Several of the high degree Module 13 genes (Table 3) were present in cyp1a’s first degree network neighborhood including sult6b1, cyp1c1, cyp1c2, gstp1, wfikkn1, foxq1a, ahrra, and slincR (Fig. 4B). Additionally, clcn2c, fgf7, and tiparp were also part of this group. The second order network neighborhood of cyp1a consisted of ahr2, gstp1, and dhrs13la, and several other genes that have not been previously associated with PAH bioactivity in zebrafish. For genes in this group that have not yet been found to have a role in this response including edn1, plvapa, and thsb1b, and several uncharacterized genes, our analysis suggests that their roles as they relate to PAH exposure should be investigated in future studies.

Network neighborhood of cyp1a. A The full network is shown with each blue circle (node) representing a zebrafish gene (2141 nodes are shown which represent the main network), and each line representing an edge (4373 edges are shown which represent the main network). The dark blue node represents cyp1a. B A subset network showing the network neighborhood of cyp1a. The cyp1a gene is in dark blue, nodes in blue represent those in the first-degree network neighborhood (directly linked to cyp1a through an edge). Nodes in light blue represent those in the second-degree network neighborhood (linked to cyp1a through one additional gene within the first network neighborhood)
We next examined the transcriptomic response of the first order network neighborhood genes of cyp1a to each chemical in our dataset. Figure 5 shows that while expression of cyp1a was increased with exposure to the AHR2 Activators, the expression of cyp1a’s network neighborhood genes showed some variability across all the chemicals, including the AHR2 Activators. Of note, even though cyp1a was induced by some of the other PAHs that were included in this study, the expression profiles of many of the first order network neighborhood genes were noticeably different from the AHR2 Activators (Fig. 5). Additionally, while most of the genes had increased expression with exposure to the AHR2 Activators, one exception was clcn2, which was decreased in its expression. However, among the other chemicals, the expression pattern of clcn2 was similar to other genes in the cyp1a network neighborhood. Genes generally also showed a much stronger increase in response to the AHR2 Activators compared to their decrease in response to other chemicals. This was especially true for cyp1a, cyp1c1, and cyp1c2, showing that AHR2 Activators strongly induce increased expression of these genes. As with centrality analysis, we also compared how many chemical treatments each gene in the network neighborhood of cyp1a responded to (Supplementary Table S4). We found that many genes responded to only a few chemical treatments. Thus, their role in chemical response can be missed by fundamental treatment to control comparisons and only emerges when we take a network approach with a compendium of data as presented here. Interestingly, we also noted that of the 25 cyp1a network neighborhood genes, 21 responded to at least one FRC, with three genes (foxq1a, NA_145, and si.ch211.229d2.5) responding to at least six of the ten FRCs.

Heatmap of chemical response of genes in the first-degree network neighborhood of cyp1a. Genes are shown on the right (y-axis) and chemicals below (x-axis). Both genes and chemicals are clustered by similarity of response. Yellow indicates higher expression of each gene in chemical treatment compared to respective DMSO control, blue indicates lower expression in chemical treatment compared to respective DMSO control
A combination of the presence in cyp1a’s network neighborhood (Fig. 4B) and the consistent induction of expression across all the AHR2 Activators (Fig. 5) led us to a subset of genes (fgf7, mamdc2b, pkhd1l1, sult6b1, NA_632, NA_732, NA_145, and NA_928) that we hypothesized would be closely associated with the AHR2 signaling pathway. To confirm the AHR2 dependence of these genes, we validated their expression upon TCDD exposure in both wildtype and AHR2-null zebrafish using qRT-PCR. We confirmed significant induction of all genes by TCDD, compared to vehicle-treated 48-hpf wildtype zebrafish (Fig. 6). Additionally, while cyp1a was significantly induced in TCDD-treated AHR2-null zebrafish, albeit significantly lower than TCDD-treated wildtype zebrafish, expression of none of the other transcripts changed in TCDD-treated AHR2-null zebrafish. These results confirm that our network analysis identifies multiple genes in the AHR2 signaling pathway.

Validation of cyp1a network neighborhood genes identified from the network analysis. Comparative gene expression of cyp1a and selected cyp1a network neighborhood genes in 48-hpf wildtype and AHR2 mutant zebrafish developmentally exposed to 0.1% DMSO or 1 ng/mL TCDD (n = 3–4 biological replicates). Beta-actin was used as the normalization control. Error bars indicate SD of the mean. * = p-value < 0.05 compared to the wildtype vehicle control (DMSO). Statistical significance was determined using a Kruskal Wallis rank sum test followed by a Dunnett’s test for data that were not normal (NA_732), or a two-way ANOVA followed by a Tukey test for data that were normal (all other genes)
Chemical type analysis in networks
We next determined the relative contribution of chemical type towards the global functional pathways identified in our network by calculating how tightly network genes remain co-expressed when AHR2 Activator samples or FRC samples were removed from the dataset. This analysis reveals how genes of certain functional pathways are organized based on response to either the AHR2 Activators or the FRCs. When AHR2 Activator data was removed from our dataset and a new network was inferred, we found several functions whose constituent genes were less tightly co-expressed (based on their average co-expression values) when comparing to a network lacking random data (Table 5). This included negative regulation of vascular development, regeneration, and metabolic processes as well as carboxylic and oxoacid metabolic processes, and actin and cytoskeletal processes. The lower co-expression of genes within these functions when AHR2 Activators are removed indicates that they are particularly important to the transcriptomic response of AHR2 Activators. We also found functions whose constituent genes were more tightly co-expressed (suggesting lower importance to AHR2 Activators compared to all chemicals). This included bone, skeletal and cartilage development, as well as eye development.
Functions showed even greater changes in co-expression when examining FRC data (Table 5). As above, FRC data was removed from our dataset and a new network was inferred and compared to a network lacking a random amount of data. The functions showing the greatest decrease in co-expression in a network lacking FRCs included regulation of vascular development and neurogenesis. Interestingly, many of the functions that showed higher co-expression were also related to neuronal development. These included brain, forebrain, head, and central nervous system development. This observation suggests that while vascular development and neurogenesis generally are pathways responding strongly to FRC exposure, the specific genes and sub-roles within these broad pathways may respond differently to FRC exposure.
Discussion
The main goal of our study was to identify the important genes and biologically relevant pathways at the same zebrafish developmental timepoint that were associated with two groups of chemicals, the AHR2 Activators and the Flame Retardant Chemicals (FRCs), using a comparative network approach. We took advantage of a large compendium of 48-hpf RNA sequencing data from zebrafish exposed to 33 unique chemical treatments to conduct the first meta-analysis of zebrafish chemical transcriptomic data using gene co-expression networks. Using functional pathway enrichment paired with the systematic removal of the individual chemical groups from the network, we identified pathways associated with each of the chemical groups. The network revealed that the AHR2 Activators were associated with specific xenobiotic metabolism-related pathways while FRC exposure corresponded to broader, more general pathways related to perturbation of normal development, with an emphasis on neurogenesis.
While the genes in our full co-expression network formed into 23 distinct modules, genes responding to the FRCs or AHR2 Activators were found primarily in only three modules and a single module, respectively. Furthermore, while there was some overlap in modules associated with the two subsets of chemicals, most modules were associated with a certain chemical class suggesting largely distinct and highly specific molecular signaling events for these two chemical types. Our hierarchical clustering analysis reflected a similar pattern, where there was a strong separation between the FRCs and the rest of the chemicals in the study, but there was no separation between the AHR2 Activators and the remaining PAHs. Additionally, developmental toxicity phenotypes identified in previous studies were not a driver of our clustering. While high throughput screening for developmental toxicity is the necessary first step for determining perturbation to development [53], the toxicology field is now moving towards leveraging these data for mechanistic studies to reveal chemical modes of action. Thus, the network analysis here, showing that chemical types induce very specific transcriptional responses, provides a platform to characterize gene expression changes associated with a diverse group of chemicals and emphasizes one of the limitations of describing chemical toxicity with only phenotypic endpoints.
For our network analysis, we collected RNA sequencing datasets that had been previously published, and combined them with unpublished data gathered in this study. The resulting larger dataset has the advantage of examining gene expression changes associated with multiple chemicals at the same time point (48 hpf), and all at phenotypically anchored exposure concentrations. Such a dataset has never been analyzed before in zebrafish, allowing new conclusions to be gained here that describe transcriptomic response to chemicals. In addition to the strength of the dataset itself, we apply co-expression network analysis, an approach that is well designed for such a compendium of data. Network analyses can identify responses that lie outside the detection limit of more traditional pairwise comparisons of control and treatment conditions, and can also integrate data from a number of different studies and research groups [30, 54]. The variations in data collected across studies is an advantage for the network analysis we apply here as it represents additional biologically relevant variations in gene expression that can be used to infer edges between genes, leading to a more robust and accurate network. The combined strengths in this study (use of a comprehensive but coordinated dataset, and network analysis to move beyond traditional transcriptomic analysis) means that we were able to ask and answer questions that could not be queried in previous studies related to pathways responding to specific chemical treatments. This is reflected, for example, in our identification of high centrality genes that do not respond to specific chemical treatments but rather emerge only when the complete data set is analyzed with a network approach. The same is true for genes in Module 13; the close association of these genes, and the strong response of this module to AHR2 Activator exposure suggest that these genes are important to AHR2 activation. However, not all of the genes in Module 13 respond to all AHR2 Activators, and their association and likely role in chemical response only emerged when we applied network analysis to our large compendium of data. It is possible that the identification of Module 13 genes that do not respond to specific AHR2 Activators reflect the subtle differences in downstream transcription following AHR2 activation, and future work should investigate the functional roles of the identified gene expression changes upon exposure to each of the AHR2 Activator chemicals. Additionally, our observation that many of the Module 13 genes were also differentially expressed upon exposure to some of the FRCs (Supplementary Table S4) points to the fact that these genes might be involved in the general disruption to zebrafish development, probably via perturbation of AHR signaling. This is conceivable since the role of AHR in normal development has been identified previously across multiple species [55,56,57]. Indeed, it is true that some FRCs have previously been shown to activate AHR [26], although AHR activation might have been a result of contaminants present in the chemical standards [58].
The success of high-level analyses of large datasets has also been used in other studies. Schuttler, et al. used an approach based on building a map of transcriptomic response of genes and then clustering those that had similar profiles to view how genes were related [59]. Our approach here differs in that we use a network approach allowing for the application of much of the mathematical tools that have been developed for network analysis (centrality, etc) in addition to collecting a larger dataset. Li, et al. used transcriptomic data to better understand chemical response with a focus on cause and effect relationships, regulatory pathways, and cardiac responses specifically [60]. Aside from cardiac chemical response, network analysis has also been used to better understand heart regeneration in zebrafish [61]. Additional network analysis focused on Weighted gene co-expression network analysis (WGCNA) has also been applied to zebrafish, but again focusing on a smaller and more chemically homogenous dataset allowing for a detailed analysis of these chemicals and responses [62]. Our inclusion here of a large number of chemicals that show a range of phenotypic and transcriptomic responses lead a more comprehensive network that can highlight many different pathways and processes.
The compendium dataset we use here also has the advantage of examining different chemical classes with roughly the same level of analysis. However, despite the similar number of datasets included in the network analysis for each chemical subgroup, we found that FRC responses drove the majority of the network structure, while the AHR2 Activator gene expression changes were more restricted in the network, which was likely due to the diversity of structures and molecular initiating events of the FRCs compared to the AHR2 Activators. Both functional enrichment of FRC-responsive modules, as well as analysis of functions shifting their co-expression after removal of FRC datasets show that the FRCs influence functions related to developmental, neurological, and signaling and transport pathways. The analysis removing the FRCs from the network identified regulation of vascular development as being particularly important to this group of chemicals. Vascular developmental abnormalities and cardiac arrhythmia have been investigated in zebrafish exposed to only few FRCs [27, 63, 64]; but our results highlight it as a critical organ system that should be more thoroughly investigated when studying FRC toxicity. Our conclusions also corroborate evidence from a number of previous studies demonstrating zebrafish FRC neurobehavioral toxicity upon exposure to multiple structurally diverse chemicals, including individual organophosphate FRCs [22, 65], DE-71 (a PBDE) [66], and TBBPA [67]. Additionally, all FRCs included in this network analysis were previously determined to induce both 24-hpf and 120-hpf behavior effects in zebrafish, albeit at varying degrees [6]. Thus, our network analysis not only confirmed observations from previous studies, but also provides guidance for future studies investigating mechanisms of FRC developmental toxicity. We recognize that even though the FRCs as a group lit up distinct parts of the network compared to the rest of the chemicals in the study, the structurally distinct chemicals within the FRCs are likely to have some unique modes of toxicity that were not captured here. Our analysis contained only a limited number of FRCs from each structural class (for example, there were two brominated phenols, TBBPA and TBBPA-DBPE), and future work should consider incorporating more FRC transcriptomic data, and using this increased FRC dataset to view how different types of FRCs induce gene modules or functional pathways. Our understanding of the mechanisms by which the structurally diverse FRCs cause developmental toxicity is still in its infancy. This is likely because several FRCs have been shown to interact with multiple nuclear receptor signaling pathways to cause gene expression changes and developmental toxicity in zebrafish [68, 69]. Thus, our discovery of some of the high centrality genes in our network such as kremen1, cacna1da, and tfe3a within Module 2 (highly associated with the FRCs), emphasizes their potentially important role in driving FRC transcriptomic responses. Tfe3 is a transcription factor, and its high co-expression with genes associated with FRCs suggests that it might be involved in regulation of the genes within this module. The identification of the other high centrality genes present in the FRC modules (Table 4) should be investigated in future research to understand their roles in the pathways enriched by their modules.
Module 13, whose genes were enriched for xenobiotic metabolism by cytochrome P450, had a prominent role in our network analysis. Although Module 13 was a small module, it was very tightly co-expressed, and contained a large number of high degree genes. This is in line with our previous studies showing that high degree genes are those that are heavily involved in a small number of specific pathways [70]. This is in contrast to high betweenness genes which are often associated with several pathways but not strongly with any one pathway, in agreement with their general position linking multiple larger groups of genes. Previous studies in centrality have found both degree and betweenness to be approximately equal in their identification of genes of importance [34]. However, those studies looked at a broader range of conditions than we do here, where we infer a network very focused on chemical response. This may suggest that in more focused networks inferred from very similar datasets using degree as the primary centrality measure may be advantageous, though further studies are needed to determine if this is an effect of network structure generally or of the network presented here in particular. However, it does suggest that for this specific network when identifying genes that respond to certain chemicals of interest, degree is a better metric of importance than betweenness.
Module 13 was primarily made up of genes responding to the AHR2 Activators, which consisted of TCDD and several PAHs, and at the center of this module was the cyp1a gene, a widely use biomarker for PAH exposure in several organisms [71, 72]. We found several AHR signaling pathway genes such as ahrra and ahrrb [73], foxq1a [13], and slincR [12] within Module 13 that were tightly co-expressed with cyp1a. More interestingly, our network analysis also identified within Module 13 other annotated and non-annotated genes, including novel long non-coding RNAs that have not been previously associated with AHR2. We hypothesized that they too were involved in the AHR signaling pathway. Our in silico network analysis conclusions are corroborated through our RT-qPCR analysis demonstrating that fgf7, mamdc2b, pkhd1l1, and sult6b1 expression is dependent on the presence of AHR2. Importantly, several novel genes, NA_632, NA_732, NA_145, and NA_928 (Entrez GeneIDs of 103,910,027, 108,182,865, 100,332,446 and 407,643 respectively) were also associated with the AHR2 signaling pathway (Fig. 6). NA_732 and NA_928 are long non-coding RNAs, while NA_632 and NA_145 are protein coding genes. Despite that fact that the functions of these genes are unknown, their presence in Module 13 linked to cyp1a strongly suggest that they are important to AHR2 signaling. Future functional studies investigating the roles of these novel AHR2-related genes will help strengthen our mechanistic understanding of AHR2 Activator toxicity.
Overall, the methods used in this study combined the strength of using an extensive transcriptomic dataset with a gene co-expression analysis approach to measure how a diverse group of chemicals altered the 48-hpf zebrafish transcriptome relative to each other. We found that the FRCs and AHR2 Activators localized to distinct regions of the network we created, highlighting very specific transcriptomic responses to each chemical group. Additionally, FRCs induced a broad response related to neurobehavior, ion signaling, and vascular development, while the AHR2 Activators centered in one module related to chemical stress and metabolism-related responses. Guided by our network, we also discovered novel genes associated with the AHR2 signaling pathway. Overall, while the FRCs and AHR2 Activators have chemical-specific gene expression changes, we also identified several candidate biomarker genes that future studies should focus on to gain a better understanding of the toxicity of these two chemical groups. While the toxicology field has thus far focused on understanding phenotypic responses associated with chemical exposure, we are now transitioning to unraveling the mechanisms of chemical hazard, which will enable more in-depth characterization of chemicals. This study provides transcriptomic biomarkers that could be used in the future for exposure determination and mixture component diagnosis as they relate to mode-of-action based risk assessment of PAHs. Additionally, the transcriptomic network that we created can be used as a resource for future studies investigating mechanisms of toxicity in developing zebrafish.
Methods
Characterization of chemical datasets
In total, we collected RNA sequencing data from 48-hpf zebrafish exposed to 33 unique chemical treatments (Supplementary Table S1). For 29 of these treatments (18 PAHs, TCDD, and 10 FRCs), the data was collected from previously published previous studies [7, 39,40,41], with the remaining 4 treatments (4 PAHs) initially analyzed in this study. Each treatment was examined with 3–8 replicates for a total of 170 RNA sequencing samples included in the study. Raw and processed RNA sequencing files for each sample have been deposited in NCBI’s GEO database (GSE171944). See Supplementary Table S1 for information on chemicals, exposure levels, and relevant references for the datasets used in this study.
Chemicals
Detailed methods for published datasets can be found in our previous studies [7, 39,40,41]. Methods for chemicals initially analyzed in this study, and TCDD exposures for RT-qPCR analysis (See below) are described here. The PAHs (benzo [a] pyrene (B [a]P), 9,10 phenanthrenequinone (9,10-PQ), and dibenzo [a,l] pyrene (DBalP)) were dissolved to 10 mM in 100% DMSO and stored in a desiccator. TCDD was purchased from Supelco (Sigma Aldrich) at 311 nM with 95.3% purity, and stored in the dark at room temperature. The chemical stocks were sonicated in a water bath sonicator for 15 min (PAHs) or 30 min (TCDD).
Zebrafish husbandry
The study’s zebrafish protocols were performed according to the relevant guidelines provided by the Oregon State University’s Institutional Animal Care and Use Committee protocols (ACUP 5143). Briefly, Tropical 5D wildtype zebrafish were housed at Oregon State University’s Sinnhuber Aquatic Research Laboratory (SARL, Corvallis, OR) in densities of 1000 fish per 100-gal tank. Fish were maintained at 28 °C on a 14:10 h light/dark cycle in recirculating filtered water, supplemented with Instant Ocean salts. Adult fish were fed GEMMA Micro 300 or 500 twice a day, and larval and juvenile fish were fed GEMMA Micro 75 and 150, respectively, three times a day [74]. Spawning funnels were placed in the tanks at night and the following morning embryos were collected and age staged [4, 75]. Embryos were maintained in embryo medium (EM) in an incubator at 28 °C until further processing. EM consisted of 15 mM NaCl, 0.5 mM KCl, 1 mMMgSO4, 0.15 mMKH2PO4, 0.05 mM Na2HPO4, and 0.7 mMNaHCO3.
Exposures and RNA-sequencing sample preparation and sequencing
Detailed exposure methodology and phenotypic endpoints identified for each chemical for the previously published datasets can be found in their respective studies [7, 39,40,41]. Briefly, for the PAHs (with the exception of 7,12-B [a] AQ and BEZO) and FRCs, an EC80 exposure concentration was selected based on a concentration-response zebrafish phenotypic screen [6, 38]. If an EC80 concentration was not attainable, the maximum concentration tested during the phenotypic screen (50 μM for PAHs, and 85 μM for the FRCs) was utilized for RNA sequencing [39, 41]. 7,12-B [a] AQ and BEZO were exposed at their EC100 concentrations [7], and 1 ng/mL TCDD was utilized, which is a concentration that ensures 99–100% of zebrafish at 120 hpf have the expected TCDD-induced toxicity endpoints [40]. All samples were processed for RNA sequencing analysis at 48 hpf, a timepoint whose transcriptome precedes and likely drives morphological and/or behavioral phenotypes observed at 120 hpf [7, 39,40,41]. Fastqc files for each chemical were re-analyzed in this study (See “Alignment and analysis of RNA-seq data” below).
For the PAHs (B [a] P, 9,10-PQ, and DBalP) initially analyzed in this study, zebrafish embryos were dechorionated using pronase at 4 hpf, and were batch-exposed to chemicals at 6 hpf in glass vials as described previously [7]. Exposure concentrations were 1 and 10 μM for B [a] P, 1.2 μM for 9,10-PQ, and 10 μM for DBalP. The vehicle control was 1% DMSO, and there were 20 embryos per glass vial in 2 mL exposure solution. Vials were incubated at 28 °C in the dark on a rocker until sample collection at 48 hpf. Following exposure, 48-hpf whole embryos were homogenized using RNAzol (Molecular Research Center, Inc.) and a bullet blender with 0.5 mM zirconium oxide beads (Next Advance), as recommended by the Next Advance. Each biological sample consisted of 20 pooled 48-hpf zebrafish. The RNA from the PAH exposures was isolated via phenol guanidine extraction. RNA integrity was assessed (RIN score > 9) using an Agilent Bioanalyzer. Total RNA samples were sent to the Oregon State University Center for Genome Research and Biocomputing Core facilities for library preparation and sequencing. This included mRNA enrichment by polyA selection. Libraries were prepared with the PrepX™ mRNA and Illumina sequencing workflow (Wafergen Biosystems). 50 bp paired-end sequencing was conducted using an Illumina HiSeq 2000 sequencer.
Alignment and analysis of RNA-seq data
All of the datasets listed in Supplementary Table S1 were analyzed (or re-analyzed if already published) using the following approach. Each fastq file was aligned to the Genome Reference Consortium Zebrafish Build 11 (GRCz11) (https://www.ncbi.nlm.nih.gov/assembly/GCF_000002035.6/) using the Star Aligner [76] with default settings. Resulting SAM files were then used to count reads aligning to genes using HTSeq (https://htseq.readthedocs.io/en/release_0.11.1/) [77] along with the gff file for the GRCz11 genome, resulting in raw read counts for 39,701 zebrafish genes. Low expression genes, defined as those with a count of ‘0’ in at least 43/170 (25%) of samples, were removed from further analysis. The final raw dataset consisted of 21,854 genes across 170 samples.
Raw counts were normalized using Bioconductor’s DESeq2 package [78]. DESeq2 was also used to calculate log2 fold changes (log2FC) and adjusted p-values (corrected for multiple hypothesis testing) for the 33 individual chemical comparisons to their respective DMSO controls. Adjusted p-values and log2FC values for all 21,854 genes are shown in Supplementary Table S5. In addition, whether the gene is a DEG (log2FC > 1 and an adjusted p-value < 0.05) is indicated. To further reduce the number of genes used to infer a network, any gene that was not differentially expressed (defined as an adjusted p-value > 0.05, no fold change cutoff) in at least 3 of the comparisons (10% of the total) was removed from further analysis. This resulted in 10,346 genes across 170 samples from 33 conditions being included in subsequent networks. Log2FC values (comparing chemicals to their respective DMSO controls) of these 10,346 genes were then used to infer the gene co-expression network.
Inferring gene co-expression networks
GENIE3 [31] was used to generate a matrix of co-expression values for all gene pairs. Regulators or targets were not pre-selected so as to allow all gene co-expressions to emerge from the analysis. The tree method was randomforest, and the number of candidate regulators randomly selected at each tree node (for the determination of the best split) was set to the square root of the total number of candidate regulators. The number of trees in an ensemble for each target gene was set to 1000. All of these settings represent the default values for GENIE3. We filtered low co-expression values (those below 0.00858) to generate a network with sufficient structure for topological analysis, as done previously [70, 79]. Networks were viewed in Cytoscape [80] by importing a .sif file with each line indicating a gene pair connected by an edge. A prefuse force directed layout was used to view the network. We also used Cytoscape to calculate centrality values by using the Analyze Network tool and analyzing as a directed network. Network neighborhoods of genes of interested were identified by selecting a gene of interest and using Cytoscape to highlight the first network neighborhood of the selected gene. This was followed by highlighting the first network neighborhood of each of the selected genes (corresponding to the second network neighborhood of the original selected gene). The 23 modules in the network were determined using the fastgreedy.community function within the igraph package in R [81] with the minimum number of genes comprising a module set to 12. Functional enrichment of modules was done using g:Profiler using default settings [44]. A function was considered to be enriched if the p-value of the enrichment was below 0.05, and the function was overrepresented in a module or network compared to the zebrafish genome as a whole. Underrepresented functions were not considered. g:Profiler outputs functions from KEGG, Gene Ontology, Reactome, and WikiPathways. We examined all enriched functions regardless of which database they were drawn from. This led to some overlap in enriched functions but also allowed us to gain the most comprehensive view of pathways enriched in certain parts of the network or in different networks.
To determine which functions and pathways may be related to certain classes of chemicals, we selected two subsets of our data, chemicals in the FRC class or chemicals in the AHR2 Activator class (Supplementary Table S1). For FRC analysis, we first inferred a GENIE3 co-expression matrix after removing FRC data (comprising 39 samples). Next, we randomly removed 39 samples of data from our dataset and calculated the resulting gene co-expression matrix. This random data removal from the full dataset and matrix calculation was repeated an additional nine times and an average gene co-expression matrix was calculated from these ten iterations of data removal. A network was then constructed from this average gene co-expression matrix. This network represents the effect of randomly removing data from our constituent dataset. This random removal network was then compared to the network inferred after specifically removing FRC data. For each of these two networks (that lack FRC data or lack an identical amount of random data), we calculated the average co-expression value between all genes that belonged to each functional enrichment identified using g:Profiler [44]. Genes of a particular function that show lower co-expression in a network that is specifically lacking FRC data compared to a network that has data randomly removed from it, suggests that this function is especially critical to the FRC response. A similar pair of networks and comparisons were made for the AHR2 Activators (comprising 46 samples).
TCDD exposure, RNA extraction, and quantitative reverse transcriptase polymerase chain reaction (qRT-PCR)
To confirm that the highly co-expressed genes associated with the AHR2 Activators in our network are in the AHR signaling pathway, we performed a qRT-PCR experiment. Wildtype and AHR2-null [56] zebrafish embryos were exposed to 0.1% DMSO or 1 ng/mL TCDD at 6 hpf, as described previously [40], in a manner similar to the TCDD RNA sequencing samples included in this study. The 1 ng/mL TCDD concentration was selected as it results in 99–100% of 120-hpf zebrafish displaying the expected TCDD-induced phenotypic malformations such as heart and cartilage malformations [82]. Briefly, exposures were conducted in 20-mL amber glass vials with 1 embryo/100 μL exposure solution for 1 h with gentle rocking. Vials were also inverted every 15 min to ensure even exposure. After exposure, embryos were rinsed three times with EM, transferred to 100-mm Petri dishes, and raised in EM at 28 °C to 48 hpf. RNA was extracted in a manner similar to the PAHs, as described above. RNA quantification and quality assessment (O.D. 260/280 ratios) was conducted using a BioTek® Synergy™ Mx microplate reader with the Gen5™ Take3™ module.
qRT-PCR was conducted in 10 μL reactions consisting of 5 μL SYBR® Green Master Mix, 0.08 μL reverse transcriptase enzyme mix (Power SYBR® Green RNA-to-CT™ 1-Step Kit; Applied Biosystems), 0.2 μL each of 10 μM forward and reverse primers, and 20 ng RNA per reaction. The gene specific primers (IDT) for qRT-PCR amplification are listed in Supplementary Table S6. The QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific) was used and the cycling parameters were as follows: reverse transcription at 48 °C for 30 min, denaturation and activation of SYBR® polymerase at 95 °C for 10 min, followed by 40 cycles of amplification (95 °C for 15 s, 60 °C for 1 min). A melt curve analysis was conducted to assess for multiple products, and it was confirmed that all primers amplified a single product. Expression values were normalized to the β-actin control, and analyzed with the 2−ΔΔCT method as described previously [83]. The data collected for each gene was tested for normality using the Shapiro-Wilk normality test. To determine significant difference compared to control (p-value < 0.05), either a two-way ANOVA with the post-hoc Tukey’s test or a Kruskal Wallis rank sum test followed by a Dunnett’s test was conducted depending on if the data that passed or failed the normality test, respectively,. Data analysis was conducted using RStudio (version 3.6.0), and data visualization was performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California, USA (www.graphpad.com).
Availability of data and materials
The raw and processed files for all datasets used in this study are in NCBI’s Gene Expression Omnibus (GEO) database (Accession number: GSE171944).
References
Tice RR, Austin CP, Kavlock RJ, Bucher JR. Improving the human hazard characterization of chemicals: a Tox21 update. Environ Health Perspect. 2013;121(7):756–65. https://doi.org/10.1289/ehp.1205784.
Dix DJ, Houck KA, Martin MT, Richard AM, Setzer RW, Kavlock RJ. The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci. 2007;95(1):5–12. https://doi.org/10.1093/toxsci/kfl103.
Raies AB, Bajic VB. In silico toxicology: computational methods for the prediction of chemical toxicity. Wiley Interdiscip Rev Comput Mol Sci. 2016;6(2):147–72. https://doi.org/10.1002/wcms.1240.
Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203(3):253–310. https://doi.org/10.1002/aja.1002030302.
Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496(7446):498–503. https://doi.org/10.1038/nature12111.
Truong L, Marvel S, Reif DM, Thomas DG, Pande P, Dasgupta S, et al. The multi-dimensional embryonic zebrafish platform predicts flame retardant bioactivity. Reprod Toxicol. 2020;96:359–69. https://doi.org/10.1016/j.reprotox.2020.08.007.
Goodale BC, la du J, Tilton SC, Sullivan CM, Bisson WH, Waters KM, et al. Ligand-specific transcriptional mechanisms underlie aryl hydrocarbon receptor-mediated developmental toxicity of oxygenated PAHs. Toxicol Sci. 2015;147(2):397–411. https://doi.org/10.1093/toxsci/kfv139.
Williams TD, Mirbahai L, Chipman JK. The toxicological application of transcriptomics and epigenomics in zebrafish and other teleosts. Brief Funct Genomics. 2014;13(2):157–71. https://doi.org/10.1093/bfgp/elt053.
Zheng M, Lu J, Zhao D. Toxicity and transcriptome sequencing (RNA-seq) analyses of adult zebrafish in response to exposure Carboxymethyl cellulose stabilized Iron sulfide nanoparticles. Sci Rep. 2018;8(1):8083. https://doi.org/10.1038/s41598-018-26499-x.
Kashyap B, Pegorsch L, Frey RA, Sun C, Shelden EA, Stenkamp DL. Eye-specific gene expression following embryonic ethanol exposure in zebrafish: roles for heat shock factor 1. Reprod Toxicol. 2014;43:111–24. https://doi.org/10.1016/j.reprotox.2013.12.002.
King-Heiden TC, et al. Molecular targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) within the zebrafish ovary: insights into TCDD-induced endocrine disruption and reproductive toxicity. Reprod Toxicol. 2008;25(1):47–57. https://doi.org/10.1016/j.reprotox.2007.07.013.
Garcia GR, Goodale BC, Wiley MW, la du JK, Hendrix DA, Tanguay RL. In vivo characterization of an AHR-dependent long noncoding RNA required for proper Sox9b expression. Mol Pharmacol. 2017;91(6):609–19. https://doi.org/10.1124/mol.117.108233.
Planchart A, Mattingly CJ. 2,3,7,8-Tetrachlorodibenzo-p-dioxin upregulates FoxQ1b in zebrafish jaw primordium. Chem Res Toxicol. 2010;23(3):480–7. https://doi.org/10.1021/tx9003165.
Kim KH, Jahan SA, Kabir E, Brown RJC. A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ Int. 2013;60:71–80. https://doi.org/10.1016/j.envint.2013.07.019.
Abdel-Shafy HI, Mansour MS. A review on polycyclic aromatic hydrocarbons: source, environmental impact, effect on human health and remediation. Egypt J Pet. 2016;25(1):107–23. https://doi.org/10.1016/j.ejpe.2015.03.011.
Shankar P, Dasgupta S, Hahn ME, Tanguay RL. A review of the functional roles of the zebrafish aryl hydrocarbon receptors. Toxicol Sci. 2020;178(2):215–38. https://doi.org/10.1093/toxsci/kfaa143.
Poland A, Kende A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin: environmental contaminant and molecular probe. Fed Proc. 1976;35(12):2404–11.
Brown DR, Clark BW, Garner LVT, di Giulio RT. Zebrafish cardiotoxicity: the effects of CYP1A inhibition and AHR2 knockdown following exposure to weak aryl hydrocarbon receptor agonists. Environ Sci Pollut Res Int. 2015;22(11):8329–38. https://doi.org/10.1007/s11356-014-3969-2.
Andreasen EA, Spitsbergen JM, Tanguay RL, Stegeman JJ, Heideman W, Peterson RE. Tissue-specific expression of AHR2, ARNT2, and CYP1A in zebrafish embryos and larvae: effects of developmental stage and 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure. Toxicol Sci. 2002;68(2):403–19. https://doi.org/10.1093/toxsci/68.2.403.
Xiong P, Yan X, Zhu Q, Qu G, Shi J, Liao C, et al. A review of environmental occurrence, fate, and toxicity of novel brominated flame retardants. Environ Sci Technol. 2019;53(23):13551–69. https://doi.org/10.1021/acs.est.9b03159.
Yang J, et al. A Review of a Class of Emerging Contaminants: The Classification, Distribution, Intensity of Consumption, Synthesis Routes, Environmental Effects and Expectation of Pollution Abatement to Organophosphate Flame Retardants (OPFRs). Int J Mol Sci. 2019;20(12):2874.
Dishaw LV, Hunter DL, Padnos B, Padilla S, Stapleton HM. Developmental exposure to organophosphate flame retardants elicits overt toxicity and alters behavior in early life stage zebrafish (Danio rerio). Toxicol Sci. 2014;142(2):445–54. https://doi.org/10.1093/toxsci/kfu194.
Kim YR, Harden FA, Toms LML, Norman RE. Health consequences of exposure to brominated flame retardants: a systematic review. Chemosphere. 2014;106:1–19. https://doi.org/10.1016/j.chemosphere.2013.12.064.
Fowles JR, Fairbrother A, Baecher-Steppan L, Kerkvliet NI. Immunologic and endocrine effects of the flame-retardant pentabromodiphenyl ether (DE-71) in C57BL/6J mice. Toxicology. 1994;86(1–2):49–61. https://doi.org/10.1016/0300-483X(94)90052-3.
Dasgupta S, Cheng V, Vliet SMF, Mitchell CA, Volz DC. Tris(1,3-dichloro-2-propyl) phosphate exposure during the early-blastula stage alters the Normal trajectory of zebrafish embryogenesis. Environ Sci Technol. 2018;52(18):10820–8. https://doi.org/10.1021/acs.est.8b03730.
Haggard DE, Das SR, Tanguay RL. Comparative Toxicogenomic responses to the flame retardant mITP in developing zebrafish. Chem Res Toxicol. 2017;30(2):508–15. https://doi.org/10.1021/acs.chemrestox.6b00423.
Mitchell CA, Dasgupta S, Zhang S, Stapleton HM, Volz DC. Disruption of nuclear receptor signaling alters Triphenyl phosphate-induced cardiotoxicity in zebrafish embryos. Toxicol Sci. 2018;163(1):307–18. https://doi.org/10.1093/toxsci/kfy037.
Dasgupta S, Vliet SM, Kupsco A, Leet JK, Altomare D, Volz DC. Tris(1,3-dichloro-2-propyl) phosphate disrupts dorsoventral patterning in zebrafish embryos. PeerJ. 2017;5:e4156. https://doi.org/10.7717/peerj.4156.
Marbach D, et al. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9(8):796–804. https://doi.org/10.1038/nmeth.2016.
Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5(1):e8. https://doi.org/10.1371/journal.pbio.0050008.
Huynh-Thu VA, et al. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS One. 2010;5(9):e12776.
Ashtiani M, Salehzadeh-Yazdi A, Razaghi-Moghadam Z, Hennig H, Wolkenhauer O, Mirzaie M, et al. A systematic survey of centrality measures for protein-protein interaction networks. BMC Syst Biol. 2018;12(1):80. https://doi.org/10.1186/s12918-018-0598-2.
Klein C, Marino A, Sagot MF, Vieira Milreu P, Brilli M. Structural and dynamical analysis of biological networks. Brief Funct Genomics. 2012;11(6):420–33. https://doi.org/10.1093/bfgp/els030.
McDermott JE, Taylor RC, Yoon H, Heffron F. Bottlenecks and hubs in inferred networks are important for virulence in salmonella typhimurium. J Comput Biol. 2009;16(2):169–80. https://doi.org/10.1089/cmb.2008.04TT.
Larkin A, Siddens LK, Krueger SK, Tilton SC, Waters KM, Williams DE, et al. Application of a fuzzy neural network model in predicting polycyclic aromatic hydrocarbon-mediated perturbations of the Cyp1b1 transcriptional regulatory network in mouse skin. Toxicol Appl Pharmacol. 2013;267(2):192–9. https://doi.org/10.1016/j.taap.2012.12.011.
Tilton SC, Siddens LK, Krueger SK, Larkin AJ, Löhr CV, Williams DE, et al. Mechanism-based classification of PAH mixtures to predict carcinogenic potential. Toxicol Sci. 2015;146(1):135–45. https://doi.org/10.1093/toxsci/kfv080.
McDermott JE, Archuleta M, Thrall BD, Adkins JN, Waters KM. Controlling the response: predictive modeling of a highly central, pathogen-targeted core response module in macrophage activation. PLoS One. 2011;6(2):e14673. https://doi.org/10.1371/journal.pone.0014673.
Geier MC, Chlebowski AC, Truong L, Massey Simonich SL, Anderson KA, Tanguay RL. Comparative developmental toxicity of a comprehensive suite of polycyclic aromatic hydrocarbons. Arch Toxicol. 2018;92(2):571–86. https://doi.org/10.1007/s00204-017-2068-9.
Shankar P, et al. Coupling Genome-wide Transcriptomics and Developmental Toxicity Profiles in Zebrafish to Characterize Polycyclic Aromatic Hydrocarbon (PAH) Hazard. Int J Mol Sci. 2019;20(10):2570.
Garcia GR, Shankar P, Dunham CL, Garcia A, la du JK, Truong L, et al. Signaling events downstream of AHR activation that contribute to toxic responses: the functional role of an AHR-dependent long noncoding RNA (slincR) using the zebrafish model. Environ Health Perspect. 2018;126(11):117002. https://doi.org/10.1289/EHP3281.
Dasgupta S, Dunham CL, Truong L, Simonich MT, Sullivan CM, Tanguay RL. Phenotypically anchored mRNA and miRNA expression profiling in zebrafish reveals flame retardant chemical toxicity networks. Front Cell Dev Biol. 2021;9:663032. https://doi.org/10.3389/fcell.2021.663032.
Goodale BC, la du JK, Bisson WH, Janszen DB, Waters KM, Tanguay RL. AHR2 mutant reveals functional diversity of aryl hydrocarbon receptors in zebrafish. PLoS One. 2012;7(1):e29346. https://doi.org/10.1371/journal.pone.0029346.
McClure RS, Wendler JP, Adkins JN, Swanstrom J, Baric R, Kaiser BLD, et al. Unified feature association networks through integration of transcriptomic and proteomic data. PLoS Comput Biol. 2019;15(9):e1007241. https://doi.org/10.1371/journal.pcbi.1007241.
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191–8. https://doi.org/10.1093/nar/gkz369.
Larigot L, Juricek L, Dairou J, Coumoul X. AhR signaling pathways and regulatory functions. Biochim Open. 2018;7:1–9. https://doi.org/10.1016/j.biopen.2018.05.001.
Dietrich C. Antioxidant functions of the aryl hydrocarbon receptor. Stem Cells Int. 2016;2016:7943495.
Haramis AP, et al. Adenomatous polyposis coli-deficient zebrafish are susceptible to digestive tract neoplasia. EMBO Rep. 2006;7(4):444–9. https://doi.org/10.1038/sj.embor.7400638.
Bacon C, Endris V, Rappold GA. The cellular function of srGAP3 and its role in neuronal morphogenesis. Mech Dev. 2013;130(6–8):391–5. https://doi.org/10.1016/j.mod.2012.10.005.
Lister JA, Lane BM, Nguyen A, Lunney K. Embryonic expression of zebrafish MiT family genes tfe3b, tfeb, and tfec. Dev Dyn. 2011;240(11):2529–38. https://doi.org/10.1002/dvdy.22743.
Duan J, Yu Y, Li Y, Li Y, Liu H, Jing L, et al. Low-dose exposure of silica nanoparticles induces cardiac dysfunction via neutrophil-mediated inflammation and cardiac contraction in zebrafish embryos. Nanotoxicology. 2016;10(5):575–85. https://doi.org/10.3109/17435390.2015.1102981.
Liu HC, et al. The cardiovascular toxicity of triadimefon in early life stage of zebrafish and potential implications to human health. Environ Pollut. 2017;231(Pt 1):1093–103. https://doi.org/10.1016/j.envpol.2017.05.072.
Carney SA, Peterson RE, Heideman W. 2,3,7,8-Tetrachlorodibenzo-p-dioxin activation of the aryl hydrocarbon receptor/aryl hydrocarbon receptor nuclear translocator pathway causes developmental toxicity through a CYP1A-independent mechanism in zebrafish. Mol Pharmacol. 2004;66(3):512–21. https://doi.org/10.1124/mol.66.3.
Bugel SM, Tanguay RL, Planchart A. Zebrafish: a marvel of high-throughput biology for 21(st) century toxicology. Curr Environ Health Rep. 2014;1(4):341–52. https://doi.org/10.1007/s40572-014-0029-5.
McClure RS, Overall CC, McDermott JE, Hill EA, Markillie LM, McCue LA, et al. Network analysis of transcriptomics expands regulatory landscapes in Synechococcus sp. PCC 7002. Nucleic Acids Res. 2016;44(18):8810–25. https://doi.org/10.1093/nar/gkw737.
Harrill JA, Hukkanen RR, Lawson M, Martin G, Gilger B, Soldatow V, et al. Knockout of the aryl hydrocarbon receptor results in distinct hepatic and renal phenotypes in rats and mice. Toxicol Appl Pharmacol. 2013;272(2):503–18. https://doi.org/10.1016/j.taap.2013.06.024.
Garcia GR, Bugel SM, Truong L, Spagnoli S, Tanguay RL. AHR2 required for normal behavioral responses and proper development of the skeletal and reproductive systems in zebrafish. PLoS One. 2018;13(3):e0193484. https://doi.org/10.1371/journal.pone.0193484.
Qin H, Powell-Coffman JA. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev Biol. 2004;270(1):64–75. https://doi.org/10.1016/j.ydbio.2004.02.004.
Brown DJ, van Overmeire I, Goeyens L, Denison MS, de Vito MJ, Clark GC. Analysis of ah receptor pathway activation by brominated flame retardants. Chemosphere. 2004;55(11):1509–18. https://doi.org/10.1016/j.chemosphere.2003.10.019.
Schuttler A, et al. Map and model-moving from observation to prediction in toxicogenomics. Gigascience. 2019;8(6):giz057.
Li R, Zupanic A, Talikka M, Belcastro V, Madan S, Dörpinghaus J, et al. Systems toxicology approach for testing chemical cardiotoxicity in larval zebrafish. Chem Res Toxicol. 2020;33(10):2550–64. https://doi.org/10.1021/acs.chemrestox.0c00095.
Rodius S, Androsova G, Götz L, Liechti R, Crespo I, Merz S, et al. Analysis of the dynamic co-expression network of heart regeneration in the zebrafish. Sci Rep. 2016;6(1):26822. https://doi.org/10.1038/srep26822.
Meng Q, et al. Toxic effects and transcriptome analyses of zebrafish (Danio rerio) larvae exposed to benzophenones. Environ Pollut. 2020;265(Pt A):114857.
Xing X, Kang J, Qiu J, Zhong X, Shi X, Zhou B, et al. Waterborne exposure to low concentrations of BDE-47 impedes early vascular development in zebrafish embryos/larvae. Aquat Toxicol. 2018;203:19–27. https://doi.org/10.1016/j.aquatox.2018.07.012.
Lema SC, Schultz IR, Scholz NL, Incardona JP, Swanson P. Neural defects and cardiac arrhythmia in fish larvae following embryonic exposure to 2,2′,4,4′-tetrabromodiphenyl ether (PBDE 47). Aquat Toxicol. 2007;82(4):296–307. https://doi.org/10.1016/j.aquatox.2007.03.002.
Sun L, Xu W, Peng T, Chen H, Ren L, Tan H, et al. Developmental exposure of zebrafish larvae to organophosphate flame retardants causes neurotoxicity. Neurotoxicol Teratol. 2016;55:16–22. https://doi.org/10.1016/j.ntt.2016.03.003.
Chen L, Huang C, Hu C, Yu K, Yang L, Zhou B. Acute exposure to DE-71: effects on locomotor behavior and developmental neurotoxicity in zebrafish larvae. Environ Toxicol Chem. 2012;31(10):2338–44. https://doi.org/10.1002/etc.1958.
Chen J, Tanguay RL, Xiao Y, Haggard DE, Ge X, Jia Y, et al. TBBPA exposure during a sensitive developmental window produces neurobehavioral changes in larval zebrafish. Environ Pollut. 2016;216:53–63. https://doi.org/10.1016/j.envpol.2016.05.059.
Liu C, Wang Q, Liang K, Liu J, Zhou B, Zhang X, et al. Effects of tris(1,3-dichloro-2-propyl) phosphate and triphenyl phosphate on receptor-associated mRNA expression in zebrafish embryos/larvae. Aquat Toxicol. 2013;128-129:147–57. https://doi.org/10.1016/j.aquatox.2012.12.010.
Dasgupta S, Vliet SMF, Cheng V, Mitchell CA, Kirkwood J, Vollaro A, et al. Complex interplay among nuclear receptor ligands, cytosine methylation, and the metabolome in driving tris (1, 3-dichloro-2-propyl) phosphate-induced epiboly defects in zebrafish. Environ Sci Technol. 2019;53(17):10497–505. https://doi.org/10.1021/acs.est.9b04127.
McClure RS, Overall CC, Hill EA, Song HS, Charania M, Bernstein HC, et al. Species-specific transcriptomic network inference of interspecies interactions. ISME J. 2018;12(8):2011–23. https://doi.org/10.1038/s41396-018-0145-6.
Payne JF, Fancey LL, Rahimtula AD, Porter EL. Review and perspective on the use of mixed-function oxygenase enzymes in biological monitoring. Comp Biochem Physiol Part C Comp Pharmacol. 1987;86(2):233–45. https://doi.org/10.1016/0742-8413(87)90074-0.
Kim RO, Kim BM, Hwang DS, Au DWT, Jung JH, Shim WJ, et al. Evaluation of biomarker potential of cytochrome P450 1A (CYP1A) gene in the marine medaka, Oryzias melastigma exposed to water-accommodated fractions (WAFs) of Iranian crude oil. Comp Biochem Physiol C-Toxicol Pharmacol. 2013;157(2):172–82. https://doi.org/10.1016/j.cbpc.2012.11.003.
Jenny MJ, Karchner SI, Franks DG, Woodin BR, Stegeman JJ, Hahn ME. Distinct roles of two zebrafish AHR repressors (AHRRa and AHRRb) in embryonic development and regulating the response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci. 2009;110(2):426–41. https://doi.org/10.1093/toxsci/kfp116.
Barton CL, Johnson EW, Tanguay RL. Facility design and health management program at the Sinnhuber aquatic research laboratory. Zebrafish. 2016;13(Suppl 1):S39–43. https://doi.org/10.1089/zeb.2015.1232.
Westerfield M, editor. The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio). 5th ed. Eugene: University of Oregon Press; 2007.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. https://doi.org/10.1093/bioinformatics/bts635.
Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. https://doi.org/10.1093/bioinformatics/btu638.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
McClure R, et al. Global Network Analysis of Neisseria gonorrhoeae Identifies Coordination between Pathways, Processes, and Regulators Expressed during Human Infection. mSystems. 2020;5(1):e00729–19.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. https://doi.org/10.1101/gr.1239303.
Csardi G, Nepusz T. The igraph software package for complex network research. InterJournal, complex systems. 2006;1695(5):1–9.
Henry TR, Spitsbergen JM, Hornung MW, Abnet CC, Peterson RE. Early life stage toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in zebrafish (Danio rerio). Toxicol Appl Pharmacol. 1997;142(1):56–68. https://doi.org/10.1006/taap.1996.8024.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–8. https://doi.org/10.1006/meth.2001.1262.
Acknowledgements
We would like to thank members of the Sinnhuber Aquatic Research Laboratory (SARL), especially Carrie Barton and Kimberly Hayward for all their help regarding fish husbandry, and Drs. Michael Simonich and Subham Dasgupta for help editing the manuscript. Pacific Northwest National Laboratory is a multi-program laboratory operated by Battelle for the U.S. Department of Energy under Contract DE-AC05-76RL01830.
Funding
This research was supported by NIEHS grants P42 ES016465, T32 ES007060, P30ES030287, and US Environmental Protection grant 83579601. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Author information
Authors and Affiliations
Contributions
P.S. designed the study, helped with data curation, assisted with methodology and data visualization, conducted validation experiments, and drafted the manuscript. R.S.M. designed the study, helped with data curation, conducted methodology and data visualization, and drafted the manuscript. K.M.W. provided supervision, and assisted with data interpretation and manuscript revision. R.L.T. conceptualized the study, acquired funding, provided supervision, and assisted with manuscript revision. All authors have read and approved the submission version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The research described in this paper was carried out and approved under the Institutional Animal Care and Use Committee (IUCUC) protocols (ACUP 5143) of Oregon State University. The study’s zebrafish protocols were performed according to the relevant guidelines provided by the Oregon State University’s Institutional Animal Care and Use Committee protocols (ACUP 5143). The study was carried out in compliance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shankar, P., McClure, R.S., Waters, K.M. et al. Gene co-expression network analysis in zebrafish reveals chemical class specific modules. BMC Genomics 22, 658 (2021). https://doi.org/10.1186/s12864-021-07940-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07940-4