
Abstract
Background
Grain weight and grain shape are important agronomic traits that affect the grain yield potential and grain quality of rice. Both grain weight and grain shape are controlled by multiple genes. The 3,000 Rice Genomes Project (3 K RGP) greatly facilitates the discovery of agriculturally important genetic variants and germplasm resources for grain weight and grain shape.
Results
Abundant natural variations and distinct phenotic differentiation among the subgroups in grain weight and grain shape were observed in a large population of 2,453 accessions from the 3 K RGP. A total of 21 stable quantitative trait nucleotides (QTNs) for the four traits were consistently identified in at least two of 3-year trials by genome-wide association study (GWAS), including six new QTNs (qTGW3.1, qTGW9, qTGW11, qGL4/qRLW4, qGL10, and qRLW1) for grain weight and grain shape. We further predicted seven candidate genes (Os03g0186600, Os09g0544400, Os11g0163600, Os04g0580700, Os10g0399700, Os10g0400100 and Os01g0171000) for the six new QTNs by high-density association and gene-based haplotype analyses. The favorable haplotypes of the seven candidate genes and five previously cloned genes in elite accessions with high TGW and RLW are also provided.
Conclusions
Our results deepen the understanding of the genetic basis of grain weight and grain shape in rice and provide valuable information for improving rice grain yield and grain quality through molecular breeding.
Similar content being viewed by others

Background
Rice (Oryza sativa L.) is one of the most widely grown crops in the world and is a staple diet for more than 3.5 billion people around the world, particularly in Asia. It is predicted that rice production must increase by ∼42 % by 2050 to keep pace with increasing global food demand [1]. As the main component of grain yield in rice, grain weight or thousand-grain weight (TGW) has always been a dominating trait for breeders. Meanwhile, grain shape is an important factor affecting physical or appearance quality, where slender grains with grain length-to-width ratio > 3 are preferred by most rice consumers [2]. Grain shape is determined by grain length (GL), grain width (GW), the ratio of length-to-width (RLW) and grain thickness, and grain weight is positively correlated with these four traits [3].
Over the past few decades, more than 60 genes associated with grain weight and grain shape have been identified in rice [4]. To date, more than 10 major quantitative trait loci (QTL) controlling grain weight and grain shape have been cloned and characterized [5,6,7,8,9]. Among them, GW2, GW7/GL7, GW8/OsSPL16 and GS9 regulate grain shape through altering cell division with influences on appearance quality like chalkiness [7, 10,11,12,13]. Moreover, several QTL for grain weight or grain shape have been identified, such as qGRL7.1 [14], qGL4b [15], qSS7 [16] and tgw11 [17], but the causal genes are yet to be characterized.
Next-generation sequencing (NGS) technologies have made genotyping more efficient and the availability of substantially increased SNP markers enables the exploration of quantitative trait nucleotides (QTNs)/genes for target traits more rapidly through genome-wide association study (GWAS), which has been widely applied in genetic dissection of agronomic traits in rice [18,19,20,21,22]. So far, several genes influencing grain weight and grain shape have already been identified by GWAS in rice, such as GLW7 for GL and TGW [23], OsLG3 for TGW and GL [24], GSE5 regulating grain width [25], and OsSNB controlling grain size [26]. Moreover, GWAS is also a powerful method to explore favorable alleles associated with important agronomic traits in rice natural populations and germplasm [27, 28]. Recently, the 3,000 Rice Genomes Project (3 K RGP) contributed 29 million single nucleotide polymorphisms (SNPs), 2.4 million small InDels and over 90,000 structural variations [29, 30]. The availability of this genomic data provides a valuable genetic resource for both scientific research and molecular breeding in rice [31, 32].
Here, we conducted a GWAS with a high-density SNP dataset using 2,453 accessions from the 3 K RGP followed by gene-based haplotype analysis to identify new candidate genes underlying rice grain weight and grain shape. The results of this study will enhance the understanding of the genetic basis of grain weight and grain shape, and provide valuable information for improving grain shape and grain yield in rice breeding.
Results
Phenotypic variations and correlations
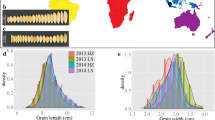
All four measured traits showed large variations in the whole population and between subgroups across the three years (Fig. 1a, Additional file 1: Table S2 and Table S3). ‘Nyao’ from Laos had the highest TGW (46.60 g), ‘GEANT W 7’ from Netherlands had the longest GL (11.82 mm), ‘DO LEUANG’ from Laos had the widest GW (4.17 mm), and ‘IET 14720’ had the largest RLW (4.55) (Additional file 1: Table S3). Among the 12 subgroups, subtropical geng had the highest mean TGW (31.38 g) primarily due to significantly longer GL (8.68 mm) and wider GW (3.51 mm) compared to the other subgroups, followed by intermediate geng and xian-3 (Additional file 1: Table S3). Moreover, xian-1B had the highest mean GL (8.73 mm) and RLW (3.34) while the overall lowest mean GW was found in the xian-1B subgroup. The basmati subgroup showed significantly lower TGW (19.86 g) and GL (7.25 mm) than all the other subgroups. Although the xian-1B, xian-3, and subtropical geng subgroups had similar GL, the two xian subgroups (xian-1B and xian-3) showed significantly larger RLW than subtropical geng.

Phenotypic distributions and correlations. a Box plots of TGW, GL, GW, and RLW in three years across 12 subgroups. TGW: thousand grain weight; GL: grain length; GW: grain width; RLW: ratio of GL to GW; G-inter: intermediate geng; G-subtrp: subtropical geng; G-tem: temperate geng; Inter: intermediate; X-1 A: xian-1 A; X-1B: xian-1B; X-2: xian-2; X-3: xian-3; X-inter: intermediate xian. b Distributions and correlations among the four traits in the whole population. c Correlations among the four traits from each subgroup. The number in the middle of the cell is the correlation coefficient; ‘*’, ‘**’, and ‘***’ refer to significant correlations (P < 0.05, P < 0.01, and P < 0.001)
TGW showed a significant positive correlation with both GL and GW in the whole population and 12 subgroups, with the strongest correlation (r = 0.85 and r = 0.71, P < 0.001) detected in the subtropical geng subgroup (Fig. 1b and c). GL was negatively correlated with GW in the whole population and most subgroups except for subtropical geng (r = 0.32, P < 0.01) and xian-3 (r = 0.13, P < 0.05). TGW was significantly negatively correlated with RLW in tropical geng subgroup (r = -0.32, P < 0.001) while a significant positive correlation between TGW and RLW was detected in the basmati (r = 0.26, P < 0.05) and subtropical geng (r = 0.22, P < 0.05) subgroups. All the four traits showed very high heritability ranging from 0.88 for GL to 0.93 for GW (Table 1).
Genome-wide LD patterns and QTN detection by GWAS
The maximum LD was 0.47, 0.64, 0.69, 0.53, 0.68, and 0.67 in the whole population, aus, basmati, xian, temperate geng and tropical geng, respectively. LD reached half of its initial value at around 300 kb in the whole population, basmati and temperate geng subgroups, 250 kb in the aus subgroup, 130 kb in the xian group and 260 kb in the tropical geng subgroup (Additional file 2: Fig. S1). Thus, the LD decay in the xian subgroup was much faster than in any of the other subgroups.
A total of 21 stable QTNs for the four traits were consistently identified in at least two years (Table 2; Additional file 2: Fig. S2A–S4F). For TGW, six QTNs were detected on chromosomes 3, 5, 7, 9, and 11. Among them, qTGW3.2 (rs3_16733441), qTGW5 (rs5_5371529), and qTGW7 (rs7_28305040) were stably detected in all three years with qTGW7 showing the strongest association signal (P = 8.12E-18) in the whole population. Two QTNs, qTGW3.1 (rs3_4504988) and qTGW9 (rs9_21404841), were detected only in the whole population while qTGW11 (rs11_3019935) was detected only in the xian subgroup.
Five QTNs for GL were mapped to chromosomes 3, 4, 5, 7, and 10. Three QTNs, qGL3 (rs3_16733441), qGL5 (rs5_5373491), and qGL7 (rs7_28289869) were consistently detected in the whole population and subgroups with qGL3 showing the strongest association signal (P = 1.97E-73) in the whole population. The QTNs, qGL4 (rs4_29308534) and qGL10 (rs10_13616240), were detected only in the whole population.
Four QTNs for GW were detected on chromosomes 3, 5, 7, and 8. Three QTNs, qGW3 (rs3_16733441), qGW5 (rs5_5371772), and qGW7 (rs7_24898274) were stably detected in all three years with qGW5 showing the strongest association signal (P = 1.84E-77) in the whole population. One QTN, qGW8 (rs8_26504638) was detected only in the aus subgroup in 2016 and 2017.
Six QTNs for RLW were identified on chromosomes 1, 3, 4, 5, 7, and 8. Four QTNs, qRLW3 (rs3_16733441), qRLW4 (rs4_29317460), qRLW5 (rs5_5371609), and qRLW7 (rs7_24533303) were consistently detected in the whole population and subgroups with qRLW5 showing the strongest association signal (P = 3.59E-69) in the whole population. Two QTNs, qRLW1 (rs1_3657795) and qRLW8 (rs8_26504638) were detected only in the aus subgroup.
We specified QTNs with overlapping regions identified for multiple traits as the same QTN, which lead to the identification of 11 different QTNs. Among them, qTGW3.2/qGL3/qGW3/qRLW3, qTGW5/qGL5/qGW5/qRLW5, qTGW7/qGL7, qGW7/qRLW7, and qGW8/qRLW8 were actually GS3, qSW5/GW5, FZP, GL7/GW7, and GW8, respectively, which are well-known genes controlling rice grain shape and grain weight. Notably, qTGW3.1, qTGW9, qTGW11, qGL4/qRLW4, qGL10, and qRLW1 were newly identified in this study (Table 2).
Candidate genes identification and haplotype analyses
The six newly identified QTNs were used for the high-density association and gene-based haplotype analyses to identify the candidate genes. In the region of qTGW3.1 (4.38–4.58 Mb on chromosome 3), 9,177 SNPs were used for high-density association analysis in the whole population. The annotated gene with the most significant hit was Os03g0186600 (Fig. 2a). Seven major haplotypes were detected among 2,066 accessions based on four SNPs in the 2-kb region upstream of the Os03g0186600 promoter, and four SNPs in the coding region (Fig. 2b). Significant differences for TGW were observed among the seven haplotypes in the whole population and several subgroups (Fig. 2c; Additional file 1: Table S4 and Table S5). In the whole population, Hap6, represented by 93 geng and intermediate accessions, had the highest mean TGW (28.68 g). Of the 73 subtropical geng accessions, 28 accessions with Hap3 had a mean TGW of 34.17 g and while a further 37 accessions had a mean TGW of 31.66 g. Hap2 showed a significantly higher TGW than the other haplotypes in the xian-1 A, xian-3, and intermediate xian subgroups (Fig. 2c).

a High-density association analysis of qTGW3.1 in 2015, 2016, and 2017. The solid line indicates the threshold to determine significant SNP. b-c Structure of candidate gene Os03g0186600 and haplotype analysis for TGW in the whole population and the 12 subgroups. Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P < 0.05)
The QTN qTGW9 was identified in a 21.24–21.55 Mb region on chromosome 9, including 9,593 SNPs used for high-density association analysis in the whole population. The most significant hit was located in Os09g0544400 (Additional file 2: Fig. S5). Three major haplotypes for Os09g0544400 were detected among the 2,270 accessions. Notably, Hap2 and Hap3 were enriched in the geng and xian subgroups, respectively, suggesting that this gene differentiated between the two subspecies. Among them, Hap2 was associated with the highest TGW of 25.76 g in the whole population (Additional file 1: Table S4 and Table S5).
qTGW11 was detected in the region from 2.82 Mb to 3.13 Mb on chromosome 11 in the xian subgroup, harboring 9,948 SNPs. Os11g0163600 was subsequently screened as the candidate gene for qTGW11 (Additional file 2: Fig. S6). A total of five haplotypes were identified in 2,056 accessions based on three SNPs in the 2-kb region upstream of the Os11g0163600 promoter, one SNP in the 5’ UTR, one SNP in the coding region, and one SNP in the 3’ UTR. Only two haplotypes, Hap2 and Hap3, were detected in the four geng subgroups while Hap2 showed a significantly higher mean TGW than Hap3 in intermediate geng and tropical geng subgroups (Additional file 1: Table S4 and Table S5).
For qGL4/qRLW4, Os04g0580700 was identified as the candidate gene (Fig. 3a). Five haplotypes were detected based on five SNPs in the 2-kb region upstream of its promoter and one SNP in the coding region (Fig. 3b). Hap5 was exclusively carried by 15 xian-3 accessions having the highest mean GL (9.72 mm) and mean RLW (3.62) (Fig. 3c). Moreover, Hap2 had a significantly higher GL and RLW than the other Haps in the aus, basmati, intermediate geng, and tropical geng subgroups (Fig. 3c; Additional file 1: Table S4 and Table S5).

a High-density association analysis of qGL4/qRLW4 in 2015, 2016, and 2017. The solid line indicates the threshold to determine significant SNP. b-c Structure of candidate gene Os04g0580700 and haplotype analysis for GL and RLW in the whole population and the 12 subgroups. Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P < 0.05)
Two candidate genes, Os10g0399700 and Os10g0400100, were identified for qGL10 (Additional file 2, Fig. S7). Four and six SNPs were used to classify haplotypes for Os10g0399700 and Os10g0400100, resulting in five and six haplotypes, respectively (Additional file 2, Fig. S7). Interestingly, Hap4 of Os10g0399700 and Hap5 of Os10g0400100 both had a significantly higher mean GL than the other haplotypes in the whole population, which were only carried by the accessions of the four geng subgroups (Additional file 1: Table S4 and Table S5).
qRLW1 was detected as an association peak of the region 3.57–3.80 Mb on chromosome 1 in the aus subgroup, including 8,116 SNPs used for high-density association analysis. The candidate gene with the most significant hit within an LD block was Os01g0171000 (Fig. 4a). Four major haplotypes were observed in 2,224 accessions based on nine SNPs located in the 2-kb promoter and coding region of Os01g0171000. Of the four major haplotypes, Hap1 had a significantly higher mean RLW than that of Hap2 in the whole population. More notably, Hap1 was mainly carried by xian accessions while Hap2 was mainly present in the geng and aus accessions (Fig. 4c; Additional file 1: Table S4 and Table S5).

a High-density association analysis of qRLW1 in 2015, 2016, and 2017. The solid line indicates the threshold to determine significant SNP. b-c Structure of candidate gene Os01g0171000 and haplotype analysis for RLW in the whole population and the 12 subgroups. Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P < 0.05)
Discussion
Abundant variations of grain weight and grain shape in rice germplasm
Generally speaking, TGW, GL, and RLW vary greatly among rice varieties, while GW and grain thickness changed much less [5, 36,37,38]. In this study, the coefficient of variations of TGW, GL, GW, and RLW ranged from 10.38 to 22.55 %, 7.56 % to 18.61, 6.73–13.84 %, and 12.29–26.16 % in the 12 rice subgroups, respectively (Additional file 1: Table S2). The basmati subgroup showed the highest level of phenotypic variation for both grain weight and grain shape, indicating its genetic diversity can be further exploited for rice breeding [39, 40]. Two xian subgroups, Xian-1 A and Xian-1B, showed less phenotypic variation for grain weight and grain shape than the other subgroups. For TGW, xian-1B showed a mean value of 24.22 g that is close to 24.89 g in xian-1 A. For grain shape, xian-1B showed significantly higher GL (8.73 mm versus 7.71 mm) and RLW (3.34 versus 2.51), and significantly smaller GW (2.65 mm versus 3.10 mm) than xian-1 A (Additional file 1: Table S3). Interestingly, xian-1 A mainly consists of landraces and is closely related phylogenetically to xian-1B that largely consists of modern varieties [29]. These results suggest that many genes related to grain shape have also been strongly selected for in xian-1B, which is equivalent to the IndII group with various breeding signatures caused by geographic adaptation and accumulation of divergent selections in distinct breeding pools [41].
Although dominant effects have also been detected in some studies, most genes reported are predominantly additive in effect for TGW, GL, GW, and RLW [3]. Compared with other grain shape traits, grain length is the major determinant of grain weight [42]. In the current study, we found six stable QTNs that had pleiotropic effects on grain shape and grain weight. For instance, two QTNs (qTGW3.2/qGL3/qGW3/qRLW3 and qTGW5/qGL5/qGW5/qRLW5) had pleiotropic effects on all four traits, and qTGW7/qGL7 showed pleiotropic effects on grain weight and grain length. The power to obtain different combinations of alleles conferring particular grain shapes and sizes from almost every subgroup has implications for the improvement of yield and grain quality, potentially enabling breeders to develop high-yielding varieties with specific grain quality to satisfy diverse quality requirements.
Identification of the favorable alleles among the previously cloned genes
Comparisons of QTNs detected in this study with the known genes for grain weight or grain shape were performed within 1 Mb physical distance around the lead SNP of each QTN based on the Nipponbare reference genome IRGSP-1.0. Of the 11 QTNs, five were found to co-locate in the same regions as previously cloned genes related to grain weight or grain shape in rice (Table 2). qTGW3.2/qGL3/qGW3/qRLW3 was mapped close to GS3, a major gene for grain size previously identified in the xian varieties [33]. qTGW5/qGL5/qGW5/qRLW5 covered qSW5/GW5 which regulates grain width and weight [34]. qGW7/qLWR7 was co-located with GL7/GW7 which controls GL and GW [11, 12]. qTGW7/qGL7 was at the same position as the gene FZP which regulates the number of spikelets per panicle, GL, and TGW [35]. qGW8/qLWR8 was mapped close to GW8 which controls grain size and quality [13].
Notably, all the five cloned genes were detected in the whole population and multiple subgroups except GW8, which was only detected in the aus subgroup, suggesting that functionally divergent alleles/haplotypes due to abundant SNP variations should exist in these genes. Despite these several important structural and functional features have been revealed for these proteins in grain size regulation [6, 11,12,13, 34, 35, 43], it is still of great significance for breeding high yielding and good quality rice varieties to discover the favorable alleles at these loci in the different subgroups. In this study, haplotype analyses of GS3 has successfully identified the G (C) to T (A) substitution at 16,733,441 bp on chromosome 3 which causes premature termination of the GS3 protein and results in long grain [44, 45]. Other than GL, 824 accessions with Hap1 (TATG) of GS3 also showed significantly higher means for both TGW and RLW than the other haplotypes in the whole population and most subgroups (Additional file 2, Fig. S8). Notably, Hap2 (GCGA) and Hap5 (GCTG) of GS3 were exclusively present in the xian accessions while Hap3 (GATG) was rarely present indicating that GS3 may be involved in the xian–geng differentiation in rice (Additional file 2, Fig. S8). For GW5, Hap4 (CGCGC) and Hap7 (CGCG-), mainly represented by the tropical geng subgroup, showed significantly higher GL, GW, and RLW than the other haplotypes in the whole population (Additional file 2, Fig. S9). A total of 915 accessions with Hap1 (GAGACGAGA) of GL7 were predominantly assigned to the xian subgroups while 475 accessions carrying the Hap2 (ACCGACGAG) of GL7 were largely in the geng subgroups (Additional file 2, Fig. S10). In the whole population, Hap2 of GL7 showed significantly higher mean TGW, GL, and GW than Hap1 while Hap1 showed significantly higher mean RLW than that of Hap2 (Additional file 2, Fig. S10). For FZP, Hap2 (A) displayed a significantly higher mean GL and RLW and significantly lower GW than Hap1 (G) in the whole population (Additional file 2, Fig. S11). Eight haplotypes of GW8 were classified by eight SNPs (Additional file 2, Fig. S12). A total of 124 accessions belonged to the geng type were assigned in Hap4 (TGCGTGTA) which exhibits the highest TGW, GL, and GW in the whole population. So far, these genes have been widely used in breeding programs, mining of their favorable haplotypes/alleles may facilitate the rational design of grain shape and weight.
New candidate genes and their future application for improvement of grain yield and quality in rice breeding
Of the three major components (panicle number per plant, number of grains per panicle, and grain weight) of rice yield, grain weight is the most stable and heritable trait, which is measured as the TGW. Moreover, three of the main parameters (GL, GW, and RLW) of grain are positively correlated with grain weight [3]. Meanwhile, grain shape is an important quality trait that affects the market value of rice products. In the current study, seven promising genes located in the six new QTNs for grain weight and grain shape were identified using a large natural population with 2,453 accessions (Table 3). For qRLW1, Os01g0171000 encodes a BRASSINOSTEROID INSENSITIVE 1 (BRI1)-Associated receptor Kinase (BAK). Several components of the BR signaling pathway in rice, such as OsBRI1 (Brassinosteroid-Insensitive1) [46], OsBAK1 (BRI1-Associated receptor Kinase1) [47, 48], and SERK2 [49] have been proven to regulate plant architecture including grain size. Os03g0186600 underlying qTGW3.1 is annotated as OsMDP1 which negatively regulates brassinosteroid (BR) signaling [50]. Several cloned genes, such as GW5 [34], GS5 [51], and GL3.1/qGL3 [52] were also suggested to regulate grain size through the BR signalling pathway. Elucidating the molecular mechanisms of Os01g0171000 and Os03g0186600 is essential to make certain of their roles in mediating BR signalling and grain size. The candidate gene Os04g0580700 for qGL4/qRLW4 encodes a rice MADS-box transcription factor OsMADS17. Two genes related to grain size, OsLG3b (OsMADS1) and FZP, regulate the expression of OsMADS17 [53,54,55], indicating that Os04g0580700 may have latent impacts on grain shape and weight.
The haplotypes of genes related to grain weight and grain shape and their distribution in different subgroups obtained in this study can provide more options for breeding by molecular design. When combined with the Kompetitive Allele Specific PCR (KASP) technique [56], breeders can efficiently pyramid favorable alleles of multiple genes that control grain weight and grain shape in rice. Furthermore, precise genome-editing techniques in plants are an alternative important tool for molecular plant breeding [57, 58]. The application of the CRISPR/Cas9-derived system on GW5 has proven that genome-editing can verify the result of GWAS precisely in a short time [34]. Thus, the favorable haplotypes detected in this study and their further verified functional SNPs will provide useful resources for precise gene editing breeding. Here, we listed dozens of elite rice accessions with combinations of favorable haplotypes of the seven new candidate genes and five well-known genes (Additional file 1: Table S6). For instance, ‘KHAO’ NGAW’ (TGW = 42.76 g, GL = 11.33mm), which is from Thailand and belongs to the subtropical geng subgroup, carryies the favorable haplotypes of GS3, GW8, and Os03g0186600 for TGW and GL. ‘Diandun 502’ (TGW = 34.04 g, RLW = 3.41) and ‘Mengguandamagu’ (TGW = 33.12 g, RLW = 3.39) are both from China and belong to the intermediate xian subgroup, with the favorable haplotypes of GS3, Os03g0186600, Os09g0544400, and Os10g0399700 for TGW and GL. ‘UP15’ (TGW = 32.29 g, RLW = 3.62) is from Japan and belongs to basmati subgroup, carrying the favorable haplotypes of Os01g0171000 and Os09g0544400 for RLW and TGW. ‘RACHANDRABHOG’ (TGW = 31.36 g, RLW = 3.73) from India belongs to xian-2 subgroup, carrying the favorable haplotypes of GS3, FZP, and Os01g0171000 for TGW, GL and RLW. These accessions with high TGW (> 30 g) and RLW (> 3.0) could be used as donor parents for rice breeding and as genetic materials for further functional research.
Conclusions
There was significant variation in grain weight and grain shape among the 12 rice subgroups that allowed the identification of favourable genes and haplotypes influencing these important traits. Six new QTNs (qTGW3.1, qTGW9, qTGW11, qGL4/ qRLW4, qGL10, and qRLW1) were identified for grain weight and grain shape by GWAS in a large natural population, and seven candidate genes were screened via high-density association and gene-based haplotype analyses. The results enhance our understanding of the genetic basis of grain weight and grain shape in rice and provide valuable information for elucidating the molecular mechanisms underlying these traits. The exploitation of favorable haplotypes and germplasm resources will be useful for improving rice grain yield and grain quality by molecular breeding.
Methods
Plant materials
A total of 2,453 accessions (Additional file 1: Table S1) from the 3 K RGP were used to test the association between the SNP genotype and phenotype of grain weight and grain shape. Based on the known population structure [29], these accessions belong to 12 subgroups, including aus (182 accessions), basmati (61 accessions), intermediate (81), intermediate geng (77 accessions), subtropical geng (79 accessions), temperate geng (260 accessions), tropical geng (291 accessions), xian-1 A (206 accessions), xian-1B (181 accessions), xian-2 (246 accessions), xian-3 (257 accessions) and intermediate xian (532 accessions).
Field trials and trait measurements
All accessions were grown at Sanya, China (18.3◦N, 109.3◦E) for three consecutive years from 2015 to 2017. Seeds were surface sterilized and approximately 100 seeds of each accession were sowed on 8 November 2015, 5 November 2016, and 20 November 2017. At 25 days after sowing, the seedlings were transplanted into a three-row plot with 10 individuals in each row at a spacing of 20 cm × 25 cm. Field trials were carried out following a randomized complete block design with two replications in each year. The field management followed the local farmers’ standard practices. At the full-ripe stage (about 40 days after flowering), seeds of eight plants in the middle of each plot were bulk harvested and air-dried in the screen house until reaching a constant seed weight.
The method of trait measurement using the automatic seed counting and analyzing instrument (Model SC-G, Hangzhou Wanshen Detection Technology Co., Ltd., Hangzhou, China, http://www.wseen.com/) has been described before [6, 59]. Briefly, at least 300 fully-filled seeds of each accession were scattered evenly on a flat-bed scanner (30 cm × 25 cm) and imaged with a high-resolution camera. The seeds number, GL in mm, GW in mm and RLW were calculated by analyzing the image via the grain analyzer software using the rice model. Then the weight of these seeds was measured using a high precision electronic balance (1/1000, APTP456 series) and the TGW in grams was subsequently calculated. The scanner was calibrated with a calibration target before each measurement.
Statistical analyses of phenotypic data
The R package ‘lme4’ [60] was used to obtain the best linear unbiased estimate (BLUE) for each genotype-environment combination and variance components using linear mixed models. Genotype and replication were treated as a fixed effect and a random effect for single-environment analysis, respectively. The BLUEs of each year were subsequently calculated and used for analysis of variance (ANOVA) and GWAS. Variance components were estimated using multi-environment analysis with genotype treated as a fixed effect while environment, replicate within an environment and genotype-by-environment interaction treated as random effects. The narrow-sense heritability (h2) was estimated as: h2 = Vg/(Vg +Vgei/s + Ve/sr), where Vg, Vgei, and Ve are the variance contributed by genotype, genotype-by-environment interaction, and residual error, respectively, while s is the number of environments and r is the number of replicates. Then, the BLUEs of three years for each accession were calculated and used for computing the Pearson’s correlation coefficients among traits with the ‘Hmisc’ package in R. One-way ANOVA followed by Duncan’s multiple range test were used for statistical comparisons across multiple means of TGW, GL, GW, and RLW among the 12 rice subgroups by the ‘agricolae’ package in R.
Genome-wide association mapping
We conducted association studies to identify genome-wide signals associated with grain weight and grain shape in the whole population and five major subgroups, aus, basmati, xian (including xian-1A, xian-1B, xian-2, xian-3, and intermediate xian), temperate geng, and tropical geng, to minimize the impact of population structure on the power of GWAS. The 4.8 M SNP dataset of 3K RGP was downloaded from the Rice SNP-Seek Database [61]. After filtering out SNPs with a missing rate of more than 20 % or with a minor allele frequency (MAF) less than 5 % using PLINK software [62] with the parameter ‘--geno 0.2’, and ‘ --maf 0.05’, a total of 3,343,302, 2,240,362, 865,777, 1,548,277, 1,884,822,and 1,728,815 SNPs were retained for GWAS in the whole population, xian, temperate geng, tropical geng, aus and basmati subgroups, respectively.
A single-locus GWAS was performed with a linear mixed model to determine the association between each SNP and the measured phenotypes by an efficient mixed-model analysis with EMMA eXpedited (EMMAX) software [63]. The kinship matrix was generated using an identical-by-state matrix based on the subset of genome-wide SNP data with the ‘indep-pairwise 50 10 0.1’ parameter in PLINK to account for the relatedness among accessions. The first three principal components were used as covariates (Q-matrix) to control population structure. The effective number of independent markers (N) was calculated using the GEC software [64] and suggestive P-value thresholds of association (1/N) were 1.76E-6, 2.40E-6, 1.03E-5, 5.63E-6, 4.08E-6, and 5.93E-6 for the whole population, xian, temperate geng, tropical geng, aus, and basmati, respectively. These suggestive P-value thresholds were used to claim significant SNP-trait associations/QTNs for the whole population and the five subgroups, respectively. Manhattan and quantile-quantile (Q–Q) plots of GWAS were created by the R package ‘qqman’ [65].
Linkage Disequilibrium (LD) decay estimates
We calculated r2, an estimation of LD, using PLINK software with the parameter ‘–r2 –ld-window-kb 1000 –ld-window 99999 –ld-window-r2 0’. The LD decay rate was measured as the chromosomal distance at which the average r2 dropped to half of its maximum value. The LD block harboring significant trait-associated SNPs was defined as the candidate region for each QTN, and the SNP with the minimum P-value within an LD block was considered as the lead SNP.
Candidate gene identification
High-density association and gene-based haplotype analyses were carried out to detect candidate genes for stable QTNs that could be detected in at least two years and were newly identified in our study. The following five steps were conducted to identify candidate genes for each QTN: 1) all available SNPs located in the QTN region were acquired from 29 M SNPs data of 3K RGP in the Rice SNP-Seek Database [61]; 2) the SNPs were filtered with the aforementioned parameters and the remaining high-quality SNPs were used to perform high-density association analyses through EMMAX software; 3) the gene with the most significant hit within a local LD block constructed around the stable QTNs was screened as the candidate gene. The R package ‘LDheatmap’ [66] was used to draw the heatmap of pairwise LDs; 4) the annotated genes from the Rice Genome Annotation Project Database (RAP-DB) [67] that harbor the significant SNPs were selected as the candidate genes; 5) gene-based haplotype analysis was carried out for each candidate gene. The SNPs within 2 kb of the upstream promoter region, 3’ untranslated region (UTR), and 5’ UTR, as well as non-synonymous SNPs in the coding regions of a candidate gene, were concatenated as the haplotype. Only major haplotypes of each candidate gene carried by at least 15 accessions in the whole population and at least 5 accessions in subgroups were used for multiple comparisons. BLUEs of three years were applied for the multiple comparisons of the major haplotypes. Duncan’s multiple comparison tests (5 % significance level) followed by one-way ANOVA were completed with the ‘agricolae’ package in R.
Availability of data and materials
The 4.8 M and 29 M SNP datasets used and/or analyzed during the current study are available from the Rice SNP-Seek Database (https://snp-seek.irri.org/_download.zul). The phenotype dataset used during the current study is provided in the Addational file 1: Table S7.
Abbreviations
- TGW:
-
Thousand grain weight
- GL:
-
Grain length
- GW:
-
Grain width
- RLW:
-
Ratio of grain length to grain width
- 3 K RGP:
-
3,000 Rice Genome Project
- ANOVA:
-
Analysis of variance
- GWAS:
-
Genome-wide association study
- QTL:
-
Quantitative trait locus/loci
- QTNs:
-
Quantitative trait nucleotides
- SNP:
-
Single nucleotide polymorphism
- MAF:
-
Minor allele frequency
- kb:
-
Kilobyte
- Mb:
-
Megabyte
- LD:
-
Linkage disequilibrium
- CDS:
-
Coding DNA sequence
- UTR:
-
Untranslated region
References
Ray DK, Mueller ND, West PC, Foley JA. Yield trends are insufficient to double global crop production by 2050. PLoS One. 2013;8(6):e66428.
Gong J, Miao J, Zhao Y, Zhao Q, Feng Q, Zhan Q, Cheng B, Xia J, Huang X, Yang S, et al. Dissecting the genetic basis of grain shape and chalkiness traits in hybrid rice using multiple collaborative populations. Mol Plant. 2017;10(10):1353–6.
Tan Y, Xing Y, Li J, Yu S, Xu C, Zhang Q. Genetic bases of appearance quality of rice grains in Shanyou 63, an elite rice hybrid. Theor Appl Genet. 2000;101(5):823–9.
Liu X, Mou C, Zhou C, Cheng Z, Jiang L, Wan J. Research progress on cloning and regulation mechanism of rice grain shape genes. Chin J Rice Sci. 2018;32(1):1–11.
Huang R, Jiang L, Zheng J, Wang T, Wang H, Huang Y, Hong Z. Genetic bases of rice grain shape: so many genes, so little known. Trends Plant Sci. 2013;18(4):218–26.
Zhang L, Ma B, Bian Z, Li X, Zhang C, Liu J, Li Q, Liu Q, He Z. Grain size selection using novel functional markers targeting 14 genes in rice. Rice. 2020;13(1):63.
Zhao D-S, Li Q-F, Zhang C-Q, Zhang C, Yang Q-Q, Pan L-X, Ren X-Y, Lu J, Gu M-H, Liu Q-Q. GS9 acts as a transcriptional activator to regulate rice grain shape and appearance quality. Nat Commun. 2018;9(1):1240.
Guo T, Chen K, Dong N-Q, Shi C-L, Ye W-W, Gao J-P, Shan J-X, Lin H-X. GRAIN SIZE AND NUMBER1 negatively regulates the OsMKKK10-OsMKK4-OsMPK6 cascade to coordinate the trade-off between grain number per panicle and grain size in rice. Plant Cell. 2018;30(4):871.
Zuo J, Li J. Molecular genetic dissection of quantitative trait loci regulating rice grain size. Annu Rev Genet. 2014;48:99–118.
Song X, Huang W, Shi M, Zhu M, Lin H. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet. 2007;39(5):623–30.
Wang S, Li S, Liu Q, Wu K, Zhang J, Wang S, Wang Y, Chen X, Zhang Y, Gao C, et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat Genet. 2015;47(8):949–54.
Wang Y, Xiong G, Hu J, Jiang L, Yu H, Xu J, Fang Y, Zeng L, Xu E, Xu J, et al. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat Genet. 2015;47(8):944–8.
Wang S, Wu K, Yuan Q, Liu X, Liu Z, Lin X, Zeng R, Zhu H, Dong G, Qian Q, et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet. 2012;44(8):950–4.
Singh R, Singh AK, Sharma TR, Singh A, Singh NK. Fine mapping of grain length QTLs on chromosomes 1 and 7 in Basmati rice (Oryza sativa L.). J Plant Biochem Biotechnol. 2012;21(2):157–66.
Kato T, Segami S, Toriyama M, Kono I, Ando T, Yano M, Kitano H, Miura K, Iwasaki Y. Detection of QTLs for grain length from large grain rice (Oryza sativa L.). Breed Sci. 2011;61(3):269–74.
Qiu X, Gong R, Tan Y, Yu S. Mapping and characterization of the major quantitative trait locus qSS7 associated with increased length and decreased width of rice seeds. Theor Appl Genet. 2012;125(8):1717–26.
Oh J-M, Balkunde S, Yang P, Yoon D-B, Ahn S-N. Fine mapping of grain weight QTL, tgw11 using near isogenic lines from a cross between Oryza sativa and O. grandiglumis. Genes Genomics. 2011;33(3):259–65.
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42(11):961–76.
Zhao K, Tung C-W, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J, et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun. 2011;2(1):467.
Crowell S, Korniliev P, Falcão A, Ismail A, Gregorio G, Mezey J, McCouch S. Genome-wide association and high-resolution phenotyping link Oryza sativa panicle traits to numerous trait-specific QTL clusters. Nat Commun. 2016;7(1):10527.
Huang X, Zhao Y, Wei X, Li C, Wang A, Zhao Q, Li W, Guo Y, Deng L, Zhu C, et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat Genet. 2012;44(1):32–9.
Yano K, Yamamoto E, Aya K, Takeuchi H, Lo P-c, Hu L, Yamasaki M, Yoshida S, Kitano H, Hirano K, et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat Genet. 2016;48(8):927–34.
Si L, Chen J, Huang X, Gong H, Luo J, Hou Q, Zhou T, Lu T, Zhu J, Shangguan Y, et al. OsSPL13 controls grain size in cultivated rice. Nat Genet. 2016;48(4):447–56.
Yu J, Xiong H, Zhu X, Zhang H, Li H, Miao J, Wang W, Tang Z, Zhang Z, Yao G, et al. OsLG3 contributing to rice grain length and yield was mined by Ho-LAMap. BMC Biol. 2017;15(1):28.
Duan P, Xu J, Zeng D, Zhang B, Geng M, Zhang G, Huang K, Huang L, Xu R, Ge S, et al. Natural variation in the promoter of GSE5 contributes to grain size diversity in rice. Mol Plant. 2017;10(5):685–94.
Ma X, Feng F, Zhang Y, Elesawi IE, Xu K, Li T, Mei H, Liu H, Gao N, Chen C, et al. A novel rice grain size gene OsSNB was identified by genome-wide association study in natural population. PLOS Genetics. 2019;15(5):e1008191.
McCouch SR, Wright MH, Tung C-W, Maron LG, McNally KL, Fitzgerald M, Singh N, DeClerck G, Agosto-Perez F, Korniliev P, et al. Open access resources for genome-wide association mapping in rice. Nat Commun. 2016;7(1):10532.
Zhai L, Zheng T, Wang X, Wang Y, Chen K, Wang S, Wang Y, Xu J, Li Z. QTL mapping and candidate gene analysis of peduncle vascular bundle related traits in rice by genome-wide association study. Rice. 2018;11(1):13.
Wang W, Mauleon R, Hu Z, Chebotarov D, Tai S, Wu Z, Li M, Zheng T, Fuentes RR, Zhang F, et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature. 2018;557(7703):43–9.
The-3K-rice-genomes-project. The 3,000 rice genomes project. GigaScience. 2014;3(1):2047–217X.
Zheng T, Yu H, Zhang H, Wu Z, Wang W, Tai S, Chi L, Ruan J, Wei C, Shi J. Rice functional genomics and breeding database (RFGB)-3K-rice SNP and InDel sub-database. Chin Sci Bull. 2015;60(4):367–71.
Wang C, Yu H, Huang J, Wang W, Faruquee M, Zhang F, Zhao X, Fu B, Chen K, Zhang H, et al. Towards a deeper haplotype mining of complex traits in rice with RFGB v2.0. Plant Biotechnol J. 2020;18(1):14–6.
Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet. 2006;112(6):1164–71.
Liu J, Chen J, Zheng X, Wu F, Lin Q, Heng Y, Tian P, Cheng Z, Yu X, Zhou K, et al. GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nature Plants. 2017;3(5):17043.
Bai X, Huang Y, Hu Y, Liu H, Zhang B, Smaczniak C, Hu G, Han Z, Xing Y. Duplication of an upstream silencer of FZP increases grain yield in rice. Nat Plants. 2017;3(11):885–93.
Qiu X, Pang Y, Yuan Z, Xing D, Xu J, Dingkuhn M, Li Z, Ye G. Genome-wide association study of grain appearance and milling quality in a worldwide collection of Indica rice germplasm. PLOS ONE. 2016;10(12):e0145577.
Ngangkham U, Samantaray S, Yadav MK, Kumar A, Chidambaranathan P, Katara JL. Effect of multiple allelic combinations of genes on regulating grain size in rice. PLOS ONE. 2018;13(1):e0190684.
Liu C, Song J, Wang Y, Huang X, Zhang F, Wang W, Xu J, Zhang Y, Yu H, Pang Y, et al. Rapid prediction of head rice yield and grain shape for genome-wide association study in indica rice. J Cereal Sci. 2020;96:103091.
Jain S, Jain RK, McCouch SR. Genetic analysis of Indian aromatic and quality rice (Oryza sativa L.) germplasm using panels of fluorescently-labeled microsatellite markers. Theor Appl Genet. 2004;109(5):965–77.
Roy S, Banerjee A, Mawkhlieng B, Misra AK, Pattanayak A, Harish GD, Singh SK, Ngachan SV, Bansal KC. Genetic diversity and population structure in aromatic and quality rice (Oryza sativa L.) landraces from North-eastern India. PloS one. 2015;10(6):e0129607-7.
Xie W, Wang G, Yuan M, Yao W, Lyu K, Zhao H, Yang M, Li P, Zhang X, Yuan J, et al. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc Natl Acad Sci. 2015;112(39):E5411.
Shi C, Shen Z. Additive and dominance correlation analysis of grain shape and yield traits in Indica rice. Acta Agronomica Sinica. 1996;22(1):36–42.
Mao H, Sun S, Yao J, Wang C, Yu S, Xu C, Li X, Zhang Q. Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc Natl Acad Sci USA. 2010;107(45):19579–84.
Takano-Kai N, Jiang H, Kubo T, Sweeney M, Matsumoto T, Kanamori H, Padhukasahasram B, Bustamante C, Yoshimura A, Doi K, et al. Evolutionary history of GS3, a gene conferring grain length in rice. Genetics. 2009;182(4):1323.
Fan C, Yu S, Wang C, Xing Y. A causal C–A mutation in the second exon of GS3 highly associated with rice grain length and validated as a functional marker. Theor Appl Genet. 2009;118(3):465–72.
Morinaka Y, Sakamoto T, Inukai Y, Agetsuma M, Kitano H, Ashikari M, Matsuoka M. Morphological alteration caused by brassinosteroid insensitivity increases the biomass and grain production of rice. Plant Physiol. 2006;141(3):924–31.
Li D, Wang L, Wang M, Xu Y-Y, Luo W, Liu Y-J, Xu Z-H, Li J, Chong K. Engineering OsBAK1 gene as a molecular tool to improve rice architecture for high yield. Plant Biotechnol J. 2009;7(8):791–806.
Yuan H, Fan S, Huang J, Zhan S, Wang S, Gao P, Chen W, Tu B, Ma B, Wang Y, et al. SG2/OsBAK1 regulates grain size and number, and functions differently in Indica and Japonica backgrounds in rice. Rice. 2017;08(1):25.
Dong N, Yin W, Liu D, Zhang X, Yu Z, Huang W, Liu J, Yang Y, Meng W, Niu M, et al. Regulation of brassinosteroid signaling and salt resistance by SERK2 and potential utilization for crop improvement in rice. Front Plant Sci. 2020;11:621859.
Duan K, Li L, Hu P, Xu S, Xu Z, Xue H. A brassinolide-suppressed rice MADS-box transcription factor, OsMDP1, has a negative regulatory role in BR signaling. Plant J. 2006;47(4):519–31.
Xu C, Liu Y, Li Y, Xu X, Xu C, Li X, Xiao J, Zhang Q. Differential expression of GS5 regulates grain size in rice. J Exp Bot. 2015;66(9):2611–23.
Zhang X, Wang J, Huang J, Lan H, Wang C, Yin C, Wu Y, Tang H, Qian Q, Li J, et al. Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proc Natl Acad Sci. 2012;109(52):21534–9.
Hu Y, Liang W, Yin C, Yang X, Ping B, Li A, Jia R, Chen M, Luo Z, Cai Q, et al. Interactions of OsMADS1 with floral homeotic genes in rice flower development. Mol Plant. 2015;8(9):1366–84.
Bai X, Huang Y, Mao D, Wen M, Zhang L, Xing Y. Regulatory role of FZP in the determination of panicle branching and spikelet formation in rice. Sci Rep. 2016;6(1):19022.
Yu J, Miao J, Zhang Z, Xiong H, Zhu X, Sun X, Pan Y, Liang Y, Zhang Q, Abdul Rehman RM, et al. Alternative splicing of OsLG3b controls grain length and yield in japonica rice. Plant Biotechnol J. 2018;16(9):1667–78.
Semagn K, Babu R, Hearne S, Olsen M. Single nucleotide polymorphism genotyping using Kompetitive Allele Specific PCR (KASP): overview of the technology and its application in crop improvement. Mol Breeding. 2014;33(1):1–14.
Zhang Y, Pribil M, Palmgren M, Gao C. A CRISPR way for accelerating improvement of food crops. Nature Food. 2020;1(4):200–5.
Zhang Y, Massel K, Godwin ID, Gao C. Applications and potential of genome editing in crop improvement. Genome Biol. 2018;19(1):210.
Li J, Zhao J, Li Y, Gao Y, Hua S, Nadeem M, Sun G, Zhang W, Hou J, Wang X, et al. Identification of a novel seed size associated locus SW9-1 in soybean. Crop J. 2019;7(4):548–59.
Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Statistical Software. 2015;67(1):1–48.
Alexandrov N, Tai S, Wang W, Mansueto L, Palis K, Fuentes RR, Ulat VJ, Chebotarov D, Zhang G, Li Z, et al. SNP-Seek database of SNPs derived from 3000 rice genomes. Nucleic Acids Res. 2015;43(D1):D1023–7.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Kang HM, Sul JH, Service SK, Zaitlen NA, Kong S-y, Freimer NB, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–54.
Li M-X, Yeung JMY, Cherny SS, Sham PC. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum Genet. 2012;131(5):747–56.
Turner SD. qqman: an R package for visualizing GWAS results using QQ and manhattan plots. J Open Source Software. 2018;3(25):731.
Shin J-H, Blay S, McNeney B, Graham J. LDheatmap: an R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. Journal of statistical software. 2006;16(3):1–10.
Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6(1):4.
Acknowledgements
We would like to thank Professor Meixue Zhou (Tasmanian Institute of Agriculture, University of Tasmania) for his critical reading of the manuscript and suggestions on data analysis. We also thank Guoliang Cao for field management, and Laiyuan Zhai for seed collection.
Funding
This work was funded by the National Key R&D Program of China (2017YFD0102002, 2017YFD0100100),the 863 Key Project from the Chinese Ministry of Science & Technology (Grant No. 2014AA10A601), and the Agricultural Science and Technology Innovation Program and the Cooperation and Innovation Mission (Grant No. CAAS-ZDXT202101). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
JX conceived and supervised the study; YN, KC, CS, ZW, HC, SZ collected data; YN, TC, CW, and TZ conducted analyses; YN and TC wrote the manuscript; FZ and JX revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no conflict interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Informationok
Additional file 1: Table S1.
Summary of 2,453 rice accessions used in our study. Table S2. Analysis of the variance in traits related to grain weight and grain shape. Table S3. Performance of grain weight and grain shape in different subgroups. Table S4. ANOVA for haplotypes of 7 candidate genes associated with grain weight and grain shape in the whole population and each subgroup. Table S5. Haplotype analyses of candidate genes associated with grain weight and grain shape. Table S6. Elite accessions with high TGW and RLW. Table S7. Raw phenotyping data of 2453 accessions collected in 2015, 2016 and 2017.
Additional file 2: Figure S1.
Comparison of LD decay in the whole population and five major subgroups. Y axis was the average r2 values of each 1 Mb region and X axis was physical distance between markers in unit of Mb. Figure S2. A‒F. Manhattan and QQ plots for TGW, GL, GW and RLW of the whole population (S2A), aus subgroup (S2B), basmati subgroup (S2C), xiansubgroup (S2D), temperate gengsubgroup (S2E), tropical gengsubgroup (S2F) in 2015. Figure S3. A‒F. Manhattan and QQ plots for TGW, GL, GW and RLW of the whole subgroup (S3A), aus subgroup (S3B), basmati subgroup (S3C), xian subgroup (S3D), temperate geng subgroup (S3E), tropical geng subgroup (S3F) in 2016.Figure S4. A‒F. Manhattan and QQ plots for TGW, GL, GW and RLW of the whole subgroup (S4A), aus subgroup (S4B), basmati subgroup (S4C), xian subgroup (S4D), temperate geng subgroup (S4E), tropical geng subgroup (S4F) in 2017. Figure S5. (a) High-density association analysis of qTGW9 in 2015, 2016, and 2017. The solid line indicates the threshold to determine significant SNP. (b-c) Gene structural of candidate gene Os09g0544400 and haplotype analysis for TGW in the whole population and the 12 subgroups. Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P < 0.05). Figure S6. High-density association analysis of qTGW11 in 2015, 2016, and 2017. The solid line indicates the threshold to determine significant SNP. (b-c) Gene structural of candidate geneOs11g0163600 and haplotype analysis for TGW in the whole population and the 12 subgroups. Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P < 0.05). Figure S7. (a) High-density association analysis of qGL10 in 2015, 2016, and 2017. The solid line indicates the threshold to determine significant SNP. (b‒c) Gene structural of candidate genes Os10g0399700 and Os10g0400100, and haplotype analyses for GL in the whole population and the 12 subgroups. Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P < 0.05). Figure S8. Haplotypes analysis ofGS3.Error bars, standard error of the mean (SEM). Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P< 0.05). Figure S9. Haplotypes analysis of GW5.Error bars, standard error of the mean (SEM). Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P < 0.05). Figure S10. Haplotypes analysis ofGL7.Error bars, standard error of the mean (SEM). Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P< 0.05). Figure S11. Haplotypes analysis of FZP.Error bars, standard error of the mean (SEM). Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P < 0.05). Figure S12. Haplotypes analysis ofGW8.Error bars, standard error of the mean (SEM). Characters on top of boxplots indicate significant differences based on Duncan’s multiple comparison tests (P< 0.05).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Niu, Y., Chen, T., Wang, C. et al. Identification and allele mining of new candidate genes underlying rice grain weight and grain shape by genome-wide association study. BMC Genomics 22, 602 (2021). https://doi.org/10.1186/s12864-021-07901-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07901-x