
Abstract
Background
The typical single-chromosome mitochondrial (mt) genome of animals has fragmented into multiple minichromosomes in the lineage Mitodivisia, which contains most of the parasitic lice of eutherian mammals. These parasitic lice differ from each other even among congeneric species in mt karyotype, i.e. the number of minichromosomes, and the gene content and gene order in each minichromosome, which is in stark contrast to the extremely conserved single-chromosome mt genomes across most animal lineages. How fragmented mt genomes evolved is still poorly understood. We use Polyplax sucking lice as a model to investigate how tRNA gene translocation shapes the dynamic mt karyotypes.
Results
We sequenced the full mt genome of the Asian grey shrew louse, Polyplax reclinata. We then inferred the ancestral mt karyotype for Polyplax lice and compared it with the mt karyotypes of the three Polyplax species sequenced to date. We found that tRNA genes were entirely responsible for mt karyotype variation among these three species of Polyplax lice. Furthermore, tRNA gene translocation observed in Polyplax lice was only between different types of minichromosomes and towards the boundaries with the control region. A similar pattern of tRNA gene translocation can also been seen in other sucking lice with fragmented mt genomes.
Conclusions
We conclude that inter-minichromosomal tRNA gene translocation orientated towards the boundaries with the control region is a major contributing factor to the highly dynamic mitochondrial genome organization in the parasitic lice of mammals.
Similar content being viewed by others


Background
Extensive fragmentation of mitochondrial (mt) genome was discovered first in three species of human lice, in which the single mt chromosome typical of animals evolved into 14 and 20 minichromosomes; each minichromosome has 1–5 genes and is 1.8–4 kb in size [1, 2]. Since then, 26 more species of parasitic lice have been sequenced for mt genomes [3,4,5,6,7,8,9,10,11,12]. It appears that mt genome fragmentation occurred at least twice in parasitic lice: once 60–90 million years ago (MYA) in the most recent common ancestor of Mitodivisia - a newly identified clade that contains the vast majority of parasitic lice of eutherian mammals [10, 13,14,15], and later ~ 25 MYA in the feather lice of the genus Columbicola [11, 13].
Unlike the extremely conserved single-chromosome mt genomes in most animal lineages [16] but somehow resembling the nuclear genomes of eukaryotes [17,18,19], the mt genome organization is highly dynamic in parasitic lice of the Mitodivisia clade [10]. These lice differ from each other, even among closely related species in the same genus, in mt karyotype, i.e. the number of minichromosomes, and the gene content and gene order in each minichromosome [8, 12]. The pig lice, Haemapinus apri and Haemapinus suis, and the horse louse, Haemapinus asini, have nine minichromosomes [3, 6], whereas the human head louse, Pediculus humanus capitis, and the human body louse, Pediculus humanus humanus, have 20 minichromosomes [1, 2]. Between these extremes, the elephant louse, Haematomyzus elephantis, has 10 minichromosomes (four genes not identified) [8]. The rodent lice, Polyplax asiatica, Polyplax spinulosa, Hoplopleura akanezumi (nine genes not identified) and Hoplopleura kitti (three genes not identified), have 11 minichromosomes [4, 5]. The guanaco louse, Microthoracius praelongiceps, the sheep louse, Bovicola ovis, the cattle louse, Bovicola bovis, and the macaque louse, Pedicinus obtusus, have 12 minichromosomes [9, 10, 12]. The goat louse, Bovicola caprae, and the dog louse, Trichodectes canis, have 13 minichromosomes [10]. The human pubic louse, Pthirus pubis (one gene not identified), and the colobus louse, Pedicinus badii, have 14 minichromosomes [2, 12]. Each minichromosome has 1 to 8 genes; the arrangement of genes in a minichromosome varies from species to species even within a genus; the only exceptions are: (1) the human head louse and the human body louse in the genus Pediculus, which diverged 83,000-170,000 years ago at the origin of clothing [1, 2, 20,21,22]; and (2) the domestic pig louse and the wild pig louse in the genus Haemapinus, which diverged ~ 10,000 years ago when pigs were domesticated [3, 23].
Why is the fragmented mt genome organization so variable in these parasitic lice? A few comparative studies to date have suggested four models or mechanisms. First, Shao et al., (2012) [2] showed that point mutation at the third anti-codon position or homologous recombination between tRNA genes accounted for the swap of identity and location between trnL1 and trnL2 and between trnR and trnG in the human lice. Dong et al., (2014a) [4] confirmed that this mechanism accounted for the swap of identity and location between trnL1 and trnL2 in the spiny rat louse, Polyplax spinulosa. Second, Shao et al., (2012) [2] showed that one minichromosome could split into two via gene degeneration followed by deletion, based on analysis of pseudo gene sequences of the human pubic louse. Third, Song et al., (2014) [6] showed that inter-minichromosomal recombination accounted for gene translocation in the horse louse, Haematopinus asini, and mt karyotype variation among Haematopinus lice. Fourth, Shao et al., (2017) [9] showed that both split and merger of minichromosomes had occurred and contributed to the variation in mt karyotypes in sucking lice.
In the present study, we used Polyplax sucking lice, which parasitize rodents and shrews, as a model to investigate the role of tRNA gene translocation in shaping the dynamic mt karyotypes. We show that frequent directional tRNA gene translocation between different types of minichromosomes is a major contributor to the highly dynamic mt genome organization of the parasitic lice of mammals.
Materials and methods
Louse collection, DNA extraction, mitochondrial genome amplification and sequencing
Specimens of Polyplax reclinata (family Polyplacidae) were collected in Yunnan Province, China, from the Asian grey shrew, Crocidura attenuata (family Soricidae). Polyplax reclinata was identified according to Chin (1999) [24]; voucher specimens (# 364) were kept at the Institute of Pathogens and Vectors, Dali University (Additional file 1). Sucking lice collected on the body surface of Crocidura attenuata were preserved in 95 % ethanol at − 20 °C prior to DNA extraction. Capture of shrews was approved by health authorities in Yunnan Province, China. Animal capture protocols and procedures were approved by the animal ethics committees at Dali University. Genomic DNA was extracted from individual lice with DNeasy Blood & Tissue kit (QIAGEN). Two pairs of primers, 12SA–12SB [25] and 16SF–Lx16SR [9], were used to amplify fragments of mt genes rrnS (375 bp) and rrnL (360 bp) (Additional file 2). These primers target conserved gene sequence motifs among arthropods. The two gene fragments were sequenced directly using Sanger method at the Thermo Fisher Scientific Genome Sequencing Facility (Guangzhou). Two pairs of specific primers for P. reclinata, 12S364F-12S364R and 16S364F-16S364R, were designed from rrnS and rrnL sequences obtained (Additional file 2). The two specific primers in each pair go outwards with 15 and 21 bp in between, respectively. PCRs with these specific primers amplified the near full-length rrnS and rrnL minichromosomes of P. reclinata (Additional file 3A). The amplicons from rrnS and rrnL minichromosomes, 2.3 and 2.6 kb in size respectively, were sequenced using Sanger method to obtain the sequences of non-coding regions. Another pair of primers specific to P. reclinata, 364F–364R (Additional file 2), was designed from conserved sequences of the non-coding regions that flank the coding regions of the rrnS and rrnL minichromosomes. The PCR with the primer pair 364F–364R produced a mixture of amplicons ranging from 1.1 to 1.8 kb in size, expected from the coding regions of the entire set of mt minichromosomes of P. reclinata (Additional file 3A). These amplicons were sequenced from both ends (i.e. paired-end) with Illumina Miseq platform: insert size 400 bp, read length 250 bp. The PCR strategy used in this study was developed from our observations in previous studies that each mt minichromosome has a distinct coding region but also a well-conserved non-coding region [1,2,3,4,5,6, 9].
Takara Ex Taq was used in the initial short PCRs with the following cycling conditions: 94 °C for 1 min; 35 cycles of 98 °C for 10 s, 45 °C for 30 s, 72 °C for 1 min; and a final extension of 72 °C for 2 min. Takara LA Taq was used in the long PCRs with the cycling conditions: 94 °C for 1 min; 35 cycles of 98 °C for 10 s, 55–65 °C (depending on primers) for 40 s, 68 °C for 4 min; and a final extension of 72 °C for 8 min. Negative controls were executed with each PCR experiment. PCR amplicons were checked by agarose gel (1 %) electrophoresis; the sizes of amplicons were estimated by comparing with DNA markers. PCR amplicons were purified with Wizard SV Gel/PCR clean-up system (Promega). Sanger sequencing was done at the Thermo Fisher Scientific Genome Sequencing Facility (Guangzhou). Illumina sequencing was done at the Majorbio Genome Sequencing Facility (Shanghai).
Assembly of sequence reads, gene identification and minichromosome verification
Illumina sequence reads (250 bp each) were assembled into contigs with Geneious 11.1.5 [26]. The assembly parameters were minimum overlap 150 bp and minimum identity 98 %. tRNA genes were identified using tRNAscan-SE [27] and ARWEN [28]. Protein-coding genes and rRNA genes were identified by BLAST searches of GenBank [29, 30]. Sequences were aligned with Clustal X [31]. The size and circular organization of each mt minichromosome of P. reclinata were verified by PCR using outbound specific primers designed from the coding region of each minichromosome (Additional file 2). The forward primer and reverse primer in each pair were next to each other with a small gap in between. PCRs with these primers amplified each circular minichromosome in full or near full length (Additional file 3B); these amplicons were also sequenced with Illumina Miseq platform to obtain the full-length sequence of the non-coding region of each minichromosome. PCR set-up, cycling conditions, agarose gel electrophoresis and size measurement were the same as described above. Negative controls were run for each PCR test. The annotated mt genome sequence of P. reclinata was deposited in GenBank (accession numbers MW291451-MW291461).
Phylogenetic analyses
We retrieved the mt genome sequences of all of the 14 species of sucking lice available in GenBank and the elephant louse (Table 1) and combined these sequences with that of P. reclinata generated in the present study. The elephant louse was used as the outgroup because it was in the suborder Rhynchophthirina, which was most closely related to sucking lice, the suborder Anoplura [10]. The sequences of eight mt protein-coding genes (atp6, atp8, cox1, cox2, cox3, cob, nad4L, nad6) and two rRNA genes (rrnS and rrnL) of these lice were used in phylogenetic analysis. Other mt protein-coding genes (nad1, nad2, nad3, nad4, and nad5) were excluded from our analysis because their sequences were not available for all of the species above. Protein-coding gene sequences were aligned based on amino acid sequences using the MAFFT algorithm implemented in TranslatorX online platform [32]. rRNA genes were aligned using the MAFFT v7.0 online server with G-INS-i strategy [33]. Individual gene alignments were concatenated after removing poorly aligned sites using GBlocks v0.91b [34]. A concatenated alignment, PCGRNA matrix, was used in subsequent analyses; this matrix combines the sequences of the eight protein-coding genes and the two rRNA genes (4,631 bp in total). The matrix was analyzed using maximum likelihood (ML) and Bayesian methods with MEGA-X [35] and 2) Bayesian inference method (BI) with MrBayes 3.2.6 [36], respectively. For ML analysis, the number of bootstrap replicates was 500; the substitution model selected was Tamura-Nei and the heuristic method selected was Nearest-Neighbor-Interchange (NNI). For Bayesian analyses, four independent Markov chains were run for 25,000 MCMC generations, sampling a tree every 10 generations. Bayesian analysis was run until the average standard deviation of split frequencies was below 0.001. The initial 10,000 trees of each MCMC run were discarded as burn-in. ML trees were directly generated in MEGA-X and the Bayesian trees were drawn with Figtree v1.4.3.
Inference of the ancestral mitochondrial karyotype of Polyplax lice
We used a parsimony method described in Shao et al., (2017) [9] to infer the ancestral mt karyotype of Polyplax lice based on information from the complete mt genomes of three Polyplax species: P. reclinata (present study), P. asiatica [4] and P. spinulosa [4]. We mapped characters of mt karyotypes on the phylogenetic tree of sucking lice inferred from mt genome sequences to identify shared characters. We inferred an mt minichromosome character to be ancestral to Polyplax: (1) if it was present in at least one Polyplax species and also in one or more non-Polyplax species of sucking lice; or (2) if it was present in all of the three Polyplax species. We considered tRNA genes separately from protein-coding and ribosome RNA genes because protein-coding and rRNA genes were much more stable than tRNA genes [9] (Figs. 1 and 2). For each inferred ancestral minichromosome character of Polyplax lice, we counted the changes needed to explain the observed mt karyotypes of Polyplax lice. If two or more minichromosome characters conflicted with one another, the character with the least required changes was inferred to be the ancestral. If two or more characters are equally parsimonious, all characters are inferred to be equally likely to be ancestral (Figs. 1 and 2).

Inferring the ancestral mitochondrial karyotype of Polyplax lice (suborder Anoplura). tRNA genes are excluded. Plus symbol (+) is for presence, minus (-) for absence, and NA for information not available. Underlined nad1 has an opposite orientation of transcription to other genes. The phylogenetic tree was inferred with mitochondrial genome sequences in the current study

Inferring the location of tRNA genes relative to protein-coding and rRNA genes in the ancestral mitochondrial karyotype of Polyplax lice. tRNA genes are indicated with their single-letter abbreviations. Plus symbol (+) is for presence, minus (-) for absence, and NA for information not available. Underlined genes have opposite orientation of transcription to other genes. The phylogenetic tree was inferred with mitochondrial genome sequences in the current study
Results and discussion
Mitochondrial genome of the Asian grey shrew louse, Polyplax reclinata
We obtained the complete mt genome of P. reclinata by assembling the 53,915,414 clean Illumina sequence reads (paired-end, 250 bp each) generated from the minichromosome amplicons (Table 2). All of the 37 mt genes typical of bilateral animals were identified in P. reclinata; these genes were on 11 circular minichromosomes (Fig. 3). The mt minichromosomes of P. reclinata range from 2,332 to 2,869 bp in size (Table 2); each minichromosome has a coding region and a non-coding region (Fig. 3; Table 2). The coding region of each minichromosome has 2–6 genes and ranges from 847 bp for atp8-atp6 minichromosome to 1,809 bp for H-nad5-F minichromosome (minichromosomes named after the genes they contain). Seven of the 11 minichromosomes of P. reclinata contain a single protein-coding or rRNA gene each; the other four minichromosomes contain two protein-coding genes each. There are 1–4 tRNA genes in each minichromosome except atp8-atp6 minichromosome, which has no tRNA genes (Fig. 3). Each of the 37 mt genes in P. reclinata is found only in one minichromosome; all genes have the same orientation of transcription relative to the non-coding region except for nad1, which has an opposite orientation to other genes (Fig. 3).

The mitochondrial genome of the Asian grey shrew louse, Polyplax reclinata. Protein-coding genes are: atp6 and atp8 (for ATP synthase subunits 6 and 8), cox1-3 (for cytochrome c oxidase subunits 1–3), cob (for cytochrome b), nad1-6 and nad4L (for NADH dehydrogenase subunits 1–6 and 4L). rRNA genes are: rrnS and rrnL (for small and large subunits of ribosomal RNA). tRNA genes are indicated with their single-letter abbreviations. Arrow indicates transcription orientation; numbers inside inner circles indicate gene length (bp); non-coding regions (NCR) are shaded in black

Phylogenetic relationships among 15 species of sucking lice (suborder Anoplura) inferred with combined sequences of eight mitochondrial protein-coding genes (atp6, atp8, cox1, cox2, cox3, cob, nad4L, nad6) and two rRNA genes (rrnS and rrnL) with maximum likelihood method (A) and Bayesian method (B). The trees were rooted with the elephant louse, Haematomyzus elephantis (suborder Rhyncophthirina). Bootstrap support values (out of 100) and Bayesian posterior probability values (out of 1) were indicated near internal nodes
We sequenced the non-coding regions of all of the 11 minichromosomes of P. reclinata in full length. The non-coding regions range from 1,060 to 1,578 bp (Table 2); the size variation is due to the additional sequences in the middle of the non-coding regions of M-L1-rrnL-V, S1-S2-rrnS-C, K-nad4, atp8-atp6, Q-nad2-N and E-cob-I minichromosomes (Additional file 4). If the additional sequences are excluded, the non-coding regions of all 11 minichromosomes have high sequence similarity to each other. Overall, the non-coding regions have high C and G content in one end but high A and T content in the other end. Indeed, a CG-rich motif (54 bp, 59 % C and G) was found immediately downstream from the 3’-end of the coding regions and an AT-rich motif (113 bp, 68 % A and T) was found upstream from the 5’-end of the coding regions in all of the minichromosomes (Additional file 4).
Mitochondrial karyotype variation among Polyplax species
Polyplax reclinata differs in mt karyotype from the two other Polyplax species reported previously [4], although these three species all have 11 minichromosomes. Polyplax reclinata shares nine minichromosomes with P. spinulosa (louse of the Asian house rat) [4] but differs in the distribution of trnL1 and trnL2 between the other two minichromosomes (Table 3). In P. spinulosa, trnL1 is downstream from cox1 and trnL2 is upstream from rrnL [4]; in P. reclinata, however, it is the opposite (Fig. 3; Table 3). Polyplax reclinata shares only four minichromosomes with P. asiatica (louse of the greater bandicoot rat) [4] and differs in the distribution of six tRNA genes among the other seven minichromosomes: trnA, trnF, trnP, trnS1, trnS2 and trnT (Table 3). If tRNA genes are excluded, the three Polyplax species are identical to one another in the distribution of protein-coding genes and rRNA genes among the 11 minichromosomes (Table 3; Fig. 3).
Phylogeny of sucking lice inferred from mt genome sequences
To assist the inference of the ancestral mt karyotype of Polyplax lice, we inferred the phylogeny of sucking lice using mt genome sequences of 15 species of sucking lice of seven genera including Polyplax reclinata, which was sequenced in the present study. The elephant louse, which was in the suborder Rhynchophthirina, was used as the outgroup (Table 1). Rhynchophthirina is the sister suborder to the sucking lice (suborder Anoplura) [10]. We obtained two trees from PCGRNA matrix (4,631 bp of protein-coding and rRNA gene sequences) with ML and Bayesian methods. These two trees have similar topologies and consistently support the monophyly of the genus Polyplax (Fig. 4): bootstrap support value (BSV) = 100 %, posterior probability (PP) = 1. Within the Polyplax, P. reclinata is more closely related to P. spinulosa than to P. asiatica (BSV = 100 %, PP = 1), despite the fact that the latter two species are found mostly on rodents whereas P. reclinata is found on shrews [37, 38]. The other four genera, Hoplopleura, Pediculus, Pedicinus and Haematopinus, also had multiple species from each genus included in our analysis; each of these genera was monophyletic with strong support (BSV = 100 %, PP = 1) (Fig. 4). The 15 species of sucking lice were divided into three clades: Polyplax and Hoplopleura forming a clade (BSV = 76.6 %: PP = 0.998); Pediculus, Pedicinus, Pthirus and Haematopinus in another clade (BSV = 69.8 %; PP = 0.983); and Microthoracius praelongiceps alone as the earliest branch (BSV = 85.2 %; PP = 1) (Fig. 4). Each of the genera included in our analysis also represents a family (Table 1). The family level relationships revealed by our analysis are consistent with that proposed by Kim (1988) based on morphological characters [39] and partially consistent with that by Light et al. (2010), Smith et al. (2011) and Johnson et al. (2018) based on gene sequences [13,14,15].
Inferred ancestral mitochondrial karyotype of Polyplax lice
We inferred the ancestral mt karyotype of Polyplax lice based on the data available for the three Polyplax species and the other 12 species of sucking lice (Table 1; Fig. 5). We mapped mt karyotype characters on the phylogeny inferred from the mt genome sequences of these lice to identify shared characters (Figs. 1 and 2). The inferred ancestral mt karyotype of Polyplax lice by parsimony consists of 11 minichromosomes; each minichromosome has a coding region with 2–7 genes and a non-coding region (Fig. 5). Seven of the 11 minichromosomes have a single protein-coding or rRNA gene each; the other four minichromosomes have two protein-coding genes each: (1) atp6 and atp8, (2) cox2 and nad6, (3) cox3 and nad4L, and (4) nad3 and nad1 (Fig. 5). The position of all genes in the ancestral mt karyotype can be inferred without conflict except the position of a cluster of two tRNA genes, S1-S2, which is either upstream from rrnS or, equally parsimoniously, downstream from nad1 (Figs. 2 and 5).

Inferred ancestral mitochondrial karyotype of Polyplax lice (suborder Anoplura). Abbreviated gene names are the same as in Fig. 3. Arrow indicates transcription orientation; non-coding regions (NCR) are shaded in black. The two minichromosomes containing S1-S2 gene cluster are boxed in dash line because the location of this cluster is either upstream from rrnS or, equally parsimoniously, downstream from nad1
Mitochondrial tRNA gene translocation in Polyplax lice
When compared with the inferred ancestral mt karyotype of Polyplax lice (Fig. 5), all three Polyplax species sequenced to date retained 11 minichromosomes and the same distribution pattern of protein-coding and rRNA genes among the minichromosomes (Table 3). Therefore, no split nor merger of minichromosomes have occurred in any of these Polyplax species since their most recent common ancestor (MRCA) (Fig. 4). However, these three Polyplax species are different from one another and from their MRCA in the position of tRNA genes. In total, eight tRNA genes have changed their positions in the three Polyplax lice since they diverged from their MRCA: trnA, trnF, trnL1, trnL2, trnP, trnS1, trnS2 and trnT (Table 3). trnA and trnT were likely translocated (trnT also inverted) in the MRCA of P. reclinata and P. spinulosa (Fig. 4) and thus are shared by these two species (Table 3). A pseudo trnT can be found in P. reclinata (Fig. 3) and P. spinulosa [4], indicating the translocation of trnT occurred relatively recently. trnL1 and trnL2 swapped their positions in P. spinulosa [4], most likely caused by point mutations at the third anti-codon position (or by homologous recombination between trnL1 and trnL2) – a mechanism suggested by previous studies [2, 9]. trnF and trnP were translocated in P. asiatica (Table 3); as a cluster, trnS1-trnS2 was translocated either in P. asiatica or equally likely in the MRCA of P. reclinata and P. spinulosa (Table 3; Fig. 5).
tRNA gene translocation is frequent and directional towards the boundaries with control region in parasitic lice with fragmented mitochondrial genomes
We noted in the Polyplax lice above that mt tRNA gene translocation was closely associated with the control region, i.e. the non-coding region (NCR). With no exception, the destination of all of the translocated tRNA genes (i.e. trnA, trnF, trnS1-trnS2, trnP and trnT excluding trnL1 and trnL2 which swapped positions in P. spinulosa as discussed above) were either immediately next to the NCR or in the case of trnP of P. asiatica, a short distance (189 bp) inside the NCR (Table 3) [4], indicating a possible role of NCR in tRNA gene translocation. The observed probability of tRNA gene translocation to be adjacent to NCR in these Polyplax species is significantly higher (p < 0.00001) than the expected probability (Table 4).
We investigated further if this was a broader pattern for parasitic lice with fragmented mt genomes by comparing the mt karyotypes: (1) between the MRCA of sucking lice [9] and the MRCA of Polyplax (Fig. 5); and (2) between the MRCA of sucking lice and each of the 12 non-Polyplax sucking louse species that have been sequenced to date [9, 12]. The MRCA of Polyplax species retained the same distribution pattern of protein-coding and rRNA genes and the same number of minichromosomes as the MRCA of sucking lice. However, five tRNA genes have translocated in the MRCA of Polyplax relative to the MRCA of sucking lice: all of these tRNA genes were translocated immediately next to the NCR despite alternative intergenic locations available for translocation (Table 5). The observed probability of tRNA gene translocation to be adjacent to NCR in the MRCA of Polyplax species is significantly higher (p < 0.00001) than the expected probability (Table 6). When compared with the MRCA of sucking lice, 19 of the 22 tRNA genes in total have translocated in the 12 species of non-Polyplax sucking lice: 32 out of the 54 (i.e. 59 %) translocation events resulted in tRNA genes moved to be adjacent to NCR despite alternative intergenic locations available for translocation (Table 7). The observed probability of tRNA gene translocation to be adjacent to NCR in these non-Polyplax species is 13.6 % higher than the expected probability but this difference is not statistically significant (p = 0.116709) (Table 8). Most likely tRNA gene translocation to be adjacent to NCR has been under-captured in non-Polyplax species (Table 7) due to the fact that any newly translocated tRNA gene adjacent to a NCR would inevitably push the previously translocated tRNA gene away from the NCR. After all, there can only be one gene at any time adjacent to each end of the NCR. Among the three comparisons above, the third comparison has the longest evolutionary time frame. The longer the evolutionary time is, the more likely tRNA gene translocation information to be adjacent to NCR will be under-captured.
Clearly, the boundaries between coding regions and NCRs are hot spots for tRNA gene translocation in parasitic lice that have fragmented mt genomes. Of the models and mechanisms proposed in previous studies (introduced above in the Background) [2, 4, 6, 9, 10], only the inter-minichromosomal recombination model proposed by Song et al., [6] based on the mt karyotype variation among Haematopinus lice can explain tRNA gene translocation between mt minichromosomes. However, it cannot explain why translocated tRNA genes tend to be in the boundaries between coding regions and NCRs. As DNA double-strand breaks (DSBs) are apparently necessary for gene translocation between mt minichromosomes, the boundary regions may have more frequent DSBs than other regions of the mt minichromosomes, possibly due to the specific AT-rich and GC-rich sequence elements in the boundary regions; these sequence elements are conserved among all mt minichromosomes of each species of parasitic louse (Additional file 4). Apparently, DSBs do not occur randomly. Tubbs et al. showed that in mouse and human cells, DSBs occur mostly in AT-rich regions, in particular at homopolymeric (dA/dT) tracts [40]. Sinai et al. showed that integration of AT-dinucleotide rich sequences into human chromosomes created recurrent gaps and breaks at the integration sites, due to a significantly increased tendency to fold into branched secondary structures at these sites [41]. Baudat et al. showed that a histone methyl transferase, PRDM9, is a major determinant of meiotic recombination hotspots in humans and mice, and furthermore, the human PRDM9 binding sites contain a 13-mer GC-rich motif and are DSB hotspots [42]. Sundararajan et al. showed that DSB hotspots have a strong tendency to occur in genes with high GC content in maize [43]. He et al. investigated the DNA sequence of DSB hotspots and identified a 20-bp-long GC-rich degenerate DNA sequence motif (named Maize Hotspot Sequence) present in about 72 % of genic hotspots in maize [44]. In general, non-canonical DNA structures such as hairpins and G-quadruplexes render a genome prone to damage including DSBs [45]. We counted the number of translocated tRNA genes in sucking lice at each end of the mt non-coding regions (Tables 3, 5 and 7) – the distribution is rather even (20 vs. 20) between the two ends where the AT-rich and GC-rich motifs were found respectively. Our observed pattern of mt tRNA gene translocation in sucking lice aligns well with the evidence presented in these experimental studies on DSB hotspots. Future studies on DSBs and DNA repair in parasitic lice may help understand further the mechanisms of mt tRNA gene translocation observed in sucking lice in the present study.
In conclusion, we sequenced the fragmented mt genome of the Asian grey shrew louse, Polyplax reclinata, which comprises 11 circular minichromosomes. We also inferred the ancestral mt karyotype of Polyplax lice based on the data available to date. We found that tRNA genes are entirely responsible for mt karyotype variation among the three species of Polyplax lice sequenced to date. Furthermore, tRNA gene translocation in these Polyplax lice is much more frequent than that of protein-coding and rRNA genes; all tRNA gene translocation observed occurs between different types of minichromosomes and is directional towards the boundaries with the control region. A similar pattern of tRNA gene translocation is also conserved in other sucking lice with fragmented mt genomes. Our results show that inter-minichromosomal tRNA gene translocation orientated towards the two ends of the control region is a major contributing factor to the highly dynamic mitochondrial genome organization in the parasitic lice of mammals.
Availability of data and materials
The nucleotide sequence of the mitochondrial genome of the Asian grey shrew louse, Polyplax reclinata, has been deposited in GenBank (accession number MW291451-MW291461) and will be released publicly once this manuscript is accepted for publication.
Abbreviations
- µl:
-
Microliter
- atp6 and atp8 :
-
Genes for ATP synthase subunits 6 and 8
- bp:
-
Base pair
- cob :
-
Gene for cytochrome b
- cox1, cox2 and cox3 :
-
Genes for cytochrome c oxidase subunits 1, 2 and 3
- DNA:
-
Deoxyribonucleic acid
- kb:
-
Kilo base pair
- min:
-
Minute
- MRCA:
-
Most recent common ancestor
- mt:
-
Mitochondrial
- Mya:
-
Million years ago
- nad1, nad2, nad3, nad4, nad4L, nad5 and nad6 :
-
Mitochondrial genes for NADH dehydrogenase subunits 1–6 and 4L
- PCR:
-
Polymerase chain reaction
- RNA:
-
Ribonucleic acid
- rRNA:
-
Ribosomal RNA
- rrnS and rrnL :
-
Genes for small and large subunits of ribosomal RNA
- sec:
-
Second
- T:
-
Thymine
- tRNA:
-
Transfer RNA
- trnA or A :
-
tRNA gene for alanine
- trnC or C :
-
tRNA gene for cysteine
- trnD or D :
-
tRNA gene for aspartic acid
- trnE or E :
-
tRNA gene for glutamic acid
- trnF or F :
-
tRNA gene for phenylalanine
- trnG or G :
-
tRNA gene for glycine
- trnH or H :
-
tRNA gene for histidine
- trnI or I :
-
tRNA gene for isoleucine
- trnK or K :
-
tRNA gene for lysine
- trnL 1 or L 1 :
-
tRNA gene for leucine (anticodon NAG)
- trnL 2 or L 2 :
-
tRNA gene for leucine (anticodon YAA)
- trnM or M :
-
tRNA gene for methionine
- trnN or N :
-
tRNA gene for asparagine
- trnP or P :
-
tRNA gene for proline
- trnQ or Q :
-
tRNA gene for glutamine
- trnR or R :
-
tRNA gene for arginine
- trnS 1 or S 1 :
-
tRNA gene for serine (anticodon NCU)
- trnS 2 or S 2 :
-
tRNA gene for serine (anticodon NGA)
- trnT or T :
-
tRNA gene for threonine
- trnV or V :
-
tRNA gene for valine
- trnW or W :
-
tRNA gene for tryptophan
- trnY or Y :
-
tRNA gene for tyrosine
- U:
-
Uracil
References
Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009;19:904–12.
Shao R, Zhu XQ, Barker SC, Herd K. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol Evol. 2012;4:1088–101.
Jiang H, Barker SC, Shao R. Substantial variation in the extent of mitochondrial genome fragmentation among blood-sucking lice of mammals. Genome Biol Evol. 2013;5:1298–308.
Dong WG, Song S, Jin DC, Guo XG, Shao R. Fragmented mitochondrial genomes of the rat lice, Polyplax asiatica and Polyplax spinulosa: intra-genus variation in fragmentation pattern and a possible link between the extent of fragmentation and the length of life cycle. BMC Genomics. 2014;15:44.
Dong WG, Song S, Guo XG, Jin DC, Yang Q, Barker SC, Shao R. Fragmented mitochondrial genomes are present in both major clades of the blood-sucking lice (suborder Anoplura): evidence from two Hoplopleura rodent lice (family Hoplopleuridae). BMC Genomics. 2014;15:751.
Song SD, Barker SC, Shao R. Variation in mitochondrial minichromosome composition between blood-sucking lice of the genus Haematopinus that infest horses and pigs. Parasite Vector. 2014;7:144.
Herd KE, Barke SC, Shao R. The mitochondrial genome of the chimpanzee louse, Pediculus schaeffi: insights into the process of mitochondrial genome fragmentation in the blood-sucking lice of great apes. BMC Genomics. 2015;16:661.
Shao R, Barker SC, Li H, Song S, Poudel S, Su Y. Fragmented mitochondrial genomes in two suborders of parasitic lice of eutherian mammals (Anoplura and Rhynchophthirina, Insecta). Sci Rep. 2015;5:17389.
Shao R, Li H, Barker SC, Song S. The mitochondrial genome of the guanaco louse, Microthoracius praelongiceps: insights into the ancestral mitochondrial karyotype of sucking Lice (Anoplura, Insecta). Genome Biol Evol. 2017;9:431–45.
Song F, Li H, Liu GH, Wang W, James P, Colwell DD, Tran A, Gong SY, Cai WZ, Shao R. Mitochondrial genome fragmentation unites the parasitic lice of eutherian mammals. Syst Biol. 2019;68:430–40.
Sweet AD, Johnson KP, Cameron SL. Mitochondrial genomes of Columbicola feather lice are highly fragmented, indicating repeated evolution of minicircle-type genomes in parasitic lice. PeerJ. 2020;8:e8759.
Fu YT, Dong Y, Wang W, Nie Y, Liu GH, Shao R. Fragmented mitochondrial genomes evolved in opposite directions between closely related macaque louse Pedicinus obtusus and colobus louse Pedicinus badii. Genomics. 2020;112:4924–33.
Smith VS, Ford T, Johnson KP, Johnson PCD, Yoshizawa K, Light JE. Multiple lineages of lice pass through the K-Pg boundary. Biol Lett. 2011;5:782–5.
Light JE, Smith VS, Allen JM, Durden LA, Reed DL. Evolutionary history of mammalian sucking lice (Phthiraptera: Anoplura). BMC Evol Biol. 2010;10:292.
Johnson KP, Nguyen N-p, Sweet AD, Boyd BM, Warnow T, Allen JM. Simultaneous radiation of bird and mammal lice following the K-Pg boundary. Biol Lett. 2018;14:141.
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–80.
Wurster DH, Benirschke K. Indian muntjac, Muntiacus muntjak: A deer with a low diploid chromosome number. Science. 1970;168:1364–6.
Qumsiyeh MB. Evolution of number and morphology of mammalian chromosomes. J Hered. 1994;85:455–65.
Rieseberg LH. Chromosomal rearrangements and speciation.Trends Ecol Evol. 2001;16:351–8.
Kittler R, Kayser M, Stoneking M. Molecular evolution of Pediculus humanus and the origin of clothing. Curr Biol. 2003;13:1414–7.
Kittler R, Kayser M, Stoneking M. Molecular evolution of Pediculus humanus and the origin of clothing (Erratum). Curr Biol. 2004;14:2309.
Toups MA, Kitchen A, Light JE, Reed DL. Origin of clothing lice indicates early clothing use by anatomically modern humans in Africa. Mol Biol Evol. 2011;28:29–32.
Ottoni C, Girdl and Flink L, Evin A, Georg C, Cupere BD, Neer WV, Bartosiewicz L, Linderholm A, Barnett R, Peters J, Decorte R, Waelkens M, Vanderheyden N, Ricaut FX, Cakırlar C, Cevik O, Hoelzel AR, Mashkour M, Fatemeh A, Karimlu M, Seno SS, Daujat J, Brock F, Pinhasi R, Hongo H, Perez-Enciso M, Rasmussen M, Frantz L, Megens HJ, Crooijmans R, Groenen M, Arbuckle B, Benecke N, Vidarsdottir US, Burger J, Cucchi T, Dobney K, Larson G. Pig domestication and human-mediated dispersal in western eurasia revealed through ancient DNA and geometric morphometrics. Mol Biol Evol. 2013;4:824–32.
Chin TH. Taxonomy and fauna of sucking lice (Anoplura) in China. Beijing Science Press; 1999.
Kambhampati S, Smith PT. PCR primers for amplification of four insect mitochondrial gene fragments. Insect Mol Biol. 1995;4:233–6.
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–9.
Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–64.
Laslett D, Canback B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–5.
Gish W, States DJ. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3:266–72.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38:W7-W13.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–77.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolution genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Durden LA, Musser GG. The sucking lice (Insecta, Anoplura) of the world: a taxonomic checklist with records of mammalian hosts and geographical distributions. Bull Amer Mus Nat Hist. 1994;218:1–90.
Durden LA, Musser GG. The mammalian hosts of the sucking lice (Anoplura) of the world: a host-parasite list. Bull Soc Vector Ecol. 1994;19:130–68.
Kim KC. Evolutionary parallelism in Anoplura and eutherian mammals. In: Service MW, editor. Biosystematics of haematophagous insects. Oxford: Claredon Press; 1988. Vol. 37. p. 91–114.
Tubbs A, Sridharan S, Wietmarschen N, Maman Y, Callen E, Stanlie A, Wu W, Xia W, Day A, Wong N, Yin M, Canela A, Fu H, Redon C, Pruitt SC, Jaszczyszyn Y, Aladjem M, Aplan PD, Hyrien O, Nussenzweig A. Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell. 2018;174:1127–42.
Sinai MIT, Salamon A, Stanleigh N, Goldberg T, Weiss A, Wang YH, Kerem B. AT-dinucleotide rich sequences drive fragile site formation. Nucleic Acids Res. 2019;47:9685–95.
Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–40.
Sundararajan A, Dukowic-Schulze S, Kwicklis M, Engstrom K, Garcia N, Oviedo OJ, Ramaraj T, Gonzales MD, He Y, Wang M, Sun Q, Pillardy J, Kianian SF, Pawlowski WP, Chen C, Mudge J. Gene evolutionary trajectories and GC patterns driven by recombination in Zea mays. Front Plant Sci. 2016;7:1433.
He Y, Wang M, Dukowic-Schulze S, Zhou A, Tiang CL, Shilo S, Sidhu GK, Eichten S, Bradbury P, Springer NM, Buckler ES, Levy AA, Sun Q, Pillardy J, Kianian PMA, Kianian SF, Chen C, Pawlowski WP. Genomic features shaping the landscape of meiotic double-strand-break hotspots in maize. Proc Natl Acad Sci USA. 2017;114:12231–6.
Puget N, Miller KM, Legube G. Non-canonical DNA/RNA structures during transcription-coupled double-strand break repair: roadblocks or bona fide repair intermediates? DNA Repair. 2019;81:102661.
Acknowledgements
We thank Dr Fan Song for assistance with an initial phylogenetic tree analysis.
Funding
We acknowledge funding support from the National Natural Science Foundation of China (No. 31660314 and 32060143 to WGD) and the Innovation Team of Vector Biology, Dali University (No. ZKLX2019104 to XGG). The funding agencies had no roles in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
WGD, YLD, XGG and RS designed the research. WGD and RS performed the research. WGD and XGG contributed reagents and materials. WGD, YLD and RS analysed the data. WGD and RS wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Capture of shrews was approved by health authorities in Yunnan Province, China. Animal capture protocols and procedures were approved by the animal ethics committees at Dali University (approval # DLDX-20100421-001). All collection methods were carried out in accordance with the approved guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Image of the voucher specimens (# 364) of the shrew louse, Polyplax reclinata.
Additional file 2:
The primers used to amplify the mitochondrial genes, minichromosomes and coding regions of the Asian grey shrew louse, Polyplax reclinata.
Additional file 3:
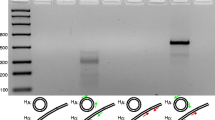
PCR amplicons generated from the mitochondrial (mt) minichromosomes of the Asian grey shrew louse, Polyplax reclinata. (A) Lane 1: amplicon of S1-S2-rrnS-C minichromosome generated with the primer pair 12S364F-12S364R. Lane 2: amplicon of M-L1-rrnL-V minichromosome generated with the primer pair 16S364F-16S364R. Lane 3: 500 bp Ladder (Tiangen) (band size in bp indicated). Lane 4: amplicons of coding regions of all mt minichromosomes generated with the primer pair 364 F-364R. Lane 5: GeneRuler 100 bp DNA Ladder (Thermo Scientific) (band size in bp indicated). The lane to the right of lane 2 is irrelevant to this manuscript. The lane to the left of lane 3 is empty. (B) Lane 1: 1 kb ladder (Tiangen) (band size in bp indicated); Lanes 2–10: amplicons of individual mt minichromosomes, T-D-Y-cox2-nad6-A, R-nad4L-P-cox3, Q-nad2-N, nad1 -G-nad3-W (gene underlined has opposite transcription orientation to other genes), K-nad4, H-nad5-F, E-cob-I, cox1-L2, atp8-atp6. Details of the primers used to amplify individual mt minichromosomes are provided in Additional file 2.
Additional file 4:
Alignment of the non-coding region (NCR) sequences of 11 mitochondrial minichromosomes of the Asian grey shrew louse, Polyplax reclinata. The primer pair, 364 F and 364R, were used to amplify the coding regions of the 11 minichromosomes (see also Additional file 3). Asterisk symbol “*” indicates conserved nucleotides; hyphen “-” indicates absent nucleotides.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dong, WG., Dong, Y., Guo, XG. et al. Frequent tRNA gene translocation towards the boundaries with control regions contributes to the highly dynamic mitochondrial genome organization of the parasitic lice of mammals. BMC Genomics 22, 598 (2021). https://doi.org/10.1186/s12864-021-07859-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07859-w