
Abstract
Background
Tilapia (Oreochromis niloticus) cultures are frequently infected by Vibrio vulnificus, causing major economic losses to production units. Previously, tilapia expressing recombinant delta-5 desaturase and delta-6 desaturase (D56) were found to be resistant to V. vulnificus infection. In this report, we profile the D56-mediated molecular changes underlying this resistance in tilapia. A comparative transcriptome analysis was performed on V. vulnificus-infected wild-type and D56-transgenic tilapia using Illumina’s sequencing-by-synthesis approach. Gene enrichment analysis on differentially expressed unigenes was performed, and the expression patterns were validated by real-time PCR.
Results
Comparative transcriptome analysis was performed on RNA-sequence profiles obtained from wild-type and D56-transgenic tilapia at 0, 6 and 24 h post-infection with V. vulnificaus. GO and KEGG gene enrichment analyses showed that D56 regulates several pathways and genes, including fatty acid (FA) metabolism associated, and inflammatory and immune response. Expression of selected FA metabolism-associated, inflammatory and immune responsive genes was validated by qPCR. The inflammatory and immune responsive genes that are modulated by FA-associated D56 likely contribute to the enhanced resistance against V. vulnificus infection in Tilapia.
Conclusions
Transcriptome profiling and filtering for two-fold change variation showed that 3795 genes were upregulated and 1839 genes were downregulated in D56-transgenic tilapia. These genes were grouped into pathways, such as FA metabolism, FA elongation, FA biosynthesis, biosynthesis of unsaturated FA, FA degradation, inflammation, immune response, and chemokines. FA-associated genes and immune-related genes were modulated by D56 at 6 h and 24 h post infection with V. vulnificus. The expression patterns of FA-related genes, inflammatory genes, antimicrobial peptide genes and immune responsive genes at 0, 3, 6, 12, 24 and 48 h post-infection suggests these genes are involved in the enhanced resistance of D56 transgenic tilapia to V. vulnificus.
Similar content being viewed by others


Background
Tilapia (Oreochrombis niloticus) is an important commercial aquaculture species throughout the world, and its production is severely affected by the pathogenic bacteria Vibrio vulnificus, which causes septicemia in fish and humans [1,2,3,4]. Omega-3 polyunsaturated fatty acids (n-3 PUFAs) are known to exert beneficial effects, such as protection of liver, reduction of cholesterol, lower blood pressure and protect from cardiovascular diseases [5, 6]. Furthermore, n-3 PUFAs show positive ionotropic effects and minimize tachyarrhythmia in animal models [7]. Many of these effects may be mediated by alterations in the pro-inflammatory cytokines, TNF-α, IL-1β, IL-6, prostaglandin (PG) E2, and PGF1α, which modulate the immune response in model organisms [8,9,10]. Dietary supplementation with eicosanoids and n-3 PUFAs is well documented to affect immune cell function and B-cell activity [11, 12], and a recent report showed that PUFA-rich food limit pathogen infection in the aquatic organisms [13]. Similarly, transgenic expression of n-3 PUFA biosynthesis genes from Atlantic salmon, i.e., Fatty acyl desaturase synthase delta (Fadsd)5 and Fadsd6, in zebrafish limits infection with Vibrio alginolyticus and V. vulnificus [5, 14].
Previously we reported the dual expression of SsFadsd5 and SsFadsd6 (D56) in tilapia [15]. The dual expression of these genes is under the control of a TRE-regulated CMV minimal promoter, which drives expression of D56 in liver and muscle [15]. Expression of D56 in tilapia enhances resistance to V. vulnificus infection [15]. In addition, the D56 transgenic tilapia exhibit altered gut microbial profiles [15]. However, the underlying molecular mechanism involved in the resistance to V. vulnificus has not been studied using a transcriptomic approach.
We compared the liver transcriptomes between V. vulnificus-susceptible wild-type tilapia and D56 transgenic tilapia with enhanced resistance to the pathogen to reveal the particular genes responsible for the resistance [15, 16]. The alterations in expression of key genes were identified by gene enrichment analysis with KEGG pathway and GO tools. We showed the involvement of fatty acid (FA)-associated genes and immunomodulatory genes in the development of resistance against V. vulnificus infection in tilapia.
Results
Expression of recombinant delta-6 desaturase and delta-5 desaturase alters the transcriptome in tilapia
Wild-type and D56-transgenic tilapia were infected with V. vulnificus, and RNA was extracted from liver at 0, 6 and 24 h post-infection (hpi). Transcriptome sequencing of six groups of samples produced a total of 275,304,348 raw reads for wild-type and D56-transgenic tilapia. After filtering the data 48,315,226, 38,578,158 and 35,079,100 clean reads were obtained for wild-type tilapia fish at 0, 6 and 24 h infected samples, respectively, representing 92.12, 89.24 and 92.19% of raw reads (Table 1). Similarly, 37,449,898, 49,652,212 and 50,987,302 clean reads (90.62, 88.48 and 91.06% of raw reads) were obtained for D56-transgenic tilapia for 0, 6 and 24 hpi samples, respectively (Table 1). A total of 42,622 unigenes were identified from the RNA-sequencing and filtered for two-fold change in expression between V. vulnificus challenged wild-type and D56-transgenic tilapia (Supplementary Figure S1). At the 0 h time-point, 3795 genes were upregulated and 1839 genes were downregulated in D56-transgenic tilapia (Fig. 1a). At 6 hpi, 4365 genes were upregulated and 1976 genes were downregulated (Fig. 1a). At 24 hpi, 4665 were upregulated and 2202 genes were downregulated (Fig. 1a). We could recognize the relevance of DEG between wild-type and D56-transgenic tilapia at different time-points. We found that 1112 DEG existed at three time-points (Fig. 1b).

Delta 5 and Delta6 transgenic (D56) tilapia fish alters the transcriptome profile. The comparative transcriptome data between wild-type / Delta 5 and Delta6 (D56) transgenic tilapia fish were mapped and differentially expressed genes were counted. a Differentially expressed genes with more than or equal to two fold change have been listed. Wild-type and D56 transgenic tilapia fish liver RNA were compared in 0, 6 and 24 h post infection with V. vulnificus. b Differentially expressed genes between wild-type and D56 transgenic tilapia fish at 0, 6 and 24 h V. vulnificus post infected liver RNA. The number of the genes unique to specific infection condition and the number of genes commonly shared by two or three infection conditions are mentioned in the respective intersections
Fatty acid-associated genes are altered in D56-transgenic tilapia according to KEGG pathway analysis
Gene enrichment analysis using the KEGG pathway database showed a total of 24, 30 and 33 pathways were affected in D56-transgenic tilapia at 0, 6, and 24 hpi, respectively (Table 2, 3 4). Immediately after infection, altered expression of various FA-associated pathways, such as FA metabolism, FA elongation, FA biosynthesis, biosynthesis of unsaturated FA, and FA degradation were observed (Table 2). Differences in the FA degradation pathway were also observed between wild-type and D56-transgenic fish at 6 h post infection (Table 3), and the FA metabolism and FA degradation pathways were identified as differentially expressed at 24 hpi (Table 4).
D56-transgenic tilapia exhibit altered immune-related gene expression in GO analysis
The GO enrichment analysis showed a total of 28, 23 and 35 gene sets that were differentially expressed in D56-transgenic and wild-type tilapia at 0, 6 and 24 hpi with V. vulnificus. Cellular component-associated GO terms, such as major histocompatibility (MHC) class II protein complexes and extracellular protein components, were altered at all the time-points examined (Table 5, 6, 7). Biological function-associated GO terms, such as defense response to bacterium, angiogenesis, immune response, antigen presenting and presentation and inflammatory response genes were also altered in D56-transgenic tilapia. Altered molecular function-related GO terms included iron ion binding protein, cytokine and chemokine activity (Table 5, 6, 7). Taken together, the GO analysis revealed that inflammatory genes, chemokine-associated genes, cytokine-associated genes, immune-related genes and iron binding protein genes are differentially regulated in D56-transgenic and wild-type tilapia after V. vulnificus infection (Table 5, 6, 7). We selected target genes with significant fold change, immune-related annotation and higher FPKM value for follow up research (Supplementary File 1).
Ectopic D56 alters FA metabolism-related genes
Since the KEGG analysis showed FA-associated pathways are altered in D56-transgenic tilapia, the expression of selected FA pathway-associated genes was analyzed by real-time PCR at 0, 3, 6, 12, 24 and 48 hpi with V. vulnificus. Since the D56 transgenes (delta-6 desaturase and delta-5 desaturase) are associated with FA biosynthesis, we analyzed the expression pattern by qPCR at many time-points. Significant alterations in the expression of FA-associated genes were observed from the qPCR data (Fig. 2). Notably, ApoA4b was downregulated by D56-transgenic tilapia at 24 hpi (Fig. 2a). CPT1 was upregulated at 24 and 48 hpi (Fig. 2b), and PCK1 was upregulated at 24 hpi (Fig. 2c). HNF4A was upregulated at 6 hpi and downregulated at 24 hpi (Fig. 2d). PPARα was upregulated at 6 and 12 hpi, but it was downregulated at 24 hpi (Fig. 2e). These results showed that FA metabolism-related genes are altered in transgenic tilapia upon V. vulnificus infection.

Delta 5 and Delta 6 transgenic (D56) tilapia fish modulates the expression of Fatty acid associated gene expression. Wild-type and D56 transgenic tilapia fish challenged with V. vulnificus and liver samples from 0, 3, 6, 12, 24 and 48 h post infected conditions were collected for qPCR. The fatty acid associated gene expression relative to EF-1α was estimated. a ApoA4b, b CPT1, c PCK1, d HNF4A, e PPARα. Values represented as Mean ± SEM (n = 5). Significance was determined by T-TEST (*P < 0.05,**P < 0.01,***P < 0.001)
Ectopic D56 modulates immune response genes
In addition to FA-associated genes, several inflammatory and immune responsive genes were altered in D56-transgenic tilapia according to the GO enrichment analysis (Table 5, 6, 7). In addition, tilapia are known to express several antimicrobial peptides (AMPs), such as Tilapia Hepcidin, LEAP2, TP3, TP4, TP5 and Progranulin (PGRN), which have been reported to exert immunomodulatory functions. Hence, expression of genes associated with pro-inflammatory cytokines, immune responsive genes and AMPs were assessed at 0, 3, 6, 12, 24 and 48 hpi with V. vulnificus in wild-type and D56-transgenic tilapia liver.
In D56-transgenic tilapia, the Complement C1q sub-component subunit B (C1qb) was upregulated at 6 hpi and downregulated at 24 hpi. Complement factor H-related protein 1 (CFHR1) was upregulated at 3, 6, 12, 24 and 48 hpi, and Complement factor D (CFD) was upregulated at 6 hpi (Fig. 3). The AMPs also showed significant differences in expression between wild-type and D56-transgenic tilapia. Tilapia Hepcidin (TH) was altered at 24 and 48 hpi; Binding protein I (BPI) was regulated at 24 and 48 hpi; liver-enriched antimicrobial peptide-2 (LEAP2) was altered at 12 and 24 hpi; Tilapia Piscidin (TP)3 was differentially expressed at 6, 24, and 48 hpi; TP4 was altered at 12 hpi; TP5 was altered at 6 hpi; PGRN expression differed at 0 and 48 hpi (Fig. 4). For the inflammatory factors, significant differences between wild-type and D56-transgenic tilapia were detected for NF-κB2 at 12 and 24 hpi; NF-κBI was altered at 24 hpi; Toll-like receptor (TLR)-2 was altered at 12 hpi; TLR-5 was altered at 6, 24 and 48 hpi; Tumor necrosis factor (TNF)-α was altered at 12 hpi; Interleukin (IL)-1β was altered at 24 hpi (Fig. 5). Other immune-related genes were also affected. Peroxiredoxin (PRDX)1 was altered at 6, 12 and 48 hpi; Atypical chemokine receptor (ACKR)4 was altered at 48 hpi; Tissue inhibitor of metalloproteinase (TIMP)2 was altered at 24 hpi (Fig. 6).

Delta 5 and Delta 6 transgenic (D56) tilapia fish alters the Complements gene expression. Wild-type and D56 transgenic tilapia fish challenged with V. vulnificus and liver samples from 0, 3, 6, 12, 24 and 48 h post infected conditions were collected for qPCR. The complements gene expression relative to EF-1α was estimated. a C1qb, b C3, c CFHR1, d CFD. Error bar is Mean ± SEM (n = 5). Significance was determined by T-TEST (*P < 0.05,**P < 0.01,***P < 0.001)

Delta 5 and Delta 6 transgenic (D56) tilapia fish alters the Antimicrobial peptide (AMP) specific gene expression. Wild-type and D56 transgenic tilapia fish challenged with V. vulnificus and liver samples from 0, 3, 6, 12, 24 and 48 h post infected conditions were collected for qPCR. The AMP specific gene expression relative to EF-1α was estimated. a TH, b BP1, c LEAP2, d TP3, e TP4, f TP5, g PGRN. Values represented as Mean ± SEM (n = 5). Significance was determined by T-TEST (*P < 0.05,**P < 0.01,***P < 0.001)

Delta 5 and Delta 6 transgenic (D56) tilapia fish modulates the pro-inflammatory cytokine gene expression. Wild-type and D56 transgenic tilapia fish challenged with V. vulnificus and liver samples from 0, 3, 6, 12, 24 and 48 h post infected conditions were collected for qPCR. Representative pro-inflammatory cytokine gene expression relative to EF-1α was estimated. a TLR-2, b TLR-5, c NF-κB2, d NF-κBI, e TNF-α, f IL-1β. Values represented as Mean ± SEM (n = 5). Significance was determined by T-TEST (*P < 0.05,**P < 0.01,***P < 0.001)

Delta 5 and Delta 6 transgenic (D56) tilapia fish alters PRDX1, ACRK4 and TIMP2 gene expression. Wild-type and D56 transgenic tilapia fish challenged with V. vulnificus and liver samples from 0, 3, 6, 12, 24 and 48 h post infected conditions were collected for qPCR. Expression of a PRDX1, b ACKR4, c TIMP2 genes relative to EF-1α was estimated. Values represented as Mean ± SEM (n = 5). Significance was determined by T-TEST (*P < 0.05,**P < 0.01,***P < 0.001)
Ectopic D56 alters pro-inflammatory cytokines and CFD in whole blood sample
We also measured gene expression in whole blood samples after challenge. Expression of cytokines, inflammatory factors and complement-related genes was analyzed by real-time qPCR. For inflammatory factors, cytokines and complement-related genes, we found that were several significant differences between wild-type and D56-transgenic tilapia whole blood. TLR-5 was altered at 24 hpi; NF-κB2 was altered at 6, 12, 24 and 48 hpi; NF-κBI was altered at 0, 6, 12, and 24 hpi; IL-1β was altered at 24 and 48 hpi; C1qb was altered at 0, 3 and 24 hpi; CFD was altered at 48 hpi (Fig. 7a-f). In whole blood samples, the expression level of CPT1 was also different at 3 and 48 hpi (Fig. 7g). According to these results, protective and immune-related genes are induced in transgenic tilapia upon V. vulnificus infection.

Delta 5 and Delta 6 transgenic (D56) tilapia fish modulates the pro-inflammatory cytokines and immune-related genes in whole blood. Wild-type and D56 transgenic tilapia fish challenged with V. vulnificus and whole blood samples from 0, 3, 6, 12, 24 and 48 h post infected conditions were collected for qPCR. Expression of a TLR-2, b IL-1β, c NF-κB2, d NF-κBI, e CFD, f C1qb, g CPT1 genes relative to EF-1α was estimated. Values represented as Mean ± SEM (n = 5). Significance was determined by T-TEST (*P < 0.05,**P < 0.01,***P < 0.001)
Discussion
Tilapia (Oreochromis niloticus) is a staple product of the aquaculture industry, with annual consumption exceeding 3.7 million metric tons, as of 2014 [17]. Presently tilapia are grown in fresh-water pond culture systems in approximately 125 countries [18]. Breeding programs were used to develop improved versions of tilapia with high biomass [18]. Availability of whole genome sequence and RNA sequence data in recent years has allowed a greater understanding of the genetic makeup and expression profiles of different strains or groups of tilapia [19]. In fresh-water and brackish-water cultures, tilapia is prone to infection with the aquatic bacterial pathogen, V. vulnificus, which severely threatens the tilapia production [4, 18, 20, 21]. V. vulnificus is a halophytic Gram-negative bacillus-type bacterium that can cause skin lesions, soft tissue dysfunction, and sepsis-induced mortality in tilapia or people who consume raw fish containing this pathogen [22, 23]. The V. vulnificus strain 93 U204 has been isolated from an infected tilapia fish and its genome was previously sequenced [3].
Regulation of gene expression plays a major role in an organisms defense against pathogens [16]. Sequencing-by-synthesis on an Illumina RNA-sequence platform has become a widely applied method for comparative transcriptome analysis [24, 25]. The primary sequence-by-synthesis data is contained in a multiplexed and mixed form with sequence information of all the groups in a Bcl file [26]. The complex Bcl form has to be converted into a readable form, such as FASTQ format prior to further analysis [14]. The comprehensive computation-based resource, Gene ontology (GO), is extensively used to analyze the large amounts of FASTQ converted reads obtained from transcriptome analysis [27, 28]. Hence, we analyzed our transcriptome data with the KEGG pathway tool to search for major pathways altered in D56 transgenic tilapia compared to wild type. The major enriched genes in identified KEGG pathways and major genes or gene groups from GO analysis were measured by real-time PCR to confirm the regulation of gene expression by D56 transgenic tilapia fish compared to wild types.
Several reports have shown that resistance to infection can be enhanced in tilapia. AMPs, such as TP3 and TP4 have been reported to decrease the bacterial counts of V. vulnificus [29, 30]. In addition, the multifunctional growth factor, PGRN, has been reported to modulate the immune response and improve survival of zebrafish infected with V. vulnificus [20]. Similarly, the granulin peptide, GRN-41, has been reported to exert anti-bacterial function against V. vulnificus [31]. When tilapia are fed with Epinecidin (Epi)-1-expressing transgenic Artemia, the mortality rate caused by V. vulnificus infection is decreased [32]. These studies demonstrate the utility of AMPs in controlling V. vulnificus infection.
D56-transgenic zebrafish and tilapia exhibit resistance to V. vulnificus infection [5, 15]. The genes expressed in D56-transgenic fish, Atlantic salmon Fadsd5 and Fadsd6, play an important role in n-3 PUFA biosynthesis. Interestingly, exogenous FAs assimilated into the vibrio species affect the swimming motility, bacterial membrane structure, permeability, and virulence [33].
A comparative analysis of liver transcriptomes of wild-type and D56-transgenic tilapia infected with V. vulnificus was performed in this study. The multiplexed data of the six groups in the Bcl file were quality controlled, and sequence data for each group was multiplexed into FASTQ format. From the short reads, 88.48–92.19% were mapped to the RNA genome in all samples. Thus, nearly 90% of the transcriptome was mapped for the comparative analysis. The KEGG pathway analysis showed several FA-associated pathways in wild-type and D56-transgenic tilapia groups at 0 hpi with V. vulnificus (Table 2). KEGG analysis also revealed that the drug metabolism pathway, amino acid pathways and Cytochrome 450 pathway were altered by D56 (Table 2). These results showed that the expression of D56 enhances FA biosynthesis, especially biosynthesis of unsaturated fatty acids, and it may also lead to the modulation of additional pathways. Several such pathways were also differentially activated between wild-type and D56-transgenic fish group 6 h and 24 h post infected with V. vulnificus (Table 3, 4). These data demonstrated the D56-mediated FA pathway may control several amino acid biosynthesis mechanisms in addition to stress response mechanisms related to pathogen resistance (Table 2, 3, 4).
The GO analysis showed that genes associated with inflammation, chemokine synthesis, iron homeostasis and immune response are altered by D56 expression in tilapia (Table 5). The iron binding genes are related to the AMP hepcidin [34,35,36]. Hence, it is possible that tilapia hepcidins (TH), such as TH1–5 and TH2–3, may be functionally affected by D56 expression. Previously, the anti-V. vulnificus activity of TH2–3 has been reported [37]. Thus, further investigations may explore the possibility that D56 regulates tilapia hepcidins. In addition, several AMPs that are secreted by tilapia fish are associated with innate immunity and immune modulatory functions. Hence, the FA-associated D56-transgene expression may also directly alter the level of AMPs in tilapia fish. To address this possibility, the expression AMP-associated genes in tilapia was studied by qPCR (Fig. 4). TH and BP1 were upregulated at 24 h and 48 hpi with V. vulnificus in D56-transgenic tilapia, suggesting the activation of these peptides was promoted by D56 to combat V. vulnificus infection (Fig. 4). The complement genes, C1qb, CFHR1 and CFD were also upregulated in D56-transgenic tilapia after V. vulnificus infection (Fig. 3). It is also possible that the plasma anti-bacterial function is stimulated by D56 expression in transgenic tilapia [38, 39]. The inflammation-associated gene expression included early upregulation of TLR-2 and TNF-α in D56-transgenic fish (Fig. 5). The expression level of NF-κB did not vary, however, it is possible that the translocation of NF-κB from cytoplasm to nucleus triggered the activation of downstream genes, such as TNF-α [40]. The CD8-related Peroxiredoxin (PRDX)1, G-protein receptor associated Atypical Chemokine Receptor (ACKR)4, Tissue inhibitor of Metalloprotease (TIMP)2 associated with extracellular matrix were also altered by the presence of D56 (Fig. 6). Probing the immunomodulatory gene expression in whole blood revealed that genes such as NF-κB2, TLR-5, IL-1β, CFD and C1qb were upregulated by D56 after V. vulnificus infection (Fig. 7). Altogether, the FA-associated pathways triggered in the D56-transgenic tilapia appear to regulate a variety of immune-related genes that may serve to enhance resistance to V. vulnificus infection.
Conclusions
We compare the results between NGS and qPCR (Supplementary Table S2). We found that TLR-2, TLR-5 and ACKR4 were induced in the D56-transgenic line. TLR-5 and TLR-2 may stimulate a pro-inflammatory response, and ACKR4 blocks chemokine signaling. Through the regulation of CFHR1 and CFD, n-3 PUFAs can modulate the complement system. Moreover, AMP-associated genes, including TP3, TP4, TP5, TH, PGRN, BPI and LEAP2, may possibly be regulated by n-3 PUFAs (Fig. 8a). In terms of downstream effects, HNF4A and PPARs may induce CPT1 and PCK1. This action would enhance ATP production from FA oxidation and help the host overcome infection by pathogenic bacteria (Fig. 8b). Furthermore, NF-κB downstream genes, such as TNF-α and IL-1β, may suppress pathogens (Fig. 8c). In this study, we measured the transcriptome-wide responses of wild-type and D56-transgenic tilapia to V. vulnificus infection. Several pathways associated with inflammation, immune response, chemokine and cytochrome were specifically altered in D56-transgenic tilapia, as revealed by KEGG pathway and GO analysis of the wild-type and D56-transgenic tilapia at the baseline (0 hpi) and after infection (6 and 24 hpi). These results suggest that D56 may modulate pro-inflammatory cytokines, AMPs and immune response genes to enhance resistance to V. vulnificus (Fig. 8).

Proposed mechanism of Delta 5 and Delta 6 transgenic (D56) mediated ω-3 fatty acid metabolism to enhance resistance against V. vulnificus in Tilapia. a The ω-3 fatty acids synthesized in D56 transgenic fish regulates or activates the extra cellular acting agents such as Antimicrobial peptide (AMP), Complement system and ACKR4 by altering their expression. b In cytosol D56 mediated ω-3 fatty acid regulates HNF4A, TLR-5 and TLR-2, PPARs, TIMP2 and PRDX1. c In nucleus transcription factors such as NF-κB is altered to regulate the proinflammatory cytokines. (Image from ourselves)
Methods
Tilapia fish and culture
Tilapia fish Oreochromis niloticus were acclimatized in FRP tanks with 2000 l capacity under the controlled conditions of 28 °C with a 13 h light/ 11 h dark cycle with Alanine rich food at 4% body weight per day for 45 days. Wild-type and transgenic tilapia fish with dual expression of delta-6 desaturase in liver and delta-5 desaturase in muscle (D56) were gift from Dr. Jen-Leih Wu’s lab from Institute of Cellular and Organismic Biology, Academia Sinica, Taipei, Taiwan [15]. After 45 days the wild-type tilapias reached an average of 8.93 ± 0.98 cm length and 11.52 ± 3.55 g weight. The transgenic tilapias were 10.06 ± 1.02 cm in length and 16.25 ± 3.61 g weight. The experimental fish were handled after getting approval from the Institutional Animal Care and Use Committee (IACUC) at Academia Sinica, Taiwan, and the IACUC guidelines were followed. According to the formula: n = (Z_1-beta * sqrt(p1q1 + p2q2) + Z_1-alpha/2 * sqrt(2 * p_avg * q_avg))^2 / delta^2, p2 = p1 + delta, p_avg = (p1 + p2) / 2, q_avg = 1 - p_avg. The minimum number of samples in each group was 5 (K = 2, α = 0.05, 1-β = 1–0.2 = 0.8, p1 = 37% and Δ = 74%). A total of 66 animals were used in this study (5 samples * 2 groups * 6 time-points = 60 for qPCR analysis; 6 animals were used for transcriptome analysis). The method for euthanasia was rapid chilling by soaking the animal in ice for 10 min. The ratio of ice to water was roughly 5:1. Ice and water were both measured volumetrically for a total volume of roughly 10 L [41].
Expression of D56 in transgenic Tilapia
The vector construct and expression of D56 has been explained previously [15]. In brief the delta-6 desaturase transgenic line driven by muscle-specific CKMb promoter express the Fadsd6 with TcFP11 reporter was created by co-injecting the plasmid into the embryo. Similarly delta-5 desaturase transgenic line driven by liver-specific Fabp10 promoter to express the Fadsd5 with TcFP13 reporter was created separately. The delta 5 and delta 6 transgenic tilapia have been crossed and the offspring carrying D56 has been selected for the experiment. Atlantic salmon (Salmon salar) delta-6 desaturase gene Fadsd6 and delta-5 desaturase gene Fadsd5 were used in the construct.
V. vulnificus culture and infection in to tilapia
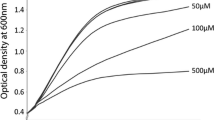
The pathogen V. vulnificus strain 93 U204 was initially cultured in a TCSB (Thiosulfate Citrate Bile Salts Sucrose, BD Difco™) agar plate for 16 h at 28 °C [3]. From overnight cultured plate a single colony was picked and cultured in 3 ml TSB (Tryptone Soy Broth, BD Difco™) with 1.5% NaCl in an incubated shaker at 28 °C with 200 rpm up to four hours until the OD600mm reached 0.7 to 0.9. At the end of acclimatization the wild-type and transgenic fish were subjected to V. vulnificus challenge by i.p. injecting 50 μl of diluted TSB containing 1.9 × 102 CFU of V. vulnificus for each fish. Thirty fishes in each group was infected with V. vulnificus and five fish per each group was sacrificed at 0, 3, 6, 12, 24 and 48 h post infection for molecular analysis.
Extraction of total RNA
The fish was sacrificed at the end of each time point of the experiment and liver tissue were collected for total RNA extraction. The liver tissue was homogenized and total RNA was extracted using Trizol® Reagent by following the company protocol (Invitrogen, USA). Purified RNA was quantified at OD260nm using a ND-1000 spectrophotometer (NanodropTechnology, USA) and qualitated by using a Bioanalyzer 2100 (Agilent Technology, USA) with RNA 6000 LabChip kit (Agilent Technology, USA).
RNA library construction and transcriptome analysis
RNA extracted from three fish in each group was used for the RNA library construction. Illumina’sTruSeq Stranded Total RNA Library Prep Gold Kit (Cat. 20,020,598) was used for library construction. AMPure XP beads (Beckman Coulter, USA) kit was used for size selection. The sequence was determined using Illumina’s sequencing-by-synthesis (SBS) technology (Illumina, USA) (150 bp, pair-end, 6Gb). Bcl2fastq v2.20 (Illumina, USA) software was used to trim adapter sequence, eliminating Unique Molecular Identifier (UMI) sequences and conversion of BCL files to the per-read FASTQ format. Both adaptor clipping and sequence quality trimming were performed using Trimmomatic v0.36 with a sliding-window approach. After FASTQC, raw reads were aligned to the downloaded tilapia reference genome using HISAT2 software. Differential expression analysis was performed using cuffdiff (cufflinks v2.2.1) with genome bias detection/correction and Welgene Biotech’s in-house pipeline (Supplementary Figure S1). Functional enrichment assay in differentially expressed genes of each experiment design was performed using clusterProfiler v3.6. Genes with low expression level (< 0.3 FPKM value) in either or both of the treated and control samples were excluded (Supplementary File S1). Genes with p value ≤0.05 and ≥ 2-fold changes were considered significantly differentially expressed.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) analysis
GO enrichment analysis of the differentially expressed genes (DEGs) was implemented using the clusterProfiler package. Pathway enrichment was used for KEGG analysis (https://www.genome.jp/kegg/), which uses public databases to examine Oreochromis niloticus biological pathways. The datasets generated and analyzed during the current study are available in the NCBI repository, https://www.ncbi.nlm.nih.gov/bioproject/705417 (Accession: PRJNA705417 ID: 705417) (Accession: PRJNA705417 ID: 705417).
Gene expression analysis with qPCR
cDNA was obtained from 0.5 μg of total RNA using ReverTra Ace® qPCR RT Master Mix with gDNA Remover kit (TOYOBO, Japan). qPCR was performed using Applied BiosystemsStepOnePlus™ System (ABI, USA) machine. The qPCR reaction mix comprise of 5 μl SYBR® Green Realtime PCR Master Mix (TOYOBO, Japan), 0.5 μl (10 μM) each of gene specific primers (Supplementary Table S1), and 4 μl of twenty times diluted cDNA. Amplification was performed with an initial 95 °C for 1 min and 40 cycles of 95 °C for 15 s, 60 °C for 15 s and 72 °C for 45 s (Supplementary Figure S2). Gene expression relative to EF-1α was estimated by 2T-△△C method as described [42].
Statistical analysis
Data are presented as the mean ± standard error of mean (SEM). Statistical analysis was performed using Student’s t-TEST (*P < 0.05, **P < 0.01, ***P < 0.001).
Availability of data and materials
The datasets generated and analyzed during the current study are available in the NCBI repository, https://www.ncbi.nlm.nih.gov/bioproject/705417 (Accession: PRJNA705417 ID: 705417).
Abbreviations
- ACKR4:
-
Atypical chemokine receptor 4
- ApoA4b:
-
Apolipoprotein A-IV-b
- BPI:
-
Bactericidal permeability-increasing protein
- CFD:
-
Complement factor D
- CFHR1:
-
Complement factor H-related protein 1
- CPT1:
-
Carnitine O-Palmitoyltransferase 1
- HNF4A:
-
Hepatocyte nuclear factor 4, Alpha
- IL-1β:
-
Interleukin 1 beta
- LEAP2:
-
Liver-expressed antimicrobial peptide 2
- NFκB2:
-
Nuclear factor kappa B subunit 2
- NFκBI:
-
NF-kappa-B inhibitor alpha
- TH:
-
Tilapia hepcidin
- TIMP2:
-
Tissue inhibitor of metalloproteinases 2
- TLR-2:
-
Toll- like receptor 2
- TLR-5:
-
Toll- like receptor 5
- TP:
-
Tilapia piscidin
- TNF-α:
-
Tumor necrosis factor alpha
- PCK1:
-
Phosphoenolpyruvate carboxykinase 1
- PGRN:
-
Progranulin
- PPARα:
-
Peroxisome proliferator activated receptor alpha
- PRDX1:
-
Peroxiredoxin-1
References
Hernandez-Cabanyero C, Sanjuan E, Fouz B, Pajuelo D, Vallejos-Vidal E, Reyes-Lopez FE, Amaro C. The effect of the environmental temperature on the adaptation to host in the zoonotic pathogen Vibrio vulnificus. Front Microbiol. 2020;11:489. https://doi.org/10.3389/fmicb.2020.00489.
Hernandez-Cabanyero C, Lee CT, Tolosa-Enguis V, Sanjuan E, Pajuelo D, Reyes-Lopez F, Tort L, Amaro C. Adaptation to host in Vibrio vulnificus, a zoonotic pathogen that causes septicemia in fish and humans. Environ Microbiol. 2019;21(8):3118–39. https://doi.org/10.1111/1462-2920.14714.
Lo WS, Chen H, Chen CY, Kuo CH. Complete Genome Sequence of Vibrio vulnificus 93U204, a Bacterium Isolated from Diseased Tilapia in Taiwan. Genome Announc. 2014;2(5):e01005-14.
Mahmud ZH, Wright AC, Mandal SC, Dai J, Jones MK, Hasan M, Rashid MH, Islam MS, Johnson JA, Gulig PA, Morris JG Jr, Ali A. Genetic characterization of Vibrio vulnificus strains from tilapia aquaculture in Bangladesh. Appl Environ Microbiol. 2010;76(14):4890–5. https://doi.org/10.1128/AEM.00636-10.
Cheng CL, Huang SJ, Wu CL, Gong HY, Ken CF, Hu SY, Wu JL. Transgenic expression of omega-3 PUFA synthesis genes improves zebrafish survival during Vibrio vulnificus infection. J Biomed Sci. 2015;22(1):103. https://doi.org/10.1186/s12929-015-0208-1.
White PJ, Mitchell PL, Schwab M, Trottier J, Kang JX, Barbier O, Marette A. Transgenic omega-3 PUFA enrichment alters morphology and gene expression profile in adipose tissue of obese mice: potential role for protectins. Metabolism. 2015;64(6):666–76. https://doi.org/10.1016/j.metabol.2015.01.017.
McLennan PL, Abeywardena MY, Charnock JS. A comparison of the long-term effects of n-3 and n-6 polyunsaturated fatty acid dietary supplements and the action of indomethacin upon the mechanical performance and susceptibility of the rat heart to dysrhythmia. Prostaglandins Leukot Med. 1987;27(2–3):183–95. https://doi.org/10.1016/0262-1746(87)90070-9.
Hardardottir I, Kinsella JE. Increasing the dietary (n-3) to (n-6) polyunsaturated fatty acid ratio increases tumor necrosis factor production by murine resident peritoneal macrophages without an effect on elicited peritoneal macrophages. J Nutr. 1992;122(10):1942–51. https://doi.org/10.1093/jn/122.10.1942.
Apour CS, Bell SJ, Forse RA. Immunologic effects of national cholesterol education panel step-2 diets with and without fish-derived N-3 fatty acid enrichment. JPEN J Parenter Enteral Nutr. 1994;18(4):381–3. https://doi.org/10.1177/014860719401800423.
Meydani SN. Effect of (n-3) polyunsaturated fatty acids on cytokine production and their biologic function. Nutrition. 1996;12(1 Suppl):S8–14.
Whelan J, Gowdy KM, Shaikh SR. N-3 polyunsaturated fatty acids modulate B cell activity in pre-clinical models: implications for the immune response to infections. Eur J Pharmacol. 2016;785:10–7. https://doi.org/10.1016/j.ejphar.2015.03.100.
Calder PC. N-3 polyunsaturated fatty acids and immune cell function. Adv Enzym Regul. 1997;37:197–237. https://doi.org/10.1016/S0065-2571(96)00004-0.
Nayak S, Al Ashhab A, Zilberg D, Khozin-Goldberg I. Dietary Supplementation with Omega-6 LC-PUFA-Rich Microalgae Regulates Mucosal Immune Response and Promotes Microbial Diversity in the Zebrafish Gut. Biology (Basel). 2020;9(6):119. https://doi.org/10.3390/biology9060119.
Wang YD, Peng KC, Wu JL, Chen JY. Transgenic expression of salmon delta-5 and delta-6 desaturase in zebrafish muscle inhibits the growth of Vibrio alginolyticus and affects fish immunomodulatory activity. Fish Shellfish Immunol. 2014;39(2):223–30. https://doi.org/10.1016/j.fsi.2014.04.021.
Chiang KY, Lin WC, Tsai TY, Lin CW, Huang SJ, Huang CY, Wu SH, Ken CF, Gong HY, Chen JY, Wu JL. Dual expression of transgenic delta-5 and delta-6 desaturase in tilapia alters gut microbiota and enhances resistance to Vibrio vulnificus infection. PLoS One. 2020;15(7):e0236601. https://doi.org/10.1371/journal.pone.0236601.
Heimes A, Brodhagen J, Weikard R, Seyfert HM, Becker D, Meyerholz MM, Petzl W, Zerbe H, Hoedemaker M, Rohmeier L, Schuberth HJ, Schmicke M, Engelmann S, Kühn C. Hepatic Transcriptome analysis identifies divergent pathogen-specific targeting-strategies to modulate the innate immune system in response to Intramammary infection. Front Immunol. 2020;11:715. https://doi.org/10.3389/fimmu.2020.00715.
Salger SA, Reza J, Deck CA, Wahab MA, Baltzegar DA, Murr AT, Borski RJ. Enhanced biodiversity of gut flora and feed efficiency in pond cultured tilapia under reduced frequency feeding strategies. Plos One. 2020;15(7):e0236100. https://doi.org/10.1371/journal.pone.0236100.
Yanez JM, Joshi R, Yoshida GM. Genomics to accelerate genetic improvement in tilapia. Anim Genet. 2020;51(5):658-74. https://doi.org/10.1111/age.12989.
Cadiz MI, Lopez ME, Diaz-Dominguez D, Caceres G, Yoshida GM, Gomez-Uchida D, Yanez JM. Whole genome re-sequencing reveals recent signatures of selection in three strains of farmed Nile tilapia (Oreochromis niloticus). Sci Rep. 2020;10(1):11514. https://doi.org/10.1038/s41598-020-68064-5.
Wu SH, Lin HJ, Lin WF, Wu JL, Gong HY. A potent tilapia secreted granulin peptide enhances the survival of transgenic zebrafish infected by Vibrio vulnificus via modulation of innate immunity. Fish Shellfish Immunol. 2018;75:74–90. https://doi.org/10.1016/j.fsi.2018.01.044.
Vinh DC, Mubareka S, Fatoye B, Plourde P, Orr P. Vibrio vulnificus septicemia after handling Tilapia species fish: a Canadian case report and review. Can J Infect Dis Med Microbiol. 2006;17(2):129–32. https://doi.org/10.1155/2006/164681.
Horseman MA, Surani S. A comprehensive review of Vibrio vulnificus: an important cause of severe sepsis and skin and soft-tissue infection. Int J Infect Dis. 2011;15(3):e157–66. https://doi.org/10.1016/j.ijid.2010.11.003.
Kim BS, Gavin HE, Satchell KJF. Variable Virulence of Biotype 3 Vibrio vulnificus due to MARTX Toxin Effector Domain Composition. mSphere. 2017;2(4):e00272-17. https://doi.org/10.1128/mSphereDirect.00272-17.
Wu Y, Xu J, Han X, Qiao G, Yang K, Wen Z, Wen X. Comparative Transcriptome Analysis Combining SMRT- and Illumina-Based RNA-Seq Identifies Potential Candidate Genes Involved in Betalain Biosynthesis in Pitaya Fruit. Int J Mol Sci. 2020;21(9):3288. https://doi.org/10.3390/ijms21093288.
Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics. 2015;13(5):278–89. https://doi.org/10.1016/j.gpb.2015.08.002.
Lindner MS, Strauch B, Schulze JM, Tausch SH, Dabrowski PW, Nitsche A, Renard BY. HiLive: real-time mapping of illumina reads while sequencing. Bioinformatics. 2017;33(6):917–319. https://doi.org/10.1093/bioinformatics/btw659.
The Gene Ontology C. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45(D1):D331–8.
O’Halloran DM. fastQ_brew: module for analysis, preprocessing, and reformatting of FASTQ sequence data. BMC Res Notes. 2017;10(1):275.
Lin WC, Chang HY, Chen JY. Electrotransfer of the tilapia piscidin 3 and tilapia piscidin 4 genes into skeletal muscle enhances the antibacterial and immunomodulatory functions of Oreochromis niloticus. Fish Shellfish Immunol. 2016;50:200–9. https://doi.org/10.1016/j.fsi.2016.01.034.
Pan CY, Tsai TY, Su BC, Hui CF, Chen JY. Study of the Antimicrobial Activity of Tilapia Piscidin 3 (TP3) and TP4 and Their Effects on Immune Functions in Hybrid Tilapia (Oreochromis spp.). Plos One. 2017;12(1):e0169678.
Wu SH, Chou HY, Liu PC, Wu JL, Gong HY. Granulin peptide GRN-41 of Mozambique tilapia is a novel antimicrobial peptide against Vibrio species. Biochem Biophys Res Commun. 2019;515(4):706–11. https://doi.org/10.1016/j.bbrc.2019.06.022.
Ting CH, Chen YC, Chen JY. Nile tilapia fry fed on antimicrobial peptide Epinecidin-1-expressing Artemia cyst exhibit enhanced immunity against acute bacterial infection. Fish Shellfish Immunol. 2018;81:37–48. https://doi.org/10.1016/j.fsi.2018.07.008.
Moravec AR, Siv AW, Hobby CR, Lindsay EN, Norbash LV, Shults DJ, Symes SJK, Giles DK. Exogenous Polyunsaturated Fatty Acids Impact Membrane Remodeling and Affect Virulence Phenotypes among Pathogenic Vibrio Species. Appl Environ Microbiol. 2017;83(22):e01415-17. https://doi.org/10.1128/AEM.01415-17.
Dhindsa S, Ghanim H, Batra M, Kuhadiya ND, Abuaysheh S, Green K, Makdissi A, Chaudhuri A, Dandona P. Effect of testosterone on hepcidin, ferroportin, ferritin and iron binding capacity in patients with hypogonadotropic hypogonadism and type 2 diabetes. Clin Endocrinol. 2016;85(5):772–80. https://doi.org/10.1111/cen.13130.
Altamura S, Kessler R, Grone HJ, Gretz N, Hentze MW, Galy B, Muckenthaler MU. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014;20(2):359–67. https://doi.org/10.1016/j.cmet.2014.07.007.
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–3. https://doi.org/10.1126/science.1104742.
Hsieh JC, Pan CY, Chen JY. Tilapia hepcidin (TH)2-3 as a transgene in transgenic fish enhances resistance to Vibrio vulnificus infection and causes variations in immune-related genes after infection by different bacterial species. Fish Shellfish Immunol. 2010;29(3):430–9. https://doi.org/10.1016/j.fsi.2010.05.001.
Hannan JP, Laskowski J, Thurman JM, Hageman GS, Holers VM. Mapping the complement factor H-related protein 1 (CFHR1):C3b/C3d interactions. Plos One. 2016;11(11):e0166200. https://doi.org/10.1371/journal.pone.0166200.
Abarrategui-Garrido C, Martinez-Barricarte R, Lopez-Trascasa M, de Cordoba SR, Sanchez-Corral P. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood. 2009;114(19):4261–71. https://doi.org/10.1182/blood-2009-05-223834.
Sode J, Bank S, Vogel U, Andersen PS, Sorensen SB, Bojesen AB, Andersen MR, Brandslund I, Dessau RB, Hoffmann HJ, et al. Genetically determined high activities of the TNF-alpha, IL23/IL17, and NFkB pathways were associated with increased risk of ankylosing spondylitis. BMC Med Genet. 2018;19(1):165. https://doi.org/10.1186/s12881-018-0680-z.
Thurman CE, Rasmussen S, Prestia KA. Effect of 3 euthanasia methods on serum yield and serum cortisol concentration in Zebrafish (Danio rerio). J Am Assoc Lab Anim Sci. 2019;58(6):823–8. https://doi.org/10.30802/AALAS-JAALAS-18-000144.
Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2^(−delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 2013;3(3):71–85.
Acknowledgements
We would like to thank Dr. Marcus J. Calkins (ICOB, Academia Sinica) for English language editing. We are grateful thank the aquaculture groups of the Marine Research Station, Institute of Cellular and Organismic Biology, Academia Sinica for their help with the tilapia maintenance.
Funding
Research funding for this study was partly from the academic committee members of Innovative Translational Agricultural Research Program (AS-109-ITAR-L081) (Academia Sinica) to Dr. Jyh-Yih Chen. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This work was financially supported by the iEGG and Animal Biotechnology Center from the Feature Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE-109-S-0023-A) in Taiwan. This work was partly financially supported by the 109–2313-B-001-007-MY3, 109–2622-B-001-002-CC1, or 108–2313-B-001-006—from the Ministry of Science and Technology (Taiwan). The funders played no part in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
P.Y.T. performed experiment and analyzed the data. P.Y.T., S.J.H., J.L.W. and J.Y.C. designed the study. P.Y.T., V. R. and J.Y.C. participated in drafting and revision of the manuscript. All authors have read and approved the final manuscript, and ensure that this is the case.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study received approval from the institutional review board of Academia Sinica.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing financial interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Figure S1.
Analysis of sample correlation.
Additional file 2: Supplementary Figure S2.
Melting curve analysis of target genes.
Additional file 3: Supplementary Table S1.
List of primers used in qPCR.
Additional file 4: Supplementary Table S2.
Comparison of RNA-seq and qRT-PCR.
Additional file 5: Supplementary File S1.
DEG between D56 and WT.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tu, PY., Huang, SJ., Rajanbabu, V. et al. Comparative transcriptome analysis reveals ectopic delta-5 and delta-6 desaturases enhance protective gene expression upon Vibrio vulnificus challenge in Tilapia (Oreochromis niloticus). BMC Genomics 22, 200 (2021). https://doi.org/10.1186/s12864-021-07521-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07521-5