
Abstract
Background
Tribenuron methyl (TBM) is an herbicide that inhibits sulfonylurea acetolactate synthase (ALS) and is one of the most widely used broad-leaved herbicides for crop production. However, soil residues or drifting of the herbicide spray might affect the germination and growth of rapeseed, Brassica napus, so it is imperative to understand the response mechanism of rape to TBM during germination. The aim of this study was to use transcriptome analysis to reveal the gene responses in herbicide-tolerant rapeseed to TBM stress during seed germination.
Results
2414, 2286, and 1068 differentially expressed genes (DEGs) were identified in TBM-treated resistant vs sensitive lines, treated vs. control sensitive lines, treated vs. control resistant lines, respectively. GO analysis showed that most DEGs were annotated to the oxidation-reduction pathways and catalytic activity. KEGG enrichment was mainly involved in plant-pathogen interactions, α-linolenic acid metabolism, glucosinolate biosynthesis, and phenylpropanoid biosynthesis. Based on GO and KEGG enrichment, a total of 137 target genes were identified, including genes involved in biotransferase activity, response to antioxidant stress and lipid metabolism. Biotransferase genes, CYP450, ABC and GST, detoxify herbicide molecules through physical or biochemical processes. Antioxidant genes, RBOH, WRKY, CDPK, MAPK, CAT, and POD regulate plant tolerance by transmitting ROS signals and triggering antioxidant enzyme expression. Lipid-related genes and hormone-related genes were also found, such as LOX3, ADH1, JAZ6, BIN2 and ERF, and they also played an important role in herbicide resistance.
Conclusions
This study provides insights for selecting TBM-tolerant rapeseed germplasm and exploring the molecular mechanism of TBM tolerance during germination.
Similar content being viewed by others


Background
Weed competition is an important limiting factor affecting crop yield [1]. Tribenuron methyl (TBM) is an herbicide that acts by inhibiting sulfonylurea acetolactate synthase (ALS), which reduces isoleucine, leucine and valine biosynthesis [2]. Rapeseed is a broad-leaved crop and therefore more sensitive to TBM, so TBM is rarely used directly for weed control in rapeseed production. However, planting methods such as rotation or intercropping can leave TBM residues in soil and herbicide spray can drift onto other areas [2, 3], which might cause physiological and biochemical changes that inhibit germination or reduce seedling quality and crop growth and development.
The biological stress response to adverse environmental factors is essentially the result of differential gene expression. Gene chip and high-throughput sequencing technologies have played a major part in the identification of genes related to the stress response at the whole genome level. The complete sequencing of the rapeseed genome also provided valuable molecular resources for studying the mechanism of stress resistance [4]. RNA-seq analysis can quickly identify differently expressed genes (DEGs) between different samples. Up to now, RNA-seq has facilitated the identification of DEGs in rapeseed under abiotic stresses such as drought [5], freezing [6] and salinity [7], as well as some candidate genes related to sclerotinia [8], seed aging [9], seed coat color [10], and flowering time [11]. Some studies on herbicide stress to plants were also carried out using RNA-seq. For example, after treatment with TBM, single nucleotide polymorphisms (SNPs) [12] and non-target-site resistance (NTSR)-related genes such as for glutathiones, peroxidases, oxidases, hydrolases, and transporter proteins were identified in Myosoton aquaticum L. (water chickweed) [13], short-awn foxtail [14], grain sorghum [12] and rye grass [15]. Application of TBM affected root and above-ground growth of cornflower [16], and reduced the biomass of foxtail millet [17]. However, there were few studies on the effect of TBM on rapeseed germination.
The germination period is the key stage of growth and development of crops, and it is highly sensitive to external stress [18]. Studies have shown that sulfonylurea herbicide stress during germination could be used to screen plants for tolerant germplasm [19], reducing the impact of TBM on crop production. Germination is a complex process involving specific gene transcription, post-translational modifications, and metabolic interactions [20] that are difficult to analyze by conventional physiological and biochemical methods. This study utilized RNA-seq to detect genes related to TBM stress during the germination stage of B. napus, characterize the physiological indices, and verify gene expression by qRT-PCR. The physiological and molecular data were combined to elucidate the response mechanism of rapeseed to TBM stress. This not only improves the accuracy of the results but also provides key information for screening and cultivating TBM-tolerant rapeseed germplasm and exploring the molecular mechanisms of TBM tolerance during germination.
Results
Comparison of germinated seed root length between S (sensitive) and R (resistant) Brassica napus lines
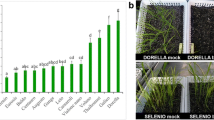
As shown in Fig. 1, the root length of the S line was significantly inhibited after exposure to TBM, while the root length of the TBM-treated R line was no different from control. This indicated that the tolerance of the S and R rapeseed lines to TBM was significantly different from each other.

Comparison of root length between different rape lines after 7 d germination. All results are expressed as the mean ± standard deviation (S.D.) of triplicate values. The symbols ‘ns’ and ‘**’ respectively represent ‘not significantly different (P > 0.05)’ and ‘an extremely significant difference (0.001 < P < 0.01)’, according to Student’s t-test
Sequencing quality and expression analysis
45,631,028, 43,758,578, 44,548,434, and 46,766,702 original reads were generated from the four RNA libraries of Sck (S line control), Rck (R line control), St (S line treatment), and Rt (R line treatment), respectively. After removing the low-quality reads, 40,034,436, 38,350,620, 39,237,176, and 42,615,278 high-quality reads were sequentially generated. The percentage alignment of the high-quality reads with the Brassica reference genome sequence was 82.28–84.6%. The percentages of single comparisons and multiple comparisons were 95.33–95.55% and 4.45–4.67%, respectively. Q20 and Q30, the percentages of bases with a correct base recognition rate greater than 99.0–99.9% were 94.43–95.5% and 88.17–88.58%, respectively, and the percentage of fuzzy bases (N) was no higher than 0.0046% (Table S1). FPKM density distribution showed that moderately expressed genes accounted for the vast majority, while weakly expressed and highly expressed genes were in the minority (Fig. 2).

FPKM density distribution of genes in the four simples
Differentially expressed gene (DEG) analysis
As shown in Fig. 3 and Fig. 4, a total of 2218 DEGs was obtained from Rck vs. Sck. The number of down-regulated DEGs (1333, 60.1%) was more than that of up-regulated DEGs (885, 39.9%). 2414 DEGs were identified in Rt vs. St, including 1594 (66.0%) up-regulated genes and 820 (34.0%) down-regulated genes, and the log2 fold-change of most DEGs was approximately + 1 to + 5. In St vs. Sck and Rt vs. Rck, 2286 and 1068 DEGs were detected, respectively. Of the 2286 DEGs in the S line, 245 (10.7%) were up-regulated and 2041 (89.3%) were down-regulated, and the log2 fold-change of most DEGs ranged from − 5 to − 1. The 1068 DEGs of the R line included 458 (42.9%) up-regulated genes and 610 (57.1%) down-regulated genes. The log2 fold-change was between − 2 and 3.

Venn diagram of the number of DEGs detected in four simples. a. Venn diagram indicated the number of up-regulated DEGs. b. Venn diagram indicated the number of down-regulated DEGs

log2fold change in the DEGs detected in Rck VS Sck, Rt VS St, St VS Sck and Rt VS Rck. a. Number of genes with a log2fold change ≤ −5. b. Number of genes with −5 < log2fold change ≤ −3; c. Number of genes with −3 < log2fold change ≤ −2. d. Number of genes with −2 < log2fold change ≤ −1. e. Number of genes with 1 ≤ log2fold change < 3; f. Number of genes with 3 ≤ log2fold change < 5; g. Number of genes with log2fold change ≥5
Enrichment analysis of DEGs in Rt vs. St, St vs. Sck and Rt vs. Rck
The DEGs in Rt vs. St, St vs. Sck and Rt vs. Rck were annotated into 19, 17 and 14 significant GO terms, respectively (Fig. 5). Under biological processes, oxidation-reduction reactions were overrepresented in Rt vs. St, St vs. Sck and Rt vs. Rck. DEGs in the S and R lines were annotated for responses to oxidative stress. Under cellular components, ubiquitin ligase complex, extracellular region, and apoplast were the most abundant terms in Rt vs. St; and DEGs in the S and R lines were mainly annotated to the extracellular region and membranes, respectively. As for molecular functions, the DEGs in the three groups were mainly related to oxidoreductase activity. In addition, DEGs in Rt vs. St were also involved in transcriptional regulation and DNA binding, and DEGs in the S and R lines participated in catalytic activity.

GO classification of DEGs. a. GO classification of DEGs in Rt VS St. b. GO classification of DEGs in St VS Sck. c. GO classification of DEGs in Rt VS Rck. BP: biological process; MF: molecular function; CC: cellular component. The x-axis represents the most abundant categories of each group, and the y-axis represents the number of the total genes in each category
KEGG enrichment was done to identify in which metabolic pathways the DEGs were involved. As shown in Table 1, the DEGs in Rt vs. St were significantly enriched in phenylpropanoid biosynthesis, cysteine and methionine metabolism, plant-pathogen interaction, MAPK signaling, alpha-linolenic acid metabolism, and linoleic acid metabolism. The DEGs in the S and R lines were significantly enriched in 18 and 9 metabolic pathways, respectively and five pathways were shared by both S and R lines, including phenylpropanoid biosynthesis, alpha-linolenic acid metabolism, tyrosine metabolism, plant hormone signal transduction, cysteine, and methionine metabolism. There were 13 unique pathways in the S line, including plant-pathogen interactions, glucosinolate biosynthesis, and MAPK signaling, while four unique pathways including valine, leucine and isoleucine degradation were found in the R line.
Functional classification of DEGs
Combining GO and KEGG enrichment analysis (Table S2), 73 DEGs were identified in Rt vs. St, and most of these genes were expressed at higher levels in Rt after TBM treatment (Fig. 6a). These genes were involved in five metabolic pathways with 59% of the genes being related to plant-pathogen interactions and 25% related to phenylpropanoid biosynthesis (Fig. 7a). Screening revealed 53 DEGs from the S line and 22 DEGs from the R line. As shown in Fig. 6b-c, the majority of DEGs were down-regulated in the S line and up-regulated in the R line after treatment. The DEGs in the S line were associated with nine metabolic pathways, including plant-pathogen interaction (21%), glucosinolate biosynthesis (17%) and plant hormone signal transduction (15%) (Fig. 7b). The DEGs in the R line were associated with eight metabolic pathways, and the top three most enriched metabolic levels were alpha-linolenic acid metabolism (27%), phenylpropanoid biosynthesis (23%) and cysteine and methionine metabolism (14%) (Fig. 7c).

Expression analysis of DEGs related to tribenuron-methyl in the four samples. a. Heatmap of DEGs in Rt VS St. b. Heatmap of DEGs in St VS Sck. c. Heatmap of DEGs in Rt VS Rck

Classification of metabolic levels of DEGs related to tribenuron-methyl. a. Classification of metabolism levels of DEGs in Rt VS St. b. Classification of metabolism levels of DEGs in St VS Sck. c Classification of metabolism levels of DEGs in Rt VS Rck. The digital numbers represent the ratios of genes in different category to all DEGs. Different colors denote different gene clusters
In Rt vs. St, there were 43 genes encoding calmodulin-like (CML) proteins, respiratory burst oxidase homolog (RBOH), WRKY DNA-binding protein, calcium-dependent protein kinase (CPK), calcium-binding EF-hand family proteins and the disease-resistance protein family, which influence plant-pathogen interactions. As shown in the metabolic pathway (Fig. 8b), CDPK affects the expression of RBOH by sensing the Ca2+ level, thereby stimulating the generation of ROS. WRKY22 and WRKY33 induce the expression of defense-related genes, eventually reorganizing the cell wall or inducing hypersensitivity. Genes encoding lipoxygenase 3 (LOX3), allene oxide cyclase 3 (AOC3), PLAT/LH2 domain-containing lipoxygenase family protein and alcohol dehydrogenase (ADH1) were enriched in α-linolenic acid metabolism (Fig. 8c), and > 4-fold changes of these genes were induced in Rt relative to St. Peroxidase-related genes were found in phenylpropanoid biosynthesis. They produced H2O2 during the defense reaction, which in turn stimulated an antioxidant stress response (Fig. 8d).

Metabolic pathway in which some DEGs related to tribenuron-methyl were involved. Boxes generally represent enzymes, small circles represent metabolites, and rounded boxes represent another metabolic pathway diagram. Green represents down-regulated gene, red represents up-regulated gene, and blue represents both up- and down-regulated gene. The expression of gene in this figure refers to comprehensive performance among in Rt VS St, St VS Sck and Rt VS Rck
The genes encoding RBOH, WRKY, LOX3, ADH1, ACO1, peroxidase, and calcium-dependent protein were down-regulated in the S line. In the R line, however, RBOH, WRKY, and calcium-dependent protein were not detected, while the genes encoding ADH1, ACO1 and peroxidase were up-regulated (Fig. 6b-c). The genes encoding CYP79F1, CYP83A1, CYP79B2, CYP79B3 and BCAT4, which are secondary metabolites that contribute to plant defense, were found in the glucosinolate biosynthetic pathway (Fig. 8a); the genes encoding MPK3 and CDPK were detected in the signal transduction and plant-pathogen interaction pathways. In these pathways, MPK family genes stimulate the expression of WRKY family members and ultimately affect the expression of related defense genes in the S line (Fig. 8b). In general, there were many DEGs between the S and R lines after TBM exposure. Combining GO and KEGG enrichment analysis, the DEGs were all down-regulated in the S line, but about 70% of the R line DEGs were up-regulated, suggesting that TBM can have an adverse reaction on rapeseed by inhibiting the biosynthesis of secondary metabolites, disrupting lipid metabolism or cell membrane structure and influencing stress signal transduction. These results also explain why the root system of S line plants was more severely inhibited compared to R line.
Verification of gene expression data by qRT-PCR analysis
To verify the RNA-seq results, 11 genes were randomly selected from the 73 genes identified above in Rt vs. St and subjected to qRT-PCR analysis. We also performed qRT-PCR to confirm expression of ALS isozyme genes (BnaC01g25380D) to distinguish expression levels between R and S lines. As shown in Fig. 9, the results of qRT-PCR analysis were consistent with the RNA sequence data, highlighting the reliability of the RNA-sequencing procedure.

qRT-PCR verification analysis. The qPCR values are presented as the averages of triplicates
Measurement of physiological parameters
Most of the genes found by screening above were redox-related, so we also analyzed a few relevant physiological indicators (Fig. 10). Carbohydrate and protein are the main raw materials of metabolism during seed germination and in this study, SUG and PRO content in the Sck and Rck were almost the same. After treatment, the content of SUG in the Rt increased significantly by 72.6% compared to control, while that in the St decreased by 33.8%. The PRO content in the St increased significantly by 37% comparing with control, and it was significantly higher than that of other treatments (p < 0.05); but, the PRO content of the R line did not change significantly. Environmental stressors can promote the production of harmful metabolites such as reactive oxygen species (ROS) in plants. Oxygen free radicals act on unsaturated fatty acids to produce lipid peroxides that break down to form toxic compounds like malondialdehyde (MDA) [7]. Compared to the control, the MDA content of the St and Rt increased by 28.9 and 13.1%, respectively. Proline protects membranes and enzymes from ROS damage [9], and superoxide dismutase (SOD), catalase (CAT), and peroxidase (POD) are important components of the antioxidant enzyme system that keeps oxidation reactions in check [21]. Our data showed that the proline content of the St and Rt lines significantly increased by 30.3 and 91.2%, respectively (p < 0.05). The SOD level in St significantly decreased by 19.58%, and the activities of CAT and POD significantly increased by 20 and 47.96%, respectively. The activity of SOD, CAT and POD in Rt was not significantly different from the control group. However, there were significant differences in CAT and POD between St and Rt. Phenylalanine ammonia lyase (PAL) is a key enzyme in the phenylpropanoid pathway [22], which is involved in the stress response. In this study, the PAL activity in St significantly increased by 29.3% (p < 0.05), while that of Rt only increased by 13.8%.

Changes of 8 physiological indicators in four simples. Each bar shows averages of triplicates. a, b and c represent significant difference (P<0.05). SUG: soluble sugar; MDA: malondialdehyde; PAL: phenylalanine ammonia-lyase; SOD: superoxide dismutase; CAT: catalase; POD: peroxidase
Discussion
The response to herbicide stress involves various antioxidant defense mechanisms [15], including enzymatic and non-enzymatic antioxidant detoxification pathways. The most common non-enzymatic antioxidant pathways include the defense response activated by plant hormones and signal transduction, as well as osmotic regulation [12]. Herbicide resistance mechanisms include mutations in target-site resistance (TSR) genes and non-target-site resistance (NTSR) changes. ALS is the first enzyme in the biosynthetic pathway of branched-chain amino acids [23]. Although we did not detect a significant change in ALS activity or that of other genes in form branched-chains amino acids of the glucosinolate biosynthetic pathway, we assume that ALS expression did occur. Studies have shown that herbicide damage to plants begins 3–8 h after herbicide application [24], and the expression level of ALS might be inhibited during that time. With increasing exposure time, the gene expression in the different lines returned to similar levels, and no difference was detected. This was confirmed by qRT-PCR for ALS isozymes. TBM mainly affects biosynthesis of branched-chain amino acids, and the two genes encoding branched-chain aminotransferase (BCA) in the S and R lines were strongly down-regulated by TBM treatment, implicating a probable negative regulation function in the adaptation to TBM.
Apart from the above-mentioned TSR genes, increasing herbicide metabolic rate is an important mechanism for enhancing herbicide resistance. The main biotransferases involved in herbicide metabolism include cytochrome P450 monooxygenase (P450), ATP binding cassette (ABC) transporter, and glutathione S-transferase (GST) [13, 25]. In this study, nine genes encoding CYP450 (CYP83A1, CYP79F1, CYP79B2, CYP79B3), which are key enzymes in the glucosinolate biosynthetic pathway, were identified as DEGs down-regulated only in the S line. Among them, CYP83A1 [26] and CYP79F1 [27] were involved in the metabolism of toxic substances and CYP79B2 and CYP79B3 could convert tryptophan to IAA (indole-3-acetic acid) [28]. Carotenoid biosynthesis in plants could be inhibited by herbicides at different steps [29]. We found four differentially expressed CYP707A3 genes that were strongly down-regulated in the S line by TBM treatment. CYP707A3 encodes ABA 8′-hydroxylase [30]. Previous studies have shown that increased expression of HvABA8’OH1 was associated with a higher germination rate [31]. This was consistent with our research, in which the expression of ABA 8′-hydroxylase was inhibited in the S line, and germination ability and root length were reduced. GST catalyzes the reaction between herbicide and glutathione, or acts as a peroxidase to reduce oxidative damage [15]. The active site, Cys-19 in GSTZ1, has an especially strong catalytic effect [32]. In previous studies, the GST gene was found to have been mostly up-regulated after herbicide treatment [13, 33, 34]. Our study identified a gene encoding GSTZ1, which was down-regulated in the R line. The reason for this difference might be that the phi-, tau- or lambda-like GSTs are much more responsive to chemical treatments, but zeta-like GST genes are not strongly induced by these treatments [35]. The ABC transporter is an efflux pump located on the cell membrane and an important protector against external stresses [36]. In general, the transcription of the ABC transporter was significantly increased after herbicide treatment [12, 13, 37], however, our results differ from these findings. Over the seven-day exposure period, TBM caused the gene encoding ABCB5 to be down-regulated in the S line but no significant change in the R line. The reason for this could be the different genetic background of the two rapeseed lines, where the expression of genes related to detoxification was suppressed in the S line, while the R line retained the capacity to breakdown TBM. This result demonstrated that the R line had a greater ability to promote expression of genes related to detoxification so that it could prevent damage from TBM.
As part of the defense mechanism, plant hormones play an important role in resistance by activating secondary signals [38, 39]. Previous researches showed that the expression of jasmonate promoted resistance [40]. BRs are critical for the induction of the detoxifying response against herbicides [41]. Overexpression of GmBIN2 in transgenic roots resulted in significantly higher relative root growth than in controls under abiotic stress [39]. In this study, no hormone-related genes such as jasmonate or brassinosteroids (BRs) were detected in the R line, but eight down-regulated genes encoding JAZ6, BIN2 and ERF were found in the S line. Auxin-based herbicides might directly induce the expression of ACC oxidase, and plant resistance could be associated with reduced ethylene synthesis [42, 43]. After TBM treatment, six genes encoding ERF2 were down-regulated in the S line, while few genes were detected in the R line. This gene expression alteration in the S line was consistent with the TBM-sensitivity phenotype. In addition, the expression of ACC oxidase was down-regulated in the S line but up-regulated in the R line, with log2 fold-changes of − 1.73 and 1.58, respectively. We inferred that expression of the ACC oxidase gene was slightly up-regulated in the R line, which might be related to the delayed response of tolerant varieties to TBM, or the existence of some other regulatory mechanism, like negative feedback regulation. A similar result was reported by Gao [44].
In addition to plant hormones, we also found some specific changes in expression of genes related to antioxidant stress. A group of 19 genes encoding WRKY and RBOH and 29 calmodulin-related genes were also found in the plant-pathogen interaction pathway that is part of the signal transduction attribute. Previous studies showed that CDPK [45], WRKY [46], Rboh [47, 48], and MAPKs could regulate plant tolerance by activating and regulating gene expression, transmitting ROS signals, and triggering hydrogen peroxide-induced antioxidant enzyme activity [14]. In the MAPK signaling pathway, two MAPK-related genes and one CAT-related gene were identified in the S line and R line, respectively. In addition, 23 genes related to POD were screened as part of the phenylpropanoid biosynthetic pathway. POD-related genes produce H2O2, which stimulates the antioxidant stress response. These genes play an important role in enhancing plant resistance. For example, WRKY is from a group of transcription factors that play a vital role in stress tolerance. Their expression was higher in Rt than in St, and similar results were obtained by other researchers who found contrasting expression pattern of genes like WRKY in resistant and susceptible genotypes [49, 50]. These genes do not work independently, as shown in the metabolic pathway (Fig. 8). CDPK alters the expression of RBOH by sensing the Ca2+level. RBOH reduces molecular oxygen to superoxide, and the latter is converted into H2O2 by superoxide dismutase. MAPK family genes stimulate the expression of WRKY family members and ultimately affect the expression of related defense genes. Previous studies have shown that the ROS scavenging system could be triggered by herbicide stress to promote the metabolism of herbicide molecules [21, 44, 51]. In this study, the activity of CAT, POD and PAL increased after TBM exposure, but the increase in the S line was higher than that of the R line. In contrast, SOD activity decreased in the S line but increased in the R line. This was consistent with the transcriptome results and phenotypic characteristics (Fig. 1), as well as previous results [38]. Taken together, these changes demonstrated that TBM stress inhibited the expression of stress-related genes and led to accumulation of toxic substances in the S line, while the R line was able to up-regulate gene expression to counteract ROS.
Herbicides have been reported to impair carbon metabolism leading to accumulation of carbohydrates [52,53,54]. In the R line, genes encoding UDP-glucose/UDP-galactose 4-epimerase (UGE4) and UDP-glucose dehydrogenase (UGD) were found, which were annotated to carbohydrate metabolism. UGE4 mediates the conversion of UDP-glucose and UDP-galactose [55], while UGD converted UDP-glucose to UDP-glucuronic acid [56], which is used for cell wall carbohydrate biosynthesis. A previous study showed that carbohydrate accumulated in roots as a result of herbicide treatment [54]. In this study, SUG content in the R line increased by 73%, but in the S line it decreased by 34%, compared to control. Another common effect on the roots of plants treated with herbicide was the induction of fermentation, but an increase in ADH activity may reduce this effect [57]. Our study showed that genes encoding ADH1 were up-regulated in Rt vs. St and the R line but down-regulated in the S line. As expected for ADH1-related genes, those encoding LOX3, ADH1, and OPC-8:0 CoA ligase1 (OPCL1) were up-regulated in Rt vs. St, but down-regulated in the S and R lines. LOX preserved intact cell membranes by delaying breakdown of polyunsaturated fatty acids, thereby reducing the formation of cytotoxic derivatives [58]. OPCL1 took part in the synthesis of JA which regulated defense-related processes in higher plants [59]. The genes mentioned above were also detected in metabolism of the unsaturated fatty acid, alpha-linolenic acid. ROS can react with unsaturated fatty acids in the cell membrane, leading to membrane lipid peroxidation, cell membrane damage, and increased permeability [9]. MDA is an intermediate product of lipid peroxidation [7, 58], and our results showed that the MDA levels in the R and S lines increased by 13 and 29%, respectively. Proline acts as a protective agent for biomembranes and as an osmotic regulator [9]. The proline concentration in the R line increased by 91%, which was much higher than the 30% of the S line. The greater amount of proline produced by the R line may have served to prevent membrane damage from lipid peroxidation and helped to maintain the intracellular osmotic balance, so that only a small amount of MDA was produced; thus, the R line showed greater resistance to TBM-induced damage.
Conclusion
After TBM stress, most DEGs were down-regulated in the S line but up-regulated in the R line. GO and KEGG analysis revealed 137 genes, including those related to antioxidant stress and herbicide detoxification such as RBOH, WRKY, CYP450, ABC, MPK3, CDPK, DOX, LOX3, and ADH. These genes were mainly enriched in the redox pathway and some metabolic pathways such as plant-pathogen interactions, α-linolenic acid metabolism, glucosinolate biosynthesis, and phenylpropanoid biosynthesis. The results were further verified by eight physiological indices. In summary, the resistant and sensitive rapeseed lines both underwent oxidative stress after TBM exposure, but, compared to the S line, the R line was better able to regulate gene expression to reduce the oxidative damage caused by the tribenuron-methyl herbicide.
Methods
Plant materials and TBM treatment
The relevant concentration of TBM was ascertained from a search of the literature [60]. Brassica napa 27,123 was selected as the sensitive (S) line while 27,085 was chosen as the resistant (R) line from the RIL population containing 172 lines. Twenty seeds of S and R lines were placed on three layers of filter paper moistened with 3 ml of 0.15 mg•kg− 1 TBM. Distilled water was used as the control. The seeds were placed into a climate chamber at 25 °C, 85% relative humidity, and 16 h/8 h of light / darkness. Each test was conducted with 20 replicates. At day 7 of treatment, the root lengths were measured and 0.1 g root samples of each biological replicate were collected into 1.5 mL centrifuge tubes, quickly frozen in liquid nitrogen and stored at − 80 °C for later determination of physiological indices and qRT-PCR. The control and treated samples of the S and R lines were labeled Sck and Rck, and St and Rt, respectively. The RIL population came from a cross between 10D130 and Zhongshuang11 (ZS11). 10D130 is a high-generation inbred line selected from the interspecific hybrids of Brassica juncea and Brassica oleracea by the Chongqing Engineering Research Center, while ZS11 is a conventional high-quality rapeseed variety selected by the Chinese Academy of Agricultural Sciences. The seeds were provided by the Chongqing Engineering Research Center. Tribenuron-methyl (TBM) was the Maifa brand produced by Hetian Chemical Co., Ltd. in Shenyang, China.
RNA extraction, cDNA library construction, and sequencing
The root samples of S and R lines under control or TBM stress were sent to Personalbio Co., Ltd. (Shanghai, China) for RNA extraction, library construction, and transcriptome sequencing on the Illumina sequencing platform. After removing the 3′-adapter, low-quality sequences (sequence quality values < Q20), the clean data were aligned to the Brassica napus reference genome (http://www.genoscope.cns.fr/Brassicanapus/cgi-bin/gbrowse/colza/) using HISAT2 [61] (http://ccb.jhu.edu/software/hisat2/index.shtml). The read count value was determined by HTSeq [62] (https://htseq.readthedocs.io/en/release_0.11.1/). Fragments per kilobase million (FPKM) values were calculated to estimate gene expression levels. DEGs between the two groups were identified using DESeq [63] based on p ≤ 0.05 and |log2 fold-change| ≥ 1. Gene ontology (GO) enrichment analysis of the DEGs was performed using topGO [64], and significantly enriched GO items were selected based on a false discovery rate (FDR) < 0.01. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using the KOBAS2.0 website (http://kobas.cbi.pku.edu.cn/home) [65], and significant enrichment was selected based on a FDR < 0.01. KEGG database is developed by Kanehisa Laboratories [66,67,68], and KEGG pathways and other KEGG materials shown in this article were copyrighted by Kanehisa Laboratories.
qRT-PCR validation
qRT-PCR was performed on a BioRad CFX96 real-time system using a kit from Vazyme Biotechnology Co., Ltd. (Nanjing, China). The reaction conditions were as follows: 95 °C for 30 s and 40 cycles (95 °C for 10 s, 56 °C for 30 s, 72 °C for 60 s). The 2-ΔΔCt method was used to evaluate the relative expression of genes based on the stable expression level of BnaActin 7 [10]. The primer pairs were designed by Vector NTI Advance 11.5.1 software and synthesized by Sangon Biotech (Shanghai, China) (Table 2).
Measurement of physiological parameters in roots
The physiological parameters, including soluble protein (PRO), soluble sugar (SUG), malondialdehyde (MDA), proline content, and phenylalanine ammonia-lyase (PAL), superoxide dismutase (SOD), catalase (CAT), and peroxidase (POD) activities were measured. All measurements were performed in triplicate and means were calculated for further analysis. The proline content was estimated using the method described by predecessors [69]. The contents of PRO, SUG, MDA, PAL, SOD, CAT, and POD were measured using kits from Sino Best Biological Technology Co., Ltd. (Shanghai, China).
Availability of data and materials
The raw sequence data are available in the NCBI Sequence Read Archive (SRA) repository. The accession number is PRJNA717988, and SRA RunSelector as follows: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA717988. All data supporting the conclusions of this article are included in the article and its additional files.
Abbreviations
- SNP:
-
single nucleotide polymorphism
- DEGs:
-
differentially expressed genes
- FPKM:
-
fragments per kilobase million
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PRO:
-
soluble protein
- SUG:
-
soluble sugar
- MDA:
-
malondialdehyde
- PAL:
-
phenylalanine ammonia-lyase
- SOD:
-
superoxide dismutase
- CAT:
-
catalase
- POD:
-
peroxidase
- DOX:
-
dioxygenases
- LOX3:
-
lipoxygenase 3
- ADH1:
-
alcohol dehydrogenase 1
- RBOH:
-
respiratory burst oxidase homologue
- WRKY:
-
WRKY DNA-binding protein
- ACO1:
-
ACC oxidase 1
- CYP450:
-
cytochrome P450
- ABC:
-
ATP binding cassette subfamily
- BCAT4:
-
branched-chain aminotransferase 4
- MPK3:
-
mitogen-activated protein kinase 3
- CDPK:
-
calcium-dependent protein kinase
- ERF2:
-
ethylene-responsive element-binding factor 2
- OPCL1:
-
OPC-8:0 CoA ligase1
References
Green JM. Current state of herbicides in herbicide-resistant crops. Pest Manag Sci. 2014;70(9):1351–7. https://doi.org/10.1002/ps.3727.
Mazur BJ, Falco SC. The development of herbicide resistant crops. Annu Rev Plant Physiol Plant Mol Biol. 1989;40(1):441–70. https://doi.org/10.1146/annurev.pp.40.060189.002301.
Qi Y, Li J, Fu G, Zhao C, Guan X, Yan B, et al. Effects of sublethal herbicides on offspring germination and seedling growth: redroot pigweed (Amaranthus retroflexus) vs. velvetleaf (Abutilon theophrasti). Sci Total Environ. 2018;645:543–9. https://doi.org/10.1016/j.scitotenv.2018.07.171.
Lu K, Wei L, Li X, Wang Y, Wu J, Liu M, et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat Commun. 2019;10(1):1154–66. https://doi.org/10.1038/s41467-019-09134-9.
Liu C, Zhang X, Zhang K, An H, Hu K, Wen J, et al. Comparative analysis of the Brassica napus root and leaf transcript profiling in response to drought stress. Int J Mol Sci. 2015;16(8):18752–77. https://doi.org/10.3390/ijms160818752.
Pu Y, Liu L, Wu J, Zhao Y, Bai J, Ma L, et al. Transcriptome profile analysis of winter rapeseed (Brassica napus L.) in response to freezing stress, reveal potentially connected events to freezing stress. Int J Mol Sci. 2019;20(11):1–25.
Wang J, Jiao J, Zhou M, Jin Z, Yu Y, Liang M. Physiological and transcriptional responses of industrial rapeseed (Brassica napus) seedlings to drought and salinity stress. Int J Mol Sci. 2019;20(22):5604–25. https://doi.org/10.3390/ijms20225604.
Qasim MU, Zhao Q, Shahid M, Samad RA, Ahmar S, Wu J, et al. Identification of QTLs containing resistance genes for sclerotinia stem rot in Brassica napus using comparative transcriptomic studies. Front Plant Sci. 2020;11:776–90. https://doi.org/10.3389/fpls.2020.00776.
Wang T, Hou L, Jian H, Di F, Li J, Liu L. Combined QTL mapping, physiological and transcriptomic analyses to identify candidate genes involved in Brassica napus seed aging. Mol Gen Genomics. 2018;293(6):1421–35. https://doi.org/10.1007/s00438-018-1468-8.
Zhao H, Basu U, Kebede B, Qu C, Li J, Rahman H. Fine mapping of the major QTL for seed coat color in Brassica rapa var. yellow Sarson by use of NIL populations and transcriptome sequencing for identification of the candidate genes. PLoS One. 2019;14(2):1–26.
Jian H, Zhang A, Ma J, Wang T, Yang B, Shuang LS, et al. Joint QTL mapping and transcriptome sequencing analysis reveal candidate flowering time genes in Brassica napus L. BMC Genomics. 2019;20(1):1–14.
Baek YS, Goodrich LV, Brown PJ, James BT, Moose SP, Lambert KN, et al. Transcriptome profiling and genome-wide association studies reveal GSTs and other defense genes involved in multiple signaling pathways induced by herbicide safener in grain sorghum. Front Plant Sci. 2019;10:192–207. https://doi.org/10.3389/fpls.2019.00192.
Bai S, Liu W, Wang H, Zhao N, Jia S, Zou N, et al. Enhanced herbicide metabolism and metabolic resistance genes identified in tribenuron-methyl resistant Myosoton aquaticum L. J Agric Food Chem. 2018;66(37):9850–7. https://doi.org/10.1021/acs.jafc.8b02740.
Zhou W, Huang Q, Wu X, Zhou Z, Ding M, Shi M, et al. Comprehensive transcriptome profiling of Salvia miltiorrhiza for discovery of genes associated with the biosynthesis of tanshinones and phenolic acids. Sci Rep. 2017;7(1):1–12.
Duhoux A, Carrère S, Gouzy J, Bonin L, Délye C. RNA-Seq analysis of rye-grass transcriptomic response to an herbicide inhibiting acetolactate-synthase identifies transcripts linked to non-target-site-based resistance. Plant Mol Biol. 2015;87(4–5):473–87. https://doi.org/10.1007/s11103-015-0292-3.
Saja D, Rys M, Stawoska I, Skoczowski A. Metabolic response of cornflower (Centaurea cyanus L.) exposed to tribenuron-methyl: one of the active substances of sulfonylurea herbicides. Acta Physiol Plant. 2016;38(7):1–13.
Shi C, Zheng Y, Geng J, Liu C, Pei H, Ren Y, et al. Identification of herbicide resistance loci using a genome-wide association study and linkage mapping in Chinese common wheat. Crop J. 2020;8(4):666–75. https://doi.org/10.1016/j.cj.2020.02.004.
Nguyen TCT, Abrams SR, Friedt W, Snowdon RJ. Quantitative trait locus analysis of seed germination, seedling vigour and seedling-regulated hormones in Brassica napus. Plant Breed. 2018;137(3):388–401. https://doi.org/10.1111/pbr.12576.
Yu X, Yang A, James AT. Selecting soybeans for sulfonylurea herbicide tolerance: a comparative proteomic study of seed germinations. Crop Pasture Sci. 2017;68(1):27–32. https://doi.org/10.1071/CP16272.
Rajjou L, Duval M, Gallardo K, Catusse J, Bally J, Job C, et al. Seed germination and vigor. Annu Rev Plant Biol. 2012;63(1):507–33. https://doi.org/10.1146/annurev-arplant-042811-105550.
Sewelam N, Kazan K, Schenk PM. Global plant stress signaling: reactive oxygen species at the cross-road. Front Plant Sci. 2016;7:1–21.
Jung YJ, Melencion SMB, Lee ES, Park JH, Alinapon CV, Oh HT, et al. Universal stress protein exhibits a redox-dependent chaperone function in Arabidopsis and enhances plant tolerance to heat shock and oxidative stress. Front Plant Sci. 2015;6:1141–52.
Zhou Q, Liu W, Zhang Y, Liu KK. Action mechanisms of acetolactate synthase-inhibiting herbicides. Pestic Biochem Physiol. 2007;89(2):89–96. https://doi.org/10.1016/j.pestbp.2007.04.004.
Délye C. Unravelling the genetic bases of non-target-site-based resistance (NTSR) to herbicides: a major challenge for weed science in the forthcoming decade. Pest Manag Sci. 2013;69(2):176–87. https://doi.org/10.1002/ps.3318.
Lampe JW, Peterson S. Brassica, biotransformation and Cancer risk: genetic polymorphisms Alter the preventive effects of cruciferous vegetables. J Nutr. 2002;132(10):2991–4. https://doi.org/10.1093/jn/131.10.2991.
Meenu AR, Majee M, Pradhan AK, Bisht NC. Genomic origin, expression differentiation and regulation of multiple genes encoding CYP83A1, a key enzyme for core glucosinolate biosynthesis, from the allotetraploid Brassica juncea. Planta. 2014;241(3):651–65. https://doi.org/10.1007/s00425-014-2205-0.
Guo L, Yang R, Gu Z. Cloning of genes related to aliphatic glucosinolate metabolism and the mechanism of sulforaphane accumulation in broccoli sprouts under jasmonic acid treatment. J Sci Food Agric. 2016;96(13):4329–36. https://doi.org/10.1002/jsfa.7629.
Kumari V, Kumar V, Bhalla TC. Functional interpretation and structural insights of Arabidopsis lyrata cytochrome P450 CYP71A13 involved in auxin synthesis. Bioinformation. 2015;11(7):330–5. https://doi.org/10.6026/97320630011330.
Vítek P, Novotná K, Hodaňová P, Rapantová B, Klem K. Detection of herbicide effects on pigment composition and PSII photochemistry in Helianthus annuus by Raman spectroscopy and chlorophyll a fluorescence. Spectrochim Acta - Part A Mol Biomol Spectrosc. 2017;170:234–41. https://doi.org/10.1016/j.saa.2016.07.025.
Umezawa T, Okamoto M, Kushiro T, Nambara E, Oono Y, Seki M, et al. CYP707A3, a major ABA 8′-hydroxylase involved in dehydration and rehydration response in Arabidopsis thaliana. Plant J. 2006;46(2):171–82. https://doi.org/10.1111/j.1365-313X.2006.02683.x.
Chono M, Matsunaka H, Seki M, Fujita M, Kiribuchi-otobe C. Isolation of a wheat (Triticum aestivum L .) mutant in ABA 8′-hydroxylase gene: effect of reduced ABA catabolism on germination inhibition under field condition. Breed Sci. 2013;115:104–15.
Thom R, Dixon DP, Edwards R, Cole DJ, Lapthorn AJ. The structure of a zeta class glutathione S-transferase from Arabidopsis thaliana: characterisation of a GST with novel active-site architecture and a putative role in tyrosine catabolism. J Mol Biol. 2001;308(5):949–62. https://doi.org/10.1006/jmbi.2001.4638.
Liu W, Bai S, Zhao N, Jia S, Li W, Zhang L, et al. Non-target site-based resistance to tribenuron-methyl and essential involved genes in Myosoton aquaticum (L.). BMC Plant Biol. 2018;18(1):1–15.
Zhao N, Li W, Bai S, Guo W, Yuan G, Wang F, et al. Transcriptome profiling to identify genes involved in mesosulfuron-methyl resistance in Alopecurus aequalis. Front Plant Sci. 2017;8:1391–407. https://doi.org/10.3389/fpls.2017.01391.
Dixon DP, Edwards R. Enzymes of tyrosine catabolism in Arabidopsis thaliana. Plant Sci. 2006;171(3):360–6. https://doi.org/10.1016/j.plantsci.2006.04.008.
Oosterhuis B, Vukman K, Vági E, Glavinas H, Jablonkai I, Krajcsi P. Specific interactions of chloroacetanilide herbicides with human ABC transporter proteins. Toxicology. 2008;248(1):45–51. https://doi.org/10.1016/j.tox.2008.03.003.
Fernanda Moreira L, Juliana Zomer S, Marta MS. Modulation of the multixenobiotic resistance mechanism in Danio rerio hepatocyte culture (ZF-L) after exposure to glyphosate and roundup®. Chemosphere. 2019;228:159–65. https://doi.org/10.1016/j.chemosphere.2019.04.140.
Smékalová V, Doskočilová A, Komis G, Šamaj J. Crosstalk between secondary messengers, hormones and MAPK modules during abiotic stress signalling in plants. Biotechnol Adv. 2014;32(1):2–11. https://doi.org/10.1016/j.biotechadv.2013.07.009.
Shuang WL, Shan CQ, Wei XD, Ming QZ, Zhang C, Nan LS, et al. Overexpression of GmBIN2, a soybean glycogen synthase kinase 3 gene, enhances tolerance to salt and drought in transgenic Arabidopsis and soybean hairy roots. J Integr Agric. 2018;17:1959–71.
Guo Q, Yoshida Y, Major IT, Wang K, Sugimoto K, Kapali G, et al. JAZ repressors of metabolic defense promote growth and reproductive fitness in Arabidopsis. Proc Natl Acad Sci U S A. 2018;115(45):10768–77.
Zhou Y, Xia X, Yu G, Wang J, Wu J, Wang M, et al. Brassinosteroids play a critical role in the regulation of pesticide metabolism in crop plants. Sci Rep. 2015;5:1–8.
Chayapakdee P, Sunohara Y, Endo M, Yamaguchi T, Fan L, Uchino A, et al. Quinclorac resistance in Echinochloa phyllopogon is associated with reduced ethylene synthesis rather than enhanced cyanide detoxification by β-cyanoalanine synthase. Pest Manag Sci. 2020;76(4):1195–204. https://doi.org/10.1002/ps.5660.
Gao Y, Pan L, Sun Y, Zhang T, Dong L, Li J. Resistance to quinclorac caused by the enhanced ability to detoxify cyanide and its molecular mechanism in Echinochloa crus-galli var. zelayensis. Pestic Biochem Physiol. 2017;143:231–8. https://doi.org/10.1016/j.pestbp.2017.08.009.
Gao Y, Li J, Pan X, Liu D, Napier R, Dong L. Quinclorac resistance induced by the suppression of the expression of 1-aminocyclopropane-1-carboxylic acid (ACC) synthase and ACC oxidase genes in Echinochloa crus-galli var. zelayensis. Pestic Biochem Physiol. 2018;146:25–32. https://doi.org/10.1016/j.pestbp.2018.02.005.
Atif RM, Shahid L, Waqas M, Ali B, Rashid MAR, Azeem F, et al. Insights on calcium-dependent protein kinases (CPKs) signaling for abiotic stress tolerance in plants. Int J Mol Sci. 2019;20(21):5298. https://doi.org/10.3390/ijms20215298.
Schluttenhofer C, Yuan L. Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol. 2015;167(2):295–306. https://doi.org/10.1104/pp.114.251769.
Kimura S, Kaya H, Kawarazaki T, Hiraoka G, Senzaki E, Michikawa M, et al. Protein phosphorylation is a prerequisite for the Ca2+-dependent activation of Arabidopsis NADPH oxidases and may function as a trigger for the positive feedback regulation of Ca2+ and reactive oxygen species. Biochim Biophys Acta - Mol Cell Res. 2012;1823(2):398–405. https://doi.org/10.1016/j.bbamcr.2011.09.011.
Ben Rejeb K, Lefebvre-De Vos D, Le Disquet I, Leprince AS, Bordenave M, Maldiney R, et al. Hydrogen peroxide produced by NADPH oxidases increases proline accumulation during salt or mannitol stress in Arabidopsis thaliana. New Phytol. 2015;208(4):1138–48. https://doi.org/10.1111/nph.13550.
Konda AK, Sabale PR, Soren KR, Subramaniam SP, Singh P, Rathod S, et al. Systems biology approaches reveal a multi-stress responsive WRKY transcription factor and stress associated gene co-expression networks in chickpea. Curr Bioinforma. 2019;14(7):591–601. https://doi.org/10.2174/1574893614666190204152500.
Yousfi FE, Makhloufi E, Marande W, Ghorbel AW, Bouzayen M, Bergès H. Comparative analysis of WRKY genes potentially involved in salt stress responses in Triticum turgidum L. ssp. durum. Front Plant Sci. 2017;7:2034–49.
Cummins I, Wortley DJ, Sabbadin F, He Z, Coxon CR, Straker HE, et al. Key role for a glutathione transferase in multiple-herbicide resistance in grass weeds. Proc Natl Acad Sci U S A. 2013;110(15):5812–7. https://doi.org/10.1073/pnas.1221179110.
Serra AA, Couée I, Heijnen D, Michon-Coudouel S, Sulmon C, Gouesbet G. Genome-wide transcriptional profiling and metabolic analysis uncover multiple molecular responses of the grass species lolium perenne under low-intensity xenobiotic stress. Front Plant Sci. 2015;6:1–22.
Orcaray L, Zulet A, Zabalza A, Royuela M. Impairment of carbon metabolism induced by the herbicide glyphosate. J Plant Physiol. 2012;169(1):27–33. https://doi.org/10.1016/j.jplph.2011.08.009.
Oyuela MER. Carbohydrate accumulation in leaves of plants treated with the herbicide chlorsulfuron or imazethapyr is due to a decrease in sink strength. J Agric Food Chem. 2004;52(25):7601–6.
Rösti J, Barton CJ, Albrecht S, Dupree P, Pauly M, Findlay K, et al. UDP-glucose 4-epimerase isoforms UGE2 and UGE4 cooperate in providing UDP-galactose for cell wall biosynthesis and growth of Arabidopsis thaliana. Plant Cell. 2007;19(5):1565–79. https://doi.org/10.1105/tpc.106.049619.
Seitz B, Klos C, Wurm M, Tenhaken R. Matrix polysaccharide precursors in Arabidopsis cell walls are synthesized by alternate pathways with organ-specific expression patterns. Plant J. 2000;21(6):537–46. https://doi.org/10.1046/j.1365-313x.2000.00696.x.
Zabalza A, González EM, Arrese-Igor C, Royuela M. Fermentative metabolism is induced by inhibiting different enzymes of the branched-chain amino acid biosynthesis pathway in pea plants. J Agric Food Chem. 2005;53(19):7486–93. https://doi.org/10.1021/jf050654x.
Xu H, Wei Y, Zhu Y, Lian L, Xie H, Cai Q, et al. Antisense suppression of LOX3 gene expression in rice endosperm enhances seed longevity. Plant Biotechnol J. 2015;13(4):526–39. https://doi.org/10.1111/pbi.12277.
Koo AJK, Howe GA. Role of peroxisomal β-oxidation in the production of plant signaling compounds. Plant Signal Behav. 2007;2(1):20–2. https://doi.org/10.4161/psb.2.1.3612.
Wang LY, Wang RL, Ye S, Gao HH, Lei W, Chen LY, et al. QTL mapping and candidate genes screening of related traits in Brassica napus l. during the germination under tribenuron-methyl stress. Sci Agric Sin. 2020;53(8):1510–23.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. https://doi.org/10.1038/nmeth.3317.
Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. https://doi.org/10.1093/bioinformatics/btu638.
Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2009;26:136–8.
Gene T, Consortium O. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9.
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:316–22.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51. https://doi.org/10.1002/pro.3715.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545–51. https://doi.org/10.1093/nar/gkaa970.
Yi Y, Fang Y, Wu K, Liu Y, Zhang W. Comprehensive gene and pathway analysis of cervical cancer progression. Oncol Lett. 2020;19(4):3316–32. https://doi.org/10.3892/ol.2020.11439.
Bates LS, Waldren RP, Teare ID. Rapid determination of free proline for water-stress studies. Plant Soil. 1973;39(1):205–7. https://doi.org/10.1007/BF00018060.
Acknowledgements
We are grateful to all the colleagues in our laboratory, and thank Chongqing Engineering Research Center for providing the seeds of Brassica napus.
Funding
This research was supported by grants from the National Key Research and Development Plan (2018YFD0100500) and Chongqing Technology Innovation and Application Development (cstc2019jscx-msxmX0383). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
CC and QYZ conceived the study. LYW, RLW and WL conducted the experiments. LYW wrote the original manuscript. JYW, CYL, QYZ and CC helped to revise the manuscript. HSS, LJM and FY collected samples and measured physiological parameters. All authors have read and agreed to the published version of the manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1 Table S1.
Quality and annotation of RNA-seq assembly.
Additional file 2 Table S2.
Genes identified by combined GO and KEGG enrichment analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, L., Wang, R., Lei, W. et al. Transcriptome analysis reveals gene responses to herbicide, tribenuron methyl, in Brassica napus L. during seed germination. BMC Genomics 22, 299 (2021). https://doi.org/10.1186/s12864-021-07614-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07614-1