
Abstract
Background
More accurate and complete reference genomes have improved understanding of gene function, biology, and evolutionary mechanisms. Hybrid genome assembly approaches leverage benefits of both long, relatively error-prone reads from third-generation sequencing technologies and short, accurate reads from second-generation sequencing technologies, to produce more accurate and contiguous de novo genome assemblies in comparison to using either technology independently. In this study, we present a novel hybrid assembly pipeline that allowed for both mitogenome de novo assembly and telomere length de novo assembly of all 7 chromosomes of the model entomopathogenic fungus, Metarhizium brunneum.
Results
The improved assembly allowed for better ab initio gene prediction and a more BUSCO complete proteome set has been generated in comparison to the eight current NCBI reference Metarhizium spp. genomes. Remarkably, we note that including the mitogenome in ab initio gene prediction training improved overall gene prediction. The assembly was further validated by comparing contig assembly agreement across various assemblers, assessing the assembly performance of each tool. Genomic synteny and orthologous protein clusters were compared between Metarhizium brunneum and three other Hypocreales species with complete genomes, identifying core proteins, and listing orthologous protein clusters shared uniquely between the two entomopathogenic fungal species, so as to further facilitate the understanding of molecular mechanisms underpinning fungal-insect pathogenesis.
Conclusions
The novel assembly pipeline may be used for other haploid fungal species, facilitating the need to produce high-quality reference fungal genomes, leading to better understanding of fungal genomic evolution, chromosome structuring and gene regulation.
Similar content being viewed by others


Background
The production of more complete and accurate genome assemblies has further improved understanding of gene function, biology, and evolutionary mechanisms [1]. High quality, accurate genome assemblies are essential for efficient genome mining, allowing for the identification of useful genes and gene clusters that drive advances in downstream applications such as metabolic engineering, synthetic biology, biotechnology-based drug development, and protein engineering [2]. The advent of second-generation sequencing technologies, such as Illumina’s sequencing by synthesis approach [3], and third generation sequencing technologies, such as Oxford Nanopore [4, 5] and Pacific Biosystems single molecule sequencing platforms [6], have reduced the cost and time of genome assembly projects in comparison to first generation Sanger (dideoxy-chain termination) sequencing [7] methods. The current state-of-the-art genome assembly approach, termed hybrid assembly, leverages benefits of both long, relatively error-prone reads from third-generation sequencing technologies, and short, accurate reads from second-generation sequencing technologies to produce more accurate and contiguous de novo genome assemblies than could be achieved using either technology independently [8]. More contiguous assemblies hold richer information about repetitive regions and chromosome structure, allowing better inferences to be made about macro-molecular genomic variations that lead to adaptation and speciation [9, 10]. Furthermore, it has been demonstrated that gene content can vary significantly between genome assemblies of differing quality made from the same read set, presumably due to the availability of new gene evidence for ab initio prediction algorithms, genome mis-assembly events and local sequence variations [11].
Fungi within the genus Metarhizium (Division: Ascomycota, Class: Sordariomycetes, Order: Hypocreales, Family: Clavicipitaceae) have a worldwide distribution. Besides being applied as biological control agents for pest control [12], species within the genus are frequently used as model organisms to investigate infection processes and host defence mechanisms of various arthropod hosts [13]. Research is also focused on their symbiotic relationship with plants, as they have been shown to improve plant growth and health through poorly understood mechanisms [14]. Additionally, some isolates of Metarhizium are capable of producing bioactive metabolites such as Swainsonine and Destruxins, compounds that have been explored as potential pharmaceuticals to treat cancer, osteoporosis, Alzheimer’s disease, and hepatitis B [15]. Given these interesting properties, there are currently only 8 species of Metarhizium with genomes deposited within GenBank, despite at least 50 species having been described within the genus. Different isolates (variants) of the same species have been found to vary greatly in their phenotypes [16], but due to the relatively small number of isolates sequenced, the extent of genomic variation between strains is poorly understood. Owing to their genomes having multiple chromosomes that contribute to their relatively large genome sizes (30–45 Mb) in comparison to bacterial microbes (around 5 Mb), de novo genome assemblies of Metarhizium spp. using first generation sequencing is very costly, and second-generation sequencing results in assemblies that are highly contiguous, falling apart around repeat rich and homologous regions of the genome. The assembled reference genomes of all 8 species currently accessible in GenBank were produced using reads from second generation sequencing technology, with some of the assemblies making use of optical mapping data to further improve assembly quality [17,18,19,20,21,22]. It is speculated that chromosome duplications and rearrangements are responsible for the differing phenotypic attributes of Metarhizium spp. strains [23], but as of yet, none of the Metarhizium genome assemblies have produced contigs or scaffolds that are chromosome length, a requirement for meaningful chromosomal macro-synteny comparisons between different strains and/or species. Karyotyping experiments carried out using pulse-field gel electrophoresis suggest the presence of 7–8 chromosomes in Metarhizium anisopliae (MAN), with chromosomes varying in size from an estimated 1.8 to 7.4 megabase pairs [23, 24]. A separate study provided evidence showing the smallest chromosome to be disposable in a strain of M. brunneum (strain V275 formerly classified as M. anisopliae) without having lethal effects [25].
In this study, we present a novel hybrid de novo assembly pipeline, incorporating Illumina and Nanopore sequencing reads, that allowed for telomere length assemblies of all 7 chromosomes of M. brunneum isolate ARSEF 4556, as well as the generation of the full circular mitochondrial genome. We benchmark this assembly against the current NCBI reference Metarhizium spp. genomes, providing evidence that the assembly is superior in terms of both standard assembly metrics, as well as gene content as determined by BUSCO scoring. Furthermore, we validate this assembly by comparing it against assemblies produced by various long read assemblers using the same read set, assessing fungal genome assembly performance. We perform genomic synteny and orthologous protein cluster comparisons of this assembly with three other complete genome assemblies of species within the Order Hypocreales, listing orthologous protein clusters shared uniquely between two of the entomopathogenic species, as well as compiling a list of core orthologous Hypocreales proteins shared across all four species. We present an improved genome sequence for the genus, as well as a hybrid assembly pipeline that could be used for other haploid fungal species, in order to facilitate efforts to produce high-quality genomes, ultimately leading to a better understanding of fungal genomic evolution.
Results
Sequencing
A total of 16,630,587 Illumina reads were produced for each pair-end read set- a theoretical coverage of around 131x of the 38 Mb sized M. brunneum genome. After end trimming, the theoretical coverage of the cumulative number of bases was reduced to around 105x. For the Nanopore sequencing run, a total of 1,839,242 raw long reads were produced. After length filtering, trimming and correction, the > 3000 bp long read dataset contained a total of 777,731 reads (N50 = 7156), containing 5,075,705,440 bases, a theoretical coverage of around 134x. The > 5000 bp long read dataset contained a total of 453,256 reads (N50 = 8530), containing 3,798,611,962 bases, a theoretical coverage of around 100x.
Genome assembly
Attempts to further reduce the number of steps in the assembly pipeline by removing individual correction steps resulted in suboptimal assemblies in comparison to using the full assembly pipeline. A tangled Flye assembly graph was produced from assembly of the FMLRC corrected long reads without the Canu trimming step (see additional file 1.A). The Flye assembly graph of the Canu trimmed long reads without the FMLRC correction step was seen to have smaller contigs, and larger contigs that failed to reach chromosome length (see additional file 1.B). The Flye assembly graph of the > 5000 bp read set with the information used to manually resolve complete chromosomes can be seen in additional file 1.C. Read assemblies of chromosomes 2, 4, 5 and 6 were found to traverse an Eulerian path, were assembled telomere to telomere, and required no further resolving. Read assemblies of chromosomes 3 and 7 were found to traverse an Eulerian path in the Flye assembly of the > 3000 bp read set (with two rounds of polishing). Chromosome 1 was deduced by subtracting chromosome 7 and using coverage depth information to deduce the correct edges between contigs, and the 5231 bp end was manually added to the end as described in the methods section. A dotplot illustrating good synteny observed between the contigs and scaffolds of the previous M. brunneum reference assembly and the 7 full length chromosomes produced in this study is presented in additional file 1.D. Tapestry output of terminal telomere counts, chromosome lengths, and long read mapping agreement can be found in Fig. 1.

Tapestry output of complete chromosomes. Terminal telomere sequence counts (CCCTAA/ TTAGGG) are given above the terminal ends (red). The green lines depict mapped long reads to each chromosome. Read mapping depths were uniform across chromosomes, with no breaks detected, however, a pile up of reads was observed around the 18 s/28 s ribosomal RNA gene cluster in chromosome
Validation of the assembly and comparison of long read assembly performance
The metrics for the various assemblers tested are listed in Table 1. The assemblers generally produced better results with the FMLRC/Canu trimmed reads used as input (as opposed to raw long reads), with the exceptions of Canu (produced a total assembly size that was three times as large as the other assemblers) and Shasta (produced a total assembly length of 104,717 bp). The Raven, Shasta and wtdbg2 assemblies suffered with telomere sequence loss irrespective of whether corrected or raw reads were used as input. The Canu assembly with raw reads produced a fragmented assembly. Necat and Flye produced the best assemblies in terms of N50, production of telomere length contigs, and telomere length presence, and Flye’s metrics were relatively robust irrespective of which corrected reads were used as input. The Flye assembly with the Ratatosk corrected reads contained 1 inter-chromosomal mis-assembly wherein a telomere repeat sequence was found in the central region of a chromosome. Aside from the Canu and Shasta assemblies with corrected reads used as input, the predicted genes and total lengths of the assemblies were moderately consistent. Assembly graphs showing TTAGGGn5 sequences detected in contigs produced by all assemblers, and colour coded blast hits of chromosomes from the final complete assembly from which mis-assemblies were inferred can be found in additional file 2.
Genome annotation
A list of each chromosome’s length, GC content, tRNA genes, rRNA genes and notable genes include; specialist entomopathogenic, endophytic and mating-type genes, are detailed in Table 2. All chromosomes were numbered according to the convention of numbering chromosomes according to size, with chromosome 1 being the largest. All chromosomes were found to be oriented in the direction of the telomere sequence CCCTAA at the 5′ chromosome end and TTAGGG at the 3′ chromosome end, further validating assembly correctness. The tRNAscan-SE tool predicted a total of 124 tRNA genes in the genome assembly and RNAmmer predicted a total of 27 rRNA genes present in the genome assembly. Table 3 lists the assembly metrics, predicted proteins and protein BUSCO scores of all NCBI Reference Metarhizium spp. Genomes, as well as the assembly produced in this study, which was found to have the highest protein BUSCO score of 99.1% (N = 4494). The protein set generated in this study was found to have a total of 4455 complete BUSCOs of which 4441 were found to be complete and single copy, 14 BUSCOs were found to be complete and duplicated, 18 BUSCOs were found to be fragmented and 21 BUSCOs were found to be missing. In contrast, the current M. brunneum NCBI reference protein set was found to have a BUSCO score of 97.0% (N = 4494), and the best Metarhizium spp. protein BUSCO score of the NCBI reference sequences was that of M. robertsii with a score of 98.5% (N = 4494). The BUSCO scores for the four ab initio gene prediction tools used are listed in Table 4. As running a native version of the latest version of GeneMarkES with the mitogenome included proved to be best, it was this gene set that was carried forward for functional analyses. A total of 11,406 genes and 11,405 proteins were predicted using this tool, of which 1251 proteins passed the SignalP5.0 threshold for containing a signal peptide sequences. A summary of the SignalP5.0 results can be found in additional file 3 and a list of the mature proteins that were found to have a signal sequence are presented in additional file 4. Comparisons of the protein sets produced in this study with the NCBI reference protein sets for M. brunneum, M. robertsii and M. anisopliae are illustrated in Fig. 2. The numbers of proteins, orthologous clusters and singletons of all four protein sets are give in Fig. 2a. In comparison to the previous M. brunneum NCBI reference protein set, the protein set generated in this study contained more predicted proteins (11,405 vs 10,689), and contained more orthologous protein clusters (10,775 vs 10,492). A Venn diagram showing the orthologous protein clusters shared between the four protein sets is depicted in Fig. 2b. In comparison to the previous M. brunneum NCBI reference protein set, the protein set generated in this study was found to share more orthologous protein clusters with both M. robertsii (10,186 vs 9948) and M. ansiopliae (9940 vs 9748). The Unicycler assembly produced a circular mtDNA genome of 24,965 base pairs (Fig. 3). Identified genes included; cox1–3, nad1–6 and nad4L, cob, atp6, atp8, atp9, rnl and rps3. A total of 25 tRNA gene sequences were identified within the mitogenome.

Comparison of orthologous gene clusters between Metarhizium protein sets. Comparison of the protein set produced in this study with the NCBI reference protein sets for M.brunneum, M.robertsii and M.anisopliae. a Number of proteins, orthologous clusters and singletons predicted for each assembly. b Venn diagram comparing orthologous protein cluster numbers between the four protein sets

Metarhizium brunneum mitogenome map. Mitochondrial gene families are colour coded as per the legend. The circle inside the inner GC content graph marks the 50% threshold
Full genome sequence-based synteny and pan-genome analyses of Hypocreales fungi
Abundant syntenic blocks were seen to be shared across C. militaris, E. festucae, Trichoderma reesei, and M. brunneum (Fig. 4). There was no discernible pattern in the sharing of these syntenic blocks amongst the chromosomes, with any individual chromosome of one species being found to share syntenic blocks with numerous other chromosomes in the other species. Assembly and annotation metrics of the C. militaris, E. festucae, and Trichoderma reesei genomes are stated in Table 5. A total of 9902, 9284, 8125 genes were predicted for C. militaris, E. festucae, and Trichoderma reesei, respectively. This is in contrast to the 11,406 genes predicted for M. brunneum long read assembly. Furthermore, the M. brunneum assembly produced in this study was found to have the highest protein BUSCO completion score of all four Hypocreales species. The results of comparing orthologous gene clusters between these species are presented in Fig. 5. There were 2449, 1939, 1654, and 943 singleton proteins detected with no ortholog/paralog for M. brunneum, C. militaris, E. festucae, and Trichoderma reesei, respectively. A total core set of 5713 clusters of proteins were found to be shared across all 4 species (see additional file 5). One hundred eighty-three unique orthologous clusters were formed between M. brunneum proteins (see additional file 6). Four hundred sixty-eight unique orthologous clusters were formed between the two entomopathogenic Hypocreales fungi in the comparison test- M. brunneum and C. militaris (see additional file 7). A list of the M. brunneum singleton proteins can be found in additional file 8. Interestingly, this number was the highest number of shared orthologous clusters between two different species in the whole comparison.

Sequence-synteny analyses between Hypocreale species. The Circo plots represent syntenic blocks greater than 1000 bp that were present in all 4 species analysed. Clear mesosynteny was observed between the different species of Hypocreales fungi, with no single chromosome showing major synteny with an individual chromosome in another species. The outer numbers indicate chromosome numbers. M. brunneum chromosomes are shown in green. T. reesei, E. festucae and C. militaris chromosomes are shown in blue

Comparison of orthologous gene clusters between the four Hypocreales fungi protein sets. Comparison of the protein set produced in this study with the chromosome length assemblies of the Hypocreales fungi; Cordyceps militaris, Epichloe festucae and Trichoderma reesei (a.) Number of proteins, orthologous clusters and singletons predicted for each assembly. (b.) Venn diagram comparing orthologous protein cluster numbers between the four protein sets
Discussion
The full genome sequence of M. brunneum has been assembled, producing telomere length sequences for all 7 chromosomes, a full mitogenome, and a more comprehensive protein set as determined by BUSCO analyses and analyses of orthologous protein clusters. The assembly and annotations are an improvement on the current M. brunneum reference assembly produced using optical mapping and mate-pair Illumina reads [18]. The seven assembled chromosomes match the number of total chromosomes predicted by pulsed-field gel electrophoresis [23, 24]. Certain genes were found to be in close proximity, as previously shown. For instance, dtx1 and dtx2 encoding Destruxins 1 and 2 were found in close proximity to dtx3 and dtx4 (which encode Destruxins 3 and 4), with the ORFs for the former being on one DNA strand and the ORFs for the latter being found on the complementary strand as previously described [29]. Furthermore, these genes were correctly placed on chromosome 7 in this assembly (the smallest chromosome), which has been shown to be dispensable, with M. brunneum losing its capacity to produce destruxins when this chromosome is lost [25]. Remarkably, chromosome 7, the smallest chromosome assembled, contained the greatest number of predicted 8 s rRNA genes. The mating-type genes MAT-1-2 and MAT_Switching were detected in full on chromosome 2. None of the MAT-1-1 type genes were detected in this assembly, excepting for a small 162 bp end segment (representing 15% of the full gene) of MAT-1-1-1, corroborating with previous work that has shown individual mating-type genes to be absent in some species of Metarhizium [20].
The circularised mtDNA matched the sequence produced by Sanger sequencing of the closely related Metarhizium anisopliae strain ME1 mtDNA, with 97.41% identity and 97% coverage. The current M. brunneum reference sequence was found to have a mitogenome of 50,066 bp, and both the mitogenome from the hybrid assembly, and the previously sequenced M. anisopliae ME1 mitogenome mapped this 50,066 bp sequence, if duplicated, with near 100% identity, signifying that it is most likely an incorrect concatemer that arose from a mis-assembly event. This further highlights the advantage of adopting hybrid assembly approaches for fungal genome assembly.
The majority of assemblers tested were found to produce assemblies in agreement with the complete genome, and further validate assembly correctness. Flye appears to be the most robust, producing telomere length chromosomes and good assembly N50 values regardless of the read correction strategy used, although the assembly with uncorrected reads produced no telomere length contigs. The other assembler found to produce good results with this fungal genome was NECAT. Raven, Shasta and wtdbg2 all suffered from loss of telomere sequences, a problem that would likely recur for all fungal assemblies. Canu performed better with raw reads, however the N50 value of the assembly was low. The Canu assembler was found to be the most customizable out of the assemblers tested, however, it also had the longest run time. Canu did not perform well when corrected reads were used as input. The Flye assembly using the NECAT corrected reads was the best assembly of the two self-corrected read sets, and this assembly pipeline was found to be best for assemblies with short reads. The results corroborate previous findings by Wick and Holt [30], who compared these assemblers with bacterial genomes. Their results agree with our findings, excepting their ranking of the Raven assembler, which we found to perform poorly with this fungal genome. However, the difference in performance of this assembler may be due to most bacterial genomes being circular. The differences in assemblies that result from differing read correction methods have been observed before by Fu, Wanf and Au [31], who produced an excellent comparative evaluation of long read correction tools.
In terms of cost, the hybrid assembly approach costs as little as €1500. Although this assembly vastly improves on the Illumina read only assemblies, further improvements could be made when conducting hybrid assembly by producing ultra-long nanopore reads [32], particularly for fungal species that contain genomic regions with large sections of tandem repeats. For this assembly, DNA was extracted using a spin column. Longer reads may be obtained by using gravimetric DNA extraction kits, a more traditional phenol-chloroform, or utilizing agarose plug DNA extractions. Given that longer reads are known to have a higher propensity to clog the nanopores, it may be beneficial to produce two sets of nanopore data, an initial run using the relatively shorter fragmented DNA to ensure good coverage, and, when good coverage is reached, perform an additional run with the ultra-long reads. The MinION sequencer is well suited for this task, as read output can be monitored in real-time. As Nanopore sequencing read accuracy continues to improve, through both software and hardware enhancements, it is unknown for how long one may need to produce short-read Illumina sequencing data to polish long read assemblies.
In comparing the whole genomes of 4 Hypocreale species, we confirmed the previous finding of the existence of mesosynteny within the Ascomycota Phylum [33]. No discernible pattern was observed between the syntenic blocks in the comparisons of any two species, with an individual chromosome sharing syntenic blocks with multiple other chromosomes of the other species. A protein list of core orthologous proteins shared across all four Hypocreales species as been compiled. This protein set may prove useful in aiding future research by narrowing the search space for molecular underpinnings of specific phenotypic functions that are unique to a Hypocreales species, as proteins in this list are unlikely to carry out unique functions given that they are shared across all four of these species, and it is know that orthologs are likely to carry out similar functions [34]. Likewise, the lists of orthologous proteins shared uniquely between the entomopathogenic species C. militaris and M. brunneum, the M. brunneum self-clusters and singletons may also aid further research into as of yet unknown molecular underpinnings related to entomopathogenesis. A list has been compiled of mature proteins resulting from removal of theoretical signal peptides, which may aid future research into M. brunneum protein function. The list may assist the recombinant production of proteins in non-fungal species, as well as allow for the production of active mature proteins as oppose to unknowingly cloning protein precursors that may not be functional.
Conclusion
In this study, we present a complete genome assembly with functional annotations, of the entomopathogenic fungi M. brunneum. This is the first Nanopore/Illumina complete de novo hybrid assembly, to our knowledge, of a fungus in the Sordariomycete class. We have demonstrated that a hybrid assembly approach can be used to cheaply produce a better genome assembly, with telomere-to-telomere chromosome assemblies that can allow for chromosomal macrosynteny comparisons between strains and species. The generation of more complete fungal genomes will lead to a better understanding of fungal evolution at a finer resolution, ultimately allowing for better understanding of the genomic underpinnings of phenotypic variation. The methodology may also prove useful for quality control purposes of commercially produced fungal-based products, given the continued decline in cost of whole genome sequencing technologies.
Methods
Insect inoculation and DNA extraction
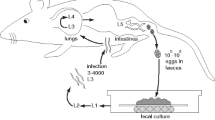
M. brunneum ARSEF strain 4556, obtained from the U.S. Department of Agriculture’s ARS Collection of Entomopathogenic Fungal Cultures (ARSEF), was cultured in SDA medium plates and incubated at 25 °C for 10 days. Sample details were deposited at the NCBI under the BioSample accession: SAMN15394350. Conidia were collected after 10 days by flooding the dish with 20 mL of 0.04% Tween 80 and scraping the surface with a scalpel. The collected conidial suspension was vortexed until complete homogenization and filtered using a sterile nylon membrane. Concentration of conidial suspension was adjusted to 1 × 108 spores mL− 1 using a hemocytometer (Neubauer, Germany). Spore viability was verified and spores were considered to have germinated if they had formed a germ-tube that was as long as spore width.
Larvae of the greater wax moth, Galleria mellonella, were immersed in 10 ml of conidial suspension for 10 s and were placed on moist filter paper in petri dishes in order to encourage sporulation and fungal growth. Controls were included with insects immersed in pure 0.04% Tween 80, in order to ensure that insect death was a result of fungal infection. Plates were incubated in the dark at 25 °C and were inspected daily. After fungal growth was observed, mycelia were collected and grown on SDA media for DNA extraction.
A total of 100 mg of conidia was scraped off the plate under a laminar flow hood, and collected into a sterile 1.5 mL DNA LoBind tube (Eppendorf, Hamburg, Germany). The conidia were ground in the tube with a micro-pestle, and DNA was extracted using the PureLink® Plant Total DNA Purification Kit (Invitrogen, Carlsbad, USA), following the manufacturer’s guidelines. The DNA was checked for purity on a Nanodrop (Thermo Scientific, USA), and DNA concentrations were measured using the Qubit broad range DNA assay kit (Thermo Scientific, USA).
Illumina sequencing
Illumina DNA library preparation and sequencing were outsourced to Eurofins Genomics GmbH, Ebersberg, Germany. Illumina paired-end reads (2 × 150 bp) were produced using the ‘INVIEW Resequencing Sequencing of Fungi 50x Coverage’ package. Illumina reads were trimmed using Trimmomatic version 0.38 [35], setting the HEADCROP configuration to 15 and the CROP configuration to 120. Read qualities were assessed with FastQC [36].
Nanopore sequencing
A total of 1 μg of genomic DNA was used for Nanopore library preparation using a 1D Ligation Sequencing Kit (SQK-LSK109, Oxford Nanopore Technologies). Sequencing was performed on a MinION device (Oxford Nanopore Technologies), equipped with a R9.4.1 MinION flow cell. Base calling was performed offline with ONT’s Guppy software pipeline version 3.4.5, enabling the --pt_scaling flag and setting the --trim_strategy flag to DNA.
Long read filtering and correction
Long read adapter trimming was performed with Porechop version 0.2.4 (www.github.com/rrwick/Porechop), setting the --adapter_threshold to 96, and enabling the --no_split flag. In order to retrieve any circular contig assemblies (e.g. mitochondrial DNA), adapter trimmed long reads and trimmed Illumina paired-end reads were used as input for Unicycler version 0.4.8-beta [37], using the default settings. The trimmed long reads were filtered to remove reads under 3000 bases in length using NanoFilt version 2.6.0 [38], and were subsequently converted from FASTQ to FASTA format using a custom AWK script- [‘BEGIN {P = 1}{if(P==1||P==2){gsub(/^[@]/,” > “);print}; if(P==4) P = 0; P++}’ in.fastq > out.fasta]. The trimmed long reads were corrected using the trimmed Illumina short reads with FMLRC version 1.0.0 [39]. These corrected reads were further trimmed with Canu version 1.9 [40], using the -trim option, setting the genome size to 38 Mb, and disabling the stop on low coverage and stop on low quality features. Two filtered read sets were generated from the Canu output using SeqKit version 0.11.0 [41], one set filtered to contain reads with > 3000 bases and the other to contain reads with > 5000 bases.
Long read assembly
One assembly was carried out per read set using Flye version 2.7 [42] using the --nano-corr flag, setting the genome size to 38 Mb and enabling the --trestle flag. Each of the two assemblies were then used to generate an additional assembly by subjecting each output to a total of two rounds of polishing with Flye (as opposed to the default of one round). Evidence from all assemblies were used to manually resolve tangles. Mapping of reads to a short contig of 5231 bp, which contained the telomere sequence TTAGGG at its terminal end, showed the contig to overlap with an end repeat region of Chromosome 1, and they were combined manually with the aid of CAP3 [43], thus producing, in combination with the manual resolving of tangles, a FASTA file containing all 7 complete chromosomes.
Validation of assembly and comparison of long read assembler performance
In order to validate the final complete assembly and compare long read assembler performance of a fungal genome, assemblies were carried out on both the adapter trimmed long reads (> 3000 bp) and the FMLRC corrected Canu trimmed long read (> 3000 bp) using various assemblers. Assemblers tested included; Canu version 2.0, Flye version 2.7, Miniasm/Minipolish version 0.1.3 [44] Raven version 1.1.10 [45], NECAT version 0.01 [46], wtdbg2 version 2.5 [47], and shasta version 0.5.1 [48]. All assemblers were run with default parameters (flagging raw or corrected reads depending on read input, Raven was run with the --weaken flag when corrected reads were used). Additional Flye assemblies were performed using both Canu and NECAT self-corrected read sets and an additional short-read corrected read set corrected with Ratatosk version 0.1 [49], in order to assess read correction strategy performance. The Ratatosk corrected reads were Canu trimmed using the same settings as for the FMLRC corrected read set. Assemblies were compared using Quast version 5.0.2 [50]. Bandage version 0.8.1 [51] was used to visualize assembly graphs and search for telomere sequences by using the built-in blast function to search the telomere sequence TTAGGGn5, as well as blast searching the complete assembly against each assembly to determine inter-chromosomal mis-assembly events.
Assembly polishing
The uncorrected, adapter trimmed > 3000 bp long reads were realigned to the manually resolved assembly with minimap2 version 2.17-r941 [52] and the resulting alignment file was used to polish the assembly with Racon version v1.4.13 [53], using default parameters with the --no-trimming flag enabled. A total of two rounds of racon polishing were performed in this manner. The corrected consensus was further polished with the same long read set using Medaka version 0.11.5 (https://github.com/nanoporetech/medaka). The trimmed short-read pair-end Illumina reads were mapped to the long-read polished contigs using BWA-mem2 version 2.0pre2 [54], and the assembly was further polished with Pilon version 1.23 [55], enabling the --fix all and --changes flags. In total, four iterations of polishing with the Illumina reads were performed in this manner, and further polishing yielded no additional changes. A summary of the full assembly pipeline is shown in Fig. 6. A dotplot comparison of the scaffolds and contigs from the NCBI reference M. brunneum ARSEF 3297 assembly (GCF_000814965.1) against the complete assembly produced in this study was made using Mummer version 3 [56].

Novel assembly pipeline used to generate telomere length de novo assembly and mitogenome assembly of Metarhizium brunneum. An overview of the steps and tools versions used to generate the complete assembly. Arrows with dashed lines represent mitogenome assembly steps. Arrows with solid lines represent the chromosomal assembly steps
Gene prediction and functional annotation
BUSCO analyses were performed with BUSCO version 4.0.2 [57], using the hypocreales_odb10 lineage gene set. Chromosomes were visualized in Tapestry version 1.0.0 (https://github.com/johnomics/tapestry) in order to determine chromosome completeness (by checking for long read mapping gaps), and setting the telomere sequence as TTAGGG- a common eukaryotic telomere repeat sequence previously shown to be present in Metarhizium telomeres [58]. All assembly annotations were performed in GenSAS version 6.0 [59], unless otherwise stated. Low complexity regions and repeats were detected and masked using RepeatModeler version 1.0.11 [60] and RepeatMasker version 4.0.7 [61], setting the DNA source to fungi and the speed/sensitivity parameter to slow. A masked consensus sequence was generated on which ab initio gene prediction was performed using the following tools; 1. GeneMarkES version 4.33 [62] with default parameters, 2. Augustus version 3.3.1 using Fusarium graminearum as the species, but otherwise keeping the default parameters, 3. GlimmerM version 2.5.1 [63] selecting Aspergillus as the organism. Two separate standalone ab initio gene predictions were conducted on the masked consensus sequence (one including the mitogenome sequence and the other without) using the latest version of GeneMarkES (4.48_3.60.lic), enabling the --ES and --Fungus flags. The highest BUSCO scoring ab initio predicted protein set was used for functional analyses using InterProScan version 5.25–68.0 [64], a native version of SignalP version 5.0 [65] setting the -org flag to eukaryote, and identifying ab initio predicted proteins with blastp [66] by conducting a protein vs protein search against the SwissProt protein data set to determine best matches. Ribosomal RNA genes were detected using RNAmmer version 1.2 [67]. tRNA genes were determined using tRNAscan-SE version 2.0.3 [68]. Comparison of orthologous gene clusters between the protein set generated in this study and the NCBI reference M. brunneum, M. anisopliae and M. robertsii protein sets was performed using OrthoVenn2 [69], with default parameters. The mitogenome, including previously described manual annotations [70], was visualized using the GeSeq tool in Chlorobox [71], selecting a circular mitochondrial sequence.
Full genome sequence-based synteny and pan-genome analyses of Hypocreales fungi
Synteny analyses were performed by comparing the M. brunneum complete genome assembly to three other species within the order Hypocreales that had genome assemblies that are designated as complete by the NCBI (full telomere length chromosomes). These included the genomes of the entomopathogenic fungus Cordyceps militaris [26], the systemic endophytic fungus Epichloe festucae [27], and the cellulolytic, endophytic fungus Trichoderma reesei [28]. Genomes were aligned with progressiveMauve v2.4.0 [72], using default settings. Alignment blocks were filtered to remove syntenic blocks that were less than 1000 bp in size, and also those which were not present in all 4 species. Synteny was inferred with i-ADHoRe v3.0 [73] running default parameters, and whole genome synteny between each species were visualized with Circos plots using Circos v2.40.1 [74]. Ab-initio gene prediction was performed on the three genome assemblies of the other Hypocreales species using GeneMarkES (4.48_3.60.lic), enabling the --ES and --Fungus flags. In order to determine the core genes shared across the 4 species, comparison of orthologous gene clusters between the protein sets for each of the Hypocreales fungi were performed with OrthoVenn2 using default parameters.
Availability of data and materials
All data generated in this study has been deposited at the NCBI under Bioproject PRJNA608152. Illumina sequencing read data can be accessed at the NCBI Sequence Reads Archive (SRA) using the accession number SRX7785787. Nanopore sequencing read data can be accessed at the NCBI SRA using the accession number SRX7785786. Sample information can be accessed at the NCBI BioSample repository using the accession number SAMN14166897. The genome assembly generated in this study can be accessed in NCBI’s GenBank database using the accession number GCA_013426205.1. Gene and protein names and functional annotations (GO terms, InterPro, PFAM) are included in GenBank entries. All output files have been deposited in the following GitHub repository- https://github.com/zacksaud/Metarhizium-Brunneum-ARSEF4556-Assembly-Project. The following genomes and/or information on the genome assemblies were retrieved from NCBI’s GenBank database; Metarhizium album ARSEF 1941 (accession number: GCA_000804445.1), Metarhizium acridum CQMa 102 (accession number: GCA_000187405.1), Metarhizium anisopliae ARSEF 549 (accession number: GCA_000814975.1), Metarhizium brunneum ARSEF 3297 (accession number: GCF_000814965.1), Metarhizium guizhouense ARSEF 977 (accession number: GCA_000814955.1), Metarhizium majus ARSEF 297 (accession number: GCA_000814945.1), Metarhizium rileyi RCEF 4871 (accession number: GCA_001636745.1), Metarhizium robertsii ARSEF 23 (accession number: GCA_000187425.2), Cordyceps militaris ATCC 34164 (accession number: GCA_008080495.1), Epichloe festucae Fl1 (accession number: GCA_003814445.1), and Trichoderma reesei QM6a (accession number: GCA_002006585.1).
Abbreviations
- ARSEF:
-
ARS Collection of Entomopathogenic Fungal Cultures
- BUSCO:
-
Benchmarking Universal Single-Copy Orthologs
- CM:
-
Cordyceps militaris
- DNA:
-
Deoxyribonucleic acid
- EF:
-
Epichloe festucae
- MAA:
-
Metarhizium robertsii
- MAN:
-
Metarhizium anisopliae
- Mb:
-
Million base pairs
- MBR:
-
Metarhizium brunneum
- NCBI:
-
National center for biotechnology information
- ORF:
-
Open reading frame
- rRNA:
-
Ribosomal ribonucleic acid
- SDA:
-
Sabouraud dextrose agar
- TR:
-
Trichoderma reesei
- tRNA:
-
Transfer ribonucleic acid
References
Worley KC, Richards S, Rogers J. The value of new genome references. Exp Cell Res. 2017;358:433–8. https://doi.org/10.1016/j.yexcr.2016.12.014.
Ziemert N, Alanjary M, Weber T. The evolution of genome mining in microbes-a review. Nat Prod Rep. 2016;33:988–1005. https://doi.org/10.1039/c6np00025h.
Bennett S. Solexa ltd. Pharmacogenomics. 2004;5(4):433–8. https://doi.org/10.1517/14622416.5.4.433.
Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Characterization of individual polynucleotide molecules using a membrane channel. Proc Natl Acad Sci U S A. 1996;93:13770–3. https://doi.org/10.1073/pnas.93.24.13770.
Jain M, Olsen HE, Paten B, Akeson M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 2016;17(1):239. https://doi.org/10.1186/s13059-016-1103-0.
Eid J, Fehr A, Gray J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–8. https://doi.org/10.1126/science.1162986.
Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci. 1977;74:5463–7. https://doi.org/10.1073/pnas.74.12.5463.
Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, Adam MP. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30:693–700. https://doi.org/10.1038/nbt.2280.
Ma J, Zhang L, Suh BB, Raney BJ, Burhans RC, Kent WJ, Blanchette M, Haussler D, Miller W. Reconstructing contiguous regions of an ancestral genome. Genome Res. 2006;16:1557–65. https://doi.org/10.1101/gr.5383506.
Lewin HA, Larkin DM, Pontius J, O’Brien SJ. Every genome sequence needs a good map. Genome Res. 2009;19:1925–8. https://doi.org/10.1101/gr.094557.109.
Florea L, Souvorov A, Kalbfleisch TS, Salzberg SL. Genome assembly has a major impact on gene content: a comparison of annotation in two Bos taurus assemblies. PLoS One. 2011;6(6):e21400. https://doi.org/10.1371/journal.pone.0021400.
de Faria MR, Wraight SP. Mycoinsecticides and mycoacaricides: a comprehensive list with worldwide coverage and international classification of formulation types. Biol Control. 2007;43(3):237–56. https://doi.org/10.1016/j.biocontrol.2007.08.001.
Leger RJ. Metarhizium anisopliae as a model for studying bioinsecticidal host pathogen interactions. In: Vurro M, Gressel J, editors. Novel biotechnologies for biocontrol agent enhancement and management. NATO security through science series. Dordrecht: Springer; 2007.
Behie SW, Moreira CC, Sementchoukova I, Barelli L, Zelisko PM, Bidochka MJ. Carbon translocation from a plant to an insect-pathogenic endophytic fungus. Nat Commun. 2017;8:14245. https://doi.org/10.1038/ncomms14245.
Wang B, Kang Q, Lu Y, Bai L, Wang C. Unveiling the biosynthetic puzzle of destruxins in Metarhizium species. Proc Natl Acad Sci U S A. 2016;109(4):1287–92. https://doi.org/10.1073/pnas.1115983109.
St. Leger RJ, May B, Allee LL, Frank DC, Staples RC, Roberts DW. Genetic differences in allozymes and in formation of infection structures among isolates of the entomopathogenic fungus Metarhizium anisopliae. J Invertebr Pathol. 1992;60(1):89–101. https://doi.org/10.1016/0022-2011(92)90159-2.
Gao Q, Jin K, Ying SH, et al. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 2011;7(1):e1001264. https://doi.org/10.1371/journal.pgen.1001264.
Hu X, Xiao G, Zheng P, et al. Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc Natl Acad Sci U S A. 2014;111(47):16796–801. https://doi.org/10.1073/pnas.1412662111.
Staats CC, Junges A, Guedes RL, et al. Comparative genome analysis of entomopathogenic fungi reveals a complex set of secreted proteins. BMC Genomics. 2014;15:822. https://doi.org/10.1186/1471-2164-15-822.
Pattemore JA, Hane JK, Williams AH, Wilson BA, Stodart BJ, Ash GJ. The genome sequence of the biocontrol fungus Metarhizium anisopliae and comparative genomics of Metarhizium species. BMC Genomics. 2014;15(1):660. https://doi.org/10.1186/1471-2164-15-660.
Shang Y, Xiao G, Zheng P, Cen K, Zhan S, Wang C. Divergent and convergent evolution of fungal pathogenicity. Genome Biol Evol. 2016;8(5):1374–87. https://doi.org/10.1093/gbe/evw082.
Cohen-Gihon I, Sharan R, Nussinov R. Processes of fungal proteome evolution and gain of function: gene duplication and domain rearrangement. Phys Biol. 2011;8:035009. https://doi.org/10.1088/1478-3975/8/3/035009.
Shimizu S, Arai Y, Matsumoto T. Electrophoretic karyotype of Metarhizium anisopliae. J Invertebr Pathol. 1992;60(2):185–7. https://doi.org/10.1016/0022-2011(92)90094-K.
Valadares-Inglis MC, Peberdy JF. Variation in the electrophoretic karyotype of Brazilian strains of Metarhizium anisopliae. Genet Mol Biol. 1998;21(1):11–4. https://doi.org/10.1590/S1415-47571998000100003.
Wang C, Skrobek A, Butt T. Concurrence of losing a chromosome and the ability to produce destruxins in a mutant of Metarhizium anisopliae. FEMS Microbiol Lett. 2003;226(2):373–8. https://doi.org/10.1016/S0378-1097(03)00640-2.
Kramer GJ, Nodwell JR. Chromosome level assembly and secondary metabolite potential of the parasitic fungus Cordyceps militaris. BMC Genomics. 2017;18(1):912. https://doi.org/10.1186/s12864-017-4307-0.
Winter DJ, Ganley ARD, Young CA, et al. Repeat elements organise 3D genome structure and mediate transcription in the filamentous fungus Epichloë festucae. PLoS Genet. 2018;14(10):e1007467. https://doi.org/10.1371/journal.pgen.1007467.
Li WC, Huang CH, Chen CL, Chuang YC, Tung SY, Wang TF. Trichoderma reesei complete genome sequence, repeat-induced point mutation, and partitioning of CAZyme gene clusters. Biotechnol Biofuels. 2017;10:170. https://doi.org/10.1186/s13068-017-0825-x.
Xu YJ, Luo F, Li B, Shang Y, Wang C. Metabolic conservation and diversification of metarhizium species correlate with fungal host-specificity. Front Microbiol. 2016;7:2020. https://doi.org/10.3389/fmicb.2016.02020.
Wick RR, Holt KE. Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Res. 2019;8:2138. https://doi.org/10.12688/f1000research.21782.2.
Fu S, Wang A, Au KF. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 2019;20:26. https://doi.org/10.1186/s13059-018-1605-z.
Jain M, Koren S, Miga KH, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36(4):338–45. https://doi.org/10.1038/nbt.4060.
Hane JK, Rouxel T, Howlett BJ, Kema GHJ, Goodwin SB, Oliver RP. A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol. 2011;12:R45. https://doi.org/10.1186/gb-2011-12-5-r45.
Fang G, Bhardwaj N, Robilotto R, Gerstein MB. Getting started in gene orthology and functional analysis. PLoS Comput Biol. 2010;6(3):e1000703. https://doi.org/10.1371/journal.pcbi.1000703.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Andrews, S: FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6):e1005595. https://doi.org/10.1371/journal.pcbi.1005595.
De Coster W, D'Hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34(15):2666–9. https://doi.org/10.1093/bioinformatics/bty149.
Wang JR, Holt J, McMillan L, Jones CD. FMLRC: hybrid long read error correction using an FM-index. BMC Bioinformatics. 2018;19(1):50. https://doi.org/10.1186/s12859-018-2051-3.
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36. https://doi.org/10.1101/gr.215087.116.
Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 2016;11(10):e0163962. https://doi.org/10.1371/journal.pone.0163962.
Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37(5):540–6. https://doi.org/10.1038/s41587-019-0072-8.
Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res. 1999;9(9):868–77. https://doi.org/10.1101/gr.9.9.868.
Wick RR, Holt KE. rrwick/Minipolish: Minipolish v0.1.3; 2020. https://doi.org/10.5281/zenodo.3752203.
Vaser R, Šikić M. Yet another de novo genome assembler 2019 11th international symposium on image and signal processing and analysis (ISPA), Dubrovnik, Croatia; 2019. p. 147–51. https://doi.org/10.1109/ISPA.2019.8868909.
Ying C, Fan N, Shang-Qian X, et al. Fast and accurate assembly of Nanopore reads via progressive error correction and adaptive read selection. bioRxiv. 2020. https://doi.org/10.1101/2020.02.01.930107.
Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17(2):155–8. https://doi.org/10.1038/s41592-019-0669-3.
Shafin K, Pesout T, Lorig-Roach R, et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat Biotechnol. 2020. https://doi.org/10.1038/s41587-020-0503-6.
Holley G, Beyter D, Ingimundardottir H, Kristmundsdottir S, Eggertsson HP, Halldorsson BV. Ratatosk – Hybrid error correction of long reads enables accurate variant calling and assembly. bioRxiv. https://doi.org/10.1101/2020.07.15.204925.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5. https://doi.org/10.1093/bioinformatics/btt086.
Wick RR, Schultz MB, Zobel J, Holt KE. Bandage: interactive visualisation of de novo genome assemblies. Bioinformatics. 2015;31(20):3350–2. https://doi.org/10.1093/bioinformatics/btv383.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100. https://doi.org/10.1093/bioinformatics/bty191.
Vaser R, Sović I, Nagarajan N, Šikić M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017;27(5):737–46. https://doi.org/10.1101/gr.214270.116.
Md V, Misra S, Li H, Aluru S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. IEEE Parallel and Distributed Processing Symposium (IPDPS); 2019.
Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. https://doi.org/10.1371/journal.pone.0112963.
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. https://doi.org/10.1186/gb-2004-5-2-r12.
Seppey M, Manni M, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness. In: Kollmar M, editor. Gene prediction. Methods in molecular biology vol 1962. New York: Humana; 2019. https://doi.org/10.1007/978-1-4939-9173-0_14.
Inglis PW, Rigden DJ, Mello LV, Louis EJ, Valadares-Inglis MC. Monomorphic subtelomeric DNA in the filamentous fungus, Metarhizium anisopliae, contains a RecQ helicase-like gene. Mol Gen Genomics. 2005;274(1):79–90. https://doi.org/10.1007/s00438-005-1154-5.
Humann JL, Lee T, Ficklin S, Main D. Structural and functional annotation of eukaryotic genomes with GenSAS. Methods Mol Biol. 1962;2019:29–51. https://doi.org/10.1007/978-1-4939-9173-0_3.
Smit AFA, Hubley R: RepeatModeler. Open-1.0. 2008–2015. (http://www.repeatmasker.org).
Smit AFA, Hubley R, Green P: RepeatMasker. Open-4.0. 2013-2015 <http://www.repeatmasker.org>.
Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18(12):1979–90. https://doi.org/10.1101/gr.081612.108.
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27(23):4636–41. https://doi.org/10.1093/nar/27.23.4636.
Jones P, Binns D, Chang HY, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–40. https://doi.org/10.1093/bioinformatics/btu031.
Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420–3. https://doi.org/10.1038/s41587-019-0036-z.
Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. https://doi.org/10.1186/1471-2105-10-421.
Lagesen K, Hallin P, Rødland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8.
Chan PP, Lowe TM. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962;2019:1–14. https://doi.org/10.1007/978-1-4939-9173-0_1.
Xu L, Dong Z, Fang L, et al. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019;47(W1):W52–8. https://doi.org/10.1093/nar/gkz333.
Kortsinoglou AM, Saud Z, Eastwood DC, Butt TM, Kouvelis VN. The mitochondrial genome contribution to the phylogeny and identification of Metarhizium species and strains. Fungal Biol (In press). https://doi.org/10.1016/j.funbio.2020.06.003.
Tillich M, Lehwark P, Pellizzer T, et al. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017;45(W1):W6–W11. https://doi.org/10.1093/nar/gkx391.
Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147. https://doi.org/10.1371/journal.pone.0011147.
Proost S, Fostier J, De Witte D, et al. i-ADHoRe 3.0--fast and sensitive detection of genomic homology in extremely large data sets. Nucleic Acids Res. 2012;40(2):e11. https://doi.org/10.1093/nar/gkr955.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;9:1639–45. https://doi.org/10.1101/gr.092759.109.
Acknowledgements
The authors thank Dr. Louela Castrillo of the USDA-ARS for providing Metarhizium brunneum ARSEF 4556. We thank Dr. Matthew Hitchings of Swansea University’s College of Medicine for suggesting various Bioinformatics tools to test. We thank Mr. James Taylor and Ms. Sophie Hocking for lab support.
Funding
Grant funding was secured from the Biotechnology and Biological Sciences Research Council, the Department for Environment, Food and Rural Affairs, the Economic and Social Research Council, the Forestry Commission, the Natural Environment Research Council and the Scottish Government, under the Tree Health and Plant Biosecurity Initiative. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
ZS, AMK, VNK and TMB conceived of the study and participated in its design and coordination. ZS and AMK carried out the laboratory work. ZS performed the bioinformatics analysis. All authors helped to draft and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Pipeline and assembly validation. A) a Flye assembly graph of the FMLRC corrected long reads without the Canu trimming step. B) a Flye assembly graph of the Canu trimmed long reads without the FMLRC correction step. Both assemblies failed to generate telomere length contigs. C) Manual resolving of tangles in Flye assembly (> 5000) graph. Evidence was used from both assemblies to resolve the final tangles. Chromosome 7 was telomere length in the > 3000 read length assembly, and, along with coverage data, allowed the tangle in the assembly graph between chromosome 1 and chromosome 7 to be resolved (blue). Chromosome 3 was also fully telomere length in the in the > 3000 read length assembly (pink). Mapping reads to the 5231 bp contig, which contained a telomere sequence at its terminal, showed the contig to overlap with the end repeat contig of chromosome one (purple). D) Dotplot comparison of the long read assembly M. brunneum reference assembly. Good synteny is observed between the 7 complete chromosomes and the contigs and scaffolds from the previous reference assembly.
Additional file 2.
Comparison of assemblers. Assembly graphs showing TTAGGGn5 sequences detected in contigs produced by all assemblers, and colour coded blast hits of chromosomes from the final complete assembly from which mis-assemblies were inferred.
Additional file 3.
Summary of SignalP results. SingalP likelihood scores, signal peptide type (if present) and signal peptide positions for all M brunneum proteins.
Additional file 4.
Mature proteins. M brunneum mature protein sequences with signal peptides removed.
Additional file 5.
Core Hypocreales protein set. A list of core proteins found to be orthologous between the four Hypocreales fungi; Metarhizum brunneum, Cordyceps militaris, Epichloe festucae and Trichoderma reesei.
Additional file 6.
Metarhizium brunneum self-cluster. The 183 unique orthologous clusters that were formed between M. brunneum proteins.
Additional file 7.
Orthologous clusters formed between the entomopathogenic fungi. The 468 unique orthologous clusters that were formed between the two entomopathogenic Hypocreales fungi in the comparison test- Metarhizium brunneum and Cordyceps militaris. Proteins within these clusters may be involved with the entomopathogenic process.
Additional file 8.
M brunneum singletons. Metarhizium brunneum proteins that did not form orthologous clusters with any other proteins.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Saud, Z., Kortsinoglou, A.M., Kouvelis, V.N. et al. Telomere length de novo assembly of all 7 chromosomes and mitogenome sequencing of the model entomopathogenic fungus, Metarhizium brunneum, by means of a novel assembly pipeline. BMC Genomics 22, 87 (2021). https://doi.org/10.1186/s12864-021-07390-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-021-07390-y