
Abstract
Background
Lactoferrampin (LFampin), Lactoferricin (LFcin), and LFchimera are three well-known antimicrobial peptides derived from Lactoferrin and proposed as alternatives for antibiotics. Although the intracellular activity of these peptides has been previously demonstrated, their mode of action is not yet fully understood. Here, we performed a molecular dynamics simulation study to understand the molecular interactions between camel Lactoferrin derived peptides, including CLFampin, CLFcin, and CLFchimera, and DNA as an important intracellular target.
Results
Our results indicate that all three peptides bind to DNA, albeit with different propensities, with CLFchimera showing the highest binding affinity. The secondary structures of the peptides, modeled on Lactoferrin, did not undergo significant changes during simulation, supporting their functional relevance. Main residues involved in the peptide-DNA interaction were identified based on binding free energy estimates calculated over 200 ns, which, as expected, confirmed strong electrostatic interactions between DNA phosphate groups and positively charged peptide side chains. Interaction between the different concentrations of CLFchimera and DNA revealed that after binding of four copies of CLFchimera to DNA, hydrogen bonds between the two strands of DNA start to break from one of the termini.
Conclusions
Importantly, our results revealed that there is no DNA-sequence preference for peptide binding, in line with a broad antimicrobial activity. Moreover, the results showed that the strength of the interaction between DNA and CLFchimera is concentration dependent. The insight provided by these results can be used for the rational redesign of natural antimicrobial peptides targeting the bacterial DNA.
Similar content being viewed by others


Background
Antibiotic resistance is becoming a serious global health problem, as infections by multidrug-resistant pathogens are increasing at an alarming pace. There is thus an urgent need to introduce new and safe antimicrobial agents, including antimicrobial peptides (AMPs), as alternatives to current antibiotics [1]. AMPs have evolved as a natural defense mechanism for fighting microbial infections [1]. They are a diverse group of innate immune system molecules that exist in all organisms [1]. AMPs usually contain 12–50 amino acid residues, have a net positive charge and an amphipathic structure [2,3,4]. One subgroup of AMPs includes peptides derived from large proteins. Lactoferrampin (LFampin) and Lactoferricin (Lfcin) are two well-known antimicrobial peptides derived from the Lactoferrin protein (LF) [5, 6]. These two cationic antimicrobial peptides have activity against a broad spectrum of microorganisms including bacteria, fungi and viruses [5, 6].
We have recently reported that a camel Lactoferrin chimera (CLFchimera) resulting from the fusion of the C-terminal ends of camel Lactoferricin 17–30 (CLFcin) and camel Lactoferrampin 265–284 (CLFampin) using the side chain of lysine as linker to the second peptide, has a broad-spectrum activity against both Gram-positive and Gram-negative bacteria [7,8,9]. Furthermore, Reyes-Cortes et al. (2016) showed that this chimeric peptide mediated its antibacterial activity by entering the cytoplasm through translocation across the bacterial membrane and possibly interacting with internal organelles [10]. To date, there has been no precise explanation for the mechanisms underlying the antimicrobial peptide function, but it is known that DNA is one of the most important intracellular targets for AMPs [11]. Thus, nucleic acids have been proven as intracellular targets for some antimicrobial peptides such as MDpep9 [11], Buforin I [12], Indolicidin [13], Cecropin PR39 [14], and NK18 [15]. Previous computational studies also showed that Buforin II (from the stomach tissue of the Asian toad bufo garagrizans) and Lasioglossin II (derived from bee venom) had considerable affinity for DNA [16, 17]. Considering these reports, Uyterhoeven et al. (2008) showed using MD simulation that Arg 2, Arg 14 and Arg 20 of Buforin II were mainly responsible for the interaction with DNA and using Fluorescent Intercalator Displacement (FID) assay indicated that disrupting Buforin-DNA interactions generally decreased the antibacterial activity of the peptide [16]. In another study, Tang et al. (2009) demonstrated that MDpep9, a recently discovered antimicrobial peptide derived from larvae of housefly (Musca domestica), a traditional food source in China, is able to form bonds with DNA phosphate groups and insert between the base pairs of the DNA helix [11].
Although the intracellular activity of Lactoferrin derived peptides has been previously demonstrated [10], the exact mechanism of action has not been yet established. As recognition of specific DNA sequences by proteins is highly complex, involving structural, energetic and dynamic aspects, the interaction cannot be easily characterized at the atomic level by experimental approaches alone [18]. The use of computational techniques such as molecular dynamics (MD) simulations provides complementary information, inaccessible experimentally, which relates directly to the thermodynamics and kinetics of the system. Herein, homology-based models were constructed for camel LFcin, LFampin and LFchimera and their interaction with DNA was analyzed using MD simulation, as a means to understand a reported intracellular mechanism of action of these peptides. The findings of this study provide basic directions for future studies regarding the function of AMPs with intracellular activity and their potential redesign with therapeutic purposes.
Methods
Molecular structure models
An arbitrary 12-bp DNA sequence adopting a canonical BDNA structure (entry 1BNA from the Protein Data Bank) was initially chosen for this study (Table 1). Since the simulations using this sequence indicated that the interaction with the peptides is DNA-sequence independent (through the backbone phosphates), no additional sequences were used in the study. The length of the DNA allows the interaction with more than one peptide, as shown in the Results and Discussion. The native structure of camel Lactoferrin was also retrieved from the PDB (entry 1DTZ). Camel Lactoferrin was used as a template structure for peptide modeling. Lactoferricin, Lactoferrampin and CLFchimera were modeled with Modeller 9.2 [19] and PEP-fold server (http://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD/) [20] using default parameters. The quality of the models was examined with PROCHECK (http://servicesn.mbi.ucla.edu/PROCHECK/) [21].
Molecular dynamics simulations
The complexes BDNA-CLFampin, BDNA-CLFcin, and BDNA-CLFchimera were studied by molecular dynamics simulation with the GROMACS 2016.1 package [22,23,24] and CHARMM27 force field [25]. Peptides and DNA were solvated in a cubic box using the Simple Point Charge (SPC) water model [26]. To neutralize the overall charge of the systems, Na and Cl ions were added as appropriate. Periodic boundary conditions were applied from this step onward. The system was first energy minimized using the steepest descent algorithm to relax high-energy contacts. After energy minimization, the system was simulated under the NPT ensemble for 500 ps, with initial velocities taken from a Maxwell-Boltzmann distribution corresponding to 100 K. During this initial simulation time, the peptide and DNA atoms were positionally restrained while the temperature was gradually increased from 100 K to 300 K at 1 atm. Bond lengths were constrained for all atoms using the LINCS algorithm (SETTLE for water), allowing a time step in the leap-frog integrator of 2 fs. Temperature and pressure were couple to the reference values using the Nosé-Hoover and Parrinello-Rahman algorithms, respectively [27,28,29]. Additional 100 ps at 300 K and 1 atm, without position-restraints, were subsequently run. In the production phase, the equilibrated systems were run in the NPT ensemble at 1 atm and 300 K for 200 ns. Long-range electrostatics were evaluated using the Particle Mesh Ewald (PME) algorithm [28]. The real space component of PME and the van der Waals interactions were calculated with a cutoff of 1.0 nm. Three replicates of 200 ns were run per system, with different initial configurations generated by insertion of the peptides at random positions. The simulations performed and their lengths are detailed in Table 1. Dynamics and stability of each peptide and BDNA, including root mean square deviation (RMSD), root-mean-square-fluctuations (RMSF), solvent accessible surface area (SASA), contacting surface area (CSA), hydrogen bonds, salt bridges, and center of mass distance were analyzed during the simulation using GROMACS built-in tools. An RMSD-based conformational clustering algorithm, using the gmx-cluster module of GROMACS, was applied to extract representative structures. The clusters were obtained using a cut-off of 1.5 Å for the RMSD to the centroid.
Binding free energy estimates
Binding free energies were estimated for BDNA-CLFampin, BDNA-CLFcin, and BDNA-CLFchimera complexes using molecular mechanics energies in combination with Poisson-Boltzmann and surface area continuum solvation (MM/PBSA). The calculations were performed with the g_mmpbsa program (https://rashmikumari.github.io/g_mmpbsa/) [30], using the single trajectory approach. The solute dielectric constant was set to 8 [31] and the ionic strength was chosen to correspond to a NaCl concentration of 150 mM. The calculation of the Gpolar solvation term was performed with the linearized Poisson-Boltzmann (PB) equation using a grid resolution of 0.05 nm and the bondi set of atomic radii. The Gnonpolar term was calculated with the SASA model using default parameters [30]. The entropic component of the binding free energy was disregarded. The average binding energy and its standard deviation were calculated with the MmPbSaStat.py python script (http://rashmikumari.github.io/g_mmpbsa/) using the second half of the simulations production phase (100 to 200 ns), by taking 1000 snapshots at 100-ps intervals. To estimate the contribution of each residue to the total binding free energy, the MmPbSaDecomp.py python script was used [30, 32]. It should be noted that this approach represents a crude estimate of the binding free energy that, most certainly, severely overestimates the real value, as noted by several authors [33]. However, the limitations of the approach are likely to affect the related systems studied here in similar ways and are therefore expected to allow for a qualitative comparison.
Results and discussion
Molecular dynamics simulation in aqueous solution of the isolated peptides and BDNA
Before simulating the interaction between the different peptides and BDNA, the individual model structures were relaxed along independent 200-ns simulations, performed in triplicate (Table 1). To that end, the homology models obtained for the peptide structures were first examined for overall quality. The Ramachandran plot for CLFampin, CLFcin and CLFchimera revealed that 93.3, 100.0 and 93.5% of the residues were situated within the most favored region, respectively, whereas the remaining residues were found within the additional allowed region.
Structural fluctuation analysis
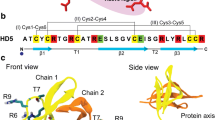
Root-mean-square deviations from the initial structure of the peptide as a function of simulation time and root-mean-square fluctuations of peptide residues are presented, for one of the 200-ns replicates, in Fig. 1. The behavior of these quantities in the remaining replicates is consistent with the observations made here (see Additional file 1: Figure S1 and Additional file 2: Figure S2). The RMSD values are stable after the initial 100 ns, the larger peptide CLFchimera showing higher RMSD and RMSF values. CLFchimera was obtained from the C-term-C-term fusion of CLFampin 265–284 and CLFcin17–30, using a lysine (Lys21) as linker [7, 8]. Figure 1b shows that the global fluctuations of the corresponding sequences in the shorter peptides are lower in general than in the fusion peptide, as expected in light of the structures shown in Fig. 1c. It is worth noting that the shorter CLFcin adopts a more stable helical structure than CLFampin when isolated in solution, to become more flexible in the fusion peptide. Structures from the stable part of the 200 ns simulations with all residues in the most favored regions of the Ramachandran plot were used as initial structures for the corresponding simulation of peptide-DNA systems.

Structural fluctuation analysis. a RMSD as a function of time; b RMSF per residue; c Cartoon structure of CLFchimera (C1), CLFampin (C2) and CLFcin (C3) at 0, 100 and 200 ns (red, blue and green, respectively); d Sequence alignment of the three peptides. RMSD and RMSF quantities were computed for structures at 0.1-ns intervals from the 200-ns simulations after least square fitting to the initial structure using the backbone atoms
Molecular dynamics simulation of the peptide-DNA systems
Simulations between CLFchimera, CLFcin and CLFampin and BDNA were performed for 200 ns in triplicate. To construct the system, the peptide was introduced in the BDNA box at a random position and orientation. Center of mass distance (COM), hydrogen bonds, salt bridges and contacting surface area between peptide and DNA were analyzed.
Center of mass distances
The center of mass distance between peptide and DNA was calculated as a function of time (Fig. 2a). Side view of snapshots of the first and last configurations are shown in Fig. 2b. In all three replicates, COM distances were initially around 3, 3 and 4 nm for CLFcin, CLFampin and CLFchimera, respectively. The peptides instantly moved toward the DNA grooves and COM distances decreased rapidly. The three replicates show some differential behavior in terms of final distance and convergence (Additional file 3: Figure S3A and Additional file 4: Figure S4A), as well as in terms of position and orientation (Additional file 3: Figure S3B and Additional file 4: Figure S4B), suggesting that the binding is not specific, as demonstrated further below.

COM distance analysis. a COM distances between CLFcin, CLFampin and CLFchimera and DNA along 200 ns. b Structures at times t = 0 (cyan) and t = 200 ns (purple): (B1) CLFcin-DNA, (B2) CLampin-DNA, and (B3) CLFchimera-DNA
Number of hydrogen bonds and salt bridges
The number of hydrogen bonds between peptide and DNA showed significant variation during simulation (Fig. 3; Additional file 5: Figure S5 and Additional file 6: Figure S6). The average number of hydrogen bonds in the second half of the three simulation replicates (100–200 ns, 300 ns in total) was 5.66 ± 0.23, 4.61 ± 0.55 and 2.63 ± 0.27 for CLFchimera, CLFcin and CLFampin, respectively (see also Additional file 7: Table S1 for details), suggesting that CLFchimera establishes more stable interactions with DNA.

Number of hydrogen bonds with DNA as a function of simulation time (200 ns). a CLFampin, b CLFcin, c CLFchimera. d Snapshot at t = 135 ns of the CLFchimera-DNA system, indicating hydrogen bonds (red lines) and salt bridges (yellow dashed)
A representative snapshot of the CLFchimera-DNA interaction is illustrated in Fig. 3d. In this frame, it can be seen that hydrogen-bonding interactions are mainly established between positively charged residues of the peptide and the DNA-backbone phosphate groups, which constitute also salt bridges.
Salt bridges also play a fundamental role in protein-ligand interactions [34, 35]. In several studies, a cutoff of 4 Å between N-O atom pairs has been used to define salt bridge formation [36, 37]. Here, we calculated salt bridges between P atoms from the nucleic-acid backbone and N atoms from Lysine and Arginine residues, and thus used 5 Å as cutoff. The average number of salt bridges in the second half of the three simulation replicates between DNA and CLFchimera, CLFcin, CLFampin were 4.09 ± 016, 3.17 ± 0.28 and 1.71 ± 0.44 (see Additional file 7: Table S1 for details). Again, CLFchimera establishes more salt bridges with DNA than the other two peptides.
Contacting surface area
The solvent-accessible surface area was calculated with the Gromacs library [38]. The contacting surface area can be then calculated using the following formula: CSA = (SASA Peptide(s) + SASA DNA – SASA Peptide(s)-DNA)/2 [39]. Initially, the CSA was close to zero due to the distance between peptides and DNA. The evolution of the CSA is shown in Fig. 4 for one of the simulation replicates (see Additional file 8: Figure S7 for the other two). In all three replicates, the CSA is stable after the initial 100 ns, indicating a stable interaction has been reached. The average CSA in the period 100–200 ns is 5.92 ± 0.41, 4.9 ± 0.1, and 4.76 ± 0.36 nm2 for the CLFchimera, CLFcin and CLFampin systems, respectively (see Additional file 7: Table S1 for details). The CSA is higher for CLFchimera than for the other two peptides, in line with the observed interactions.

Contacting surface area between peptide and DNA along a 200 ns MD simulation
MM/PBSA binding free energy estimate
The binding free energy was estimated using the MM/PBSA method. The results for the period 100–200 ns in one of the replicates are presented in Table 2. As indicated in the Methods section, particularly for this type of systems (high charge density), the single-trajectory MM/PBSA approach represents a very crude estimate of the binding free energy that, most certainly, severely overestimates the real value. Nevertheless, the calculations will be used here to qualitatively compare and rank the different systems, which should be relatively safe given that the nature of the interactions is the same in all cases. The results indicate that CLFchimera has the lowest DNA-binding energy. The plot of the binding free energy along the period 100–200 ns in one of the replicates is shown in Fig. 5 (see Additional file 9: Figure S8 for the other two replicates). No significant differences in the obtained binding free energy values were observed among replicates (− 786 ± 2.545, − 731 ± 3.521 and − 712 ± 7.801 kJ/mol for CLFchimera; − 340 ± 4.437, − 352 ± 4.437 and − 316 ± 7.215 kJ/mol for CLFcin; − 71 ± 3.063, − 78 ± 5.103 and − 62 ± 2.202 kJ/mol for CLFampin).

Estimated binding free energy for the peptide-DNA systems. Calculated with the MM/PBSA method on the 100–200 ns period of one of the simulation replicates
The free energy values for the CLFchimera-DNA system were decomposed into residue contributions using the MmPbSaDecomp.py python script. The results, presented in Fig. 6 for one of the simulation replicates, indicate that residues LYS5, LYS9, LYS13, ARG16, LYS18, ARG27, LYS34 and LYS35 are more relevant for binding. On the other hand, GLU12 and SER36 have a detrimental effect. The contributions in the other two simulation replicates follow the same trends (Additional file 10: Figure S9).

Contribution to DNA binding free energies of amino-acid residues in CLFchimera
Previous experimental studies revealed that substitution of positively charged residues such as LYS269, LYS277 and LYS282 with alanine in bovine Lactoferrampin (LYS9, LYS13 and LYS18 in CLFchimera) resulted in a dramatic decrease in antimicrobial activity [40, 41], a finding consistent with our in silico results (Fig. 6). However, Karn et al. (2006) showed that substitution of GLU276 (GLU12 in CLFchimera) with glycine in bovine Lactoferrampin had no effect on increasing antimicrobial activity [40]. Several experimental studies regarding bovine Lactoferricin indicated that the core hexapeptide “RRWQWR” in this peptide has a significant role in antimicrobial activity [42]. The first two amino acids from this central core in CLFchimera (ARG27 and ARG28) made a considerable contribution to the interaction with DNA in our simulations (Fig. 6); however, they were not as effective as other positively charged residues. Investigation of minimum distances (averaged over the three replicates) showed that LYS5 and LYS35 were closest to DNA, 0.13 ± 0.03 nm and 0.12 ± 0.02 nm, respectively (see Additional file 11: Figure S10).
As shown in Fig. 6, GLU12 and SER36 play a major inhibiting role in the interaction with DNA. Additional file 10: Figure S9 shows that they displayed also the largest minimum distance to DNA, with 0.64 ± 0.13 nm and 0.57 ± 0.09 nm, respectively.
Effect of CLFchimera concentration on DNA binding
Based on the previous sections, CLFchimera showed a considerably higher affinity than CLFcin and CLFampin for DNA. Therefore, we chose the former peptide as candidate and evaluated the effect of its concentration on DNA binding by performing simulations with 1, 2, 3 and 4 CLFchimera molecules and one DNA helix.
Number of hydrogen bonds and salt bridges
As expected, when peptide concentrations rose, the number of hydrogen bonds between DNA and peptides increased but showed saturation (Fig. 7 for one of the replicates of each system; Additional file 12: Figure S11 and Additional file 13: Figure S12 for the other two replicates). The average number of hydrogen bonds in the 100–200 ns period of the three replicates was 5.66 ± 0.23, 9.66 ± 0.56, 11.75 ± 0.45, and 12.4 ± 0.37 for 1, 2, 3 and 4 peptides, respectively (see Additional file 7: Table S1 for details). The corresponding average number of salt bridges was 4.09 ± 0.16, 5.79 ± 0.72, 6.50 ± 0.24, and 7.03 ± 0.06, respectively (see Additional file 7: Table S1 for details). The largest change in the number of hydrogen bonds and salt bridges occurs in the transition between one and two peptides, and shows saturation after three peptides.

Number of hydrogen bonds with DNA at different concentrations of CLFchimera. a 1-CLFchimera, b 2-CLFchimera, c 3-CLFchimera, d 4-CLFchimera
The average percentage of hydrogen bonds involving the DNA phosphate group revealed that nearly all hydrogen bonding was formed between peptide side chains and DNA phosphate groups (see Additional file 14: Table S2). As these groups are equal in all nucleic-acid bases, we may conclude that the binding of CLFchimera to DNA is DNA-sequence unspecific. For this reason, a potential DNA-related antimicrobial activity of this peptide could consist in the disruption of replication. Uyterhoeven et al., 2008 reported that Buforin II, another antimicrobial peptide, would probably target nucleic acids in a non-sequence-specific manner [16]. Sim et al., 2017 also examined the interaction of Buforin II and DesHDAP1 with DNA and suggested that a large percentage of hydrogen bonds were formed between peptide side chains and phosphate groups in the nucleic acid backbone (96.3 and 81.7%, respectively). Then, they demonstrated experimentally that Buforin II and DesHDAP1 did not show signs of sequence-specific DNA binding [43]. In another study on a particular AMP derived from Chinese traditional edible housefly larvae by Tang et al., 2009, it was revealed that the phosphate anion of the DNA double helix is one of the binding sites in DNA-peptide interaction [11]. The results of the present study are in accordance with all these previous findings. On the other hand, an experimental study on the interaction between bovine Indolicidin and DNA demonstrated that Indolicidin bound tightly to the ds [AT], ds [GC] and ds [AG] sequences, but formed loose bonds with ds [GT] [44].
Contacting surface area
Results from the CSA analysis at different concentrations of CLFchimera revealed an increase in CSA with peptide concentration (Fig. 8 for one of the replicates of each system and Additional file 15; Figure S13 for the other two replicates). The average CSA (three replicates) for 1, 2, 3 and 4 peptides was 5.92 ± 0.41, 10.30 ± 0.83, 11.79 ± 1.14 and 14.55 ± 1.39 nm2, respectively (see Additional file 7: Table S1 for details). The highest increase in CSA occurs for the transition between one and two peptides. However, the difference between 3 and 4 peptides is slightly larger than could be expected from the previous analysis of interactions, indicating that the increase in contact area is not proportionally translated in additional specific interactions.

Contacting surface area at different concentrations of CLFchimera
MM/PBSA binding free energy estimate
As shown in Table 3 (one of the replicates), the CLFchimera-DNA binding energy decreases with increasing peptide concentration. The decrease in binding energy per peptide is again largest in the transition from one to two peptides. The results for the different replicates are in this regard consistent: − 946 ± 4.504, − 927 ± 9.31 and − 894 ± 5.056 kJ/mol for 2-CLFchimera; − 1004 ± 4.007, − 1020 ± 9.23 and − 975 ± 8.09 kJ/mol for 3-CLFchimera; − 1027 ± 15.718, − 1071 ± 12.21 and − 1022 ± 10.12 kJ/mol for 4-CLFchimera. The saturation effect observed on going from three to four peptides could be partially due to repulsion between the highly charged peptides, in addition to crowding at the sites favorable for the interaction with DNA phosphate groups.
The binding energy contribution per amino-acid residue (Fig. 9 for one replicate and Additional file 16: Figure S14 for the other two) indicates that the residues with important contributions are the same at the different concentrations. It also shows that they are primarily responsible for the decrease in binding energy with increasing concentration.

Contribution of amino-acid residues to DNA-binding energy at different concentrations of CLFchimera
Tables 2 and 3 demonstrate that complex formation and stability was highly correlated with electrostatic interaction, as expected. Our MM/PBSA results conform to the results obtained by Pandey et al., 2018 on similar systems [18].
A computational study by Khabiri et al., 2017 reported that changes in binding free energy do not correlate strongly with salt bridges or hydrogen bonding in protein-DNA interactions [45]. Contrary to their results, we show that for our peptide-DNA systems the electrostatic energy and binding free energy are correlated with the number of hydrogen bonds and salt bridges (Fig. 10), with an R2 between binding free energy and the number of hydrogen bonds and salt bridges of − 0.92 and − 0.95, respectively, and an R2 of − 0.95 and − 0.97 between electrostatic energy and hydrogen bonds and salt bridges, respectively.

Trends in the number of hydrogen bonds and salt bridges, and electrostatic, van der Waals and binding energy in simulations of DNA with CLFampin, CLFcin and different concentrations of CLFchimera
Structural characterization of CLFchimera
To assess the conformational heterogeneity of the peptide, clustering analysis was performed for all simulation trajectories. CLFchimera was thus distributed into 22 clusters. The conformation of the top seven clusters, covering 97% of all structures, is shown in Fig. 11. Clusters one and two comprised 65% of all structures. The analysis shows that the helical segments of CLFchimera are stable along the simulations, with major conformational changes affecting the turn between them.

Top seven, most-populated conformations of CLFchimera in the simulations
Effect of CLFchimera concentration on DNA conformation
Analysis of RMSD and hydrogen bonds between the two strands of the DNA revealed that the presence of 1, 2 and 3 molecules of CLFchimera had no significant effect on DNA conformation during MD simulations. However, four copies of the peptide caused DNA partial denaturation (Fig. 12a and b), significantly increasing the RMSD values (Fig. 12c) and decreasing the number of backbone hydrogen bonds (Fig. 13) after about 50 ns of simulation. This result was observed for two (out of three) replicates of four copies of concentrations.

Monitoring of BDNA conformation during MD simulation. a changes in BDNA conformation in the absence of the peptide. b change of BDNA conformation with presence of four copies of the peptide. c RMSD analysis of the DNA without and with four copies of the peptide

Analysis of the number of DNA interstrand hydrogen bonds in the presence of zero and four copies of CLFchimera
Conclusions
To improve our understanding of the possible intracellular mechanisms of antimicrobial peptides (AMPs) derived from camel lactoferrin, we planned an in silico study based on molecular dynamics simulation. In this study, DNA was selected as a well-known intracellular target for AMPs. Overall, the simulation results indicated that the chimeric peptide CLFchimera has a higher affinity for DNA than its component peptides CLFcin and CLFampin. It is shown that the interaction between DNA and CLFchimera follows a saturation curve with increasing peptide concentration, the largest gain in binding free energy corresponding, for a 12-bp DNA, to the transition between one and two peptide copies. Saturation is reached after three peptide copies, with four copies inducing the denaturation of the DNA helix.
Binding free energy analysis revealed that the number of lysine and arginine residues (12 in CLFchimera, 4 in CLFampin, 7 in CLFcin) is largely responsible for the higher affinity of the CLFchimera-DNA complex. It is in fact concluded that electrostatic interactions and particularly salt bridges play a pivotal role in the interaction between CLFchimera and DNA. Moreover, nearly all hydrogen bonds between CLFchimera and DNA involved the backbone phosphate groups of DNA, indicating that CLFchimera targets the nucleic acids in a sequence-unspecific manner.
A conformational clustering analysis suggests that CLFchimera mostly performs its activity through the interaction of its two α-helices with DNA. These secondary structure elements, which are stable throughout the simulations, facilitate an optimal presentation of charge density and orientation for the interaction with the phosphate groups. The outcomes of this study provide insight into the structural and dynamic aspects of the interaction between Lactoferrin-derived AMPs and DNA as well as new directions for the design of novel AMPs with intracellular activity.
Availability of data and materials
Data sharing is not applicable to this article as the datasets that support the findings of this study are included within the article and its Additional files.
Abbreviations
- AMP:
-
Antimicrobial peptides
- CLFampin:
-
Camel lactoferrampin
- CLFcin:
-
Camel Lactoferricin
- COM:
-
Center of mass
- CSA:
-
Contacting surface area
- DA:
-
Deoxyadenosine
- DC:
-
Deoxycytidine
- DG:
-
Deoxyguanosine
- DNA:
-
Deoxyribonucleic acid
- DT:
-
Deoxythymidine
- LFampin:
-
Lactoferrampin
- LFcin:
-
Lactoferricin
- LINCS:
-
Linear constraint solver
- MM/PBSA:
-
Poisson-Boltzmann and surface area continuum solvation
- PB:
-
Poisson-Boltzmann
- PDB:
-
Protein data bank
- PME:
-
Particle mesh ewald
- RMSD:
-
Root mean square deviation
- RMSF:
-
Root-mean-square-fluctuations
- SASA:
-
Solvent accessible surface area
- SPC:
-
Simple point charge
References
Wang S, Zeng X, Yang Q, Qiao S. Antimicrobial peptides as potential alternatives to antibiotics in food animal industry. Int J Mol Sci. 2016;17(5):603.
Linde A, Ross C, Davis E, Dib L, Blecha F, Melgarejo T. Innate immunity and host defense peptides in veterinary medicine. J Vet Intern Med. 2008;22(2):247–65.
Skalickova S, Heger Z, Krejcova L, Pekarik V, Bastl K, Janda J, Kostolansky F, Vareckova E, Zitka O, Adam V. Perspective of use of antiviral peptides against influenza virus. Viruses. 2015;7(10):5428–42.
Pirkhezranian Z, Tanhaeian A, Mirzaii M, Sekhavati MH. Expression of Enterocin-P in HEK platform: evaluation of its cytotoxic effects on cancer cell lines and its potency to interact with cell-surface glycosaminoglycan by molecular modeling. Int J Pept Res Ther. 2019;19(4):1–10.
Sang Y, Blecha F. Porcine host defense peptides: expanding repertoire and functions. Dev Comp Immunol. 2009;33(3):334–43.
Baloglu S, Boyle SM, Vemulapalli R, Sriranganathan N, Schurig GG, Toth TE. Immune responses of mice to vaccinia virus recombinants expressing either listeria monocytogenes partial listeriolysin or Brucella abortus ribosomal L7/L12 protein. Vet Microbiol. 2005;109(1–2):11–7.
Tanhaiean A, Azghandi M, Razmyar J, Mohammadi E, Sekhavati MH. Recombinant production of a chimeric antimicrobial peptide in E. coli and assessment of its activity against some avian clinically isolated pathogens. Microb Pathog. 2018;122(5):73–8.
Tanhaeian A, Ahmadi FS, Sekhavati MH, Mamarabadi M. Expression and purification of the Main component contained in camel Milk and its antimicrobial activities against bacterial plant pathogens. Probiotics Antimicrob Proteins. 2018;10(4):1–7.
Daneshmand A, Kermanshahi H, Sekhavati MH, Javadmanesh A, Ahmadian M. Antimicrobial peptide, cLF36, affects performance and intestinal morphology, microflora, junctional proteins, and immune cells in broilers challenged with E. coli. Sci Rep. 2019;9(1):1–9.
Reyes-Cortes R, Acosta-Smith E, Mondragón-Flores R, Nazmi K, Bolscher JG, Canizalez-Roman A, Leon-Sicairos N. Antibacterial and cell penetrating effects of LFcin17–30, LFampin265–284, and LF chimera on enteroaggregative Escherichia coli. Biochem Cell Biol. 2016;95(1):76–81.
Tang Y-L, Shi Y-H, Zhao W, Hao G, Le G-W. Interaction of MDpep9, a novel antimicrobial peptide from Chinese traditional edible larvae of housefly, with Escherichia coli genomic DNA. Food Chem. 2009;115(3):867–72.
Park CB, Kim HS, Kim SC. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun. 1998;244(1):253–7.
Hartmann M, Berditsch M, Hawecker J, Ardakani MF, Gerthsen D, Ulrich AS. Damage of the bacterial cell envelope by antimicrobial peptides gramicidin S and PGLa as revealed by transmission and scanning electron microscopy. Antimicrob Agents Chemother. 2010;54(8):3132–42.
Ho Y-H, Sung T-C, Chen C-S. Lactoferricin B inhibits the phosphorylation of the two-component system response regulators BasR and CreB. Mol Cell Proteomics. 2012;11(4):M111 014720.
Yan J, Wang K, Dang W, Chen R, Xie J, Zhang B, Song J, Wang R. Two hits are better than one: membrane-active and DNA binding-related double-action mechanism of NK-18, a novel antimicrobial peptide derived from mammalian NK-lysin. Antimicrob Agents Chemother. 2013;57(1):220–8.
Uyterhoeven ET, Butler CH, Ko D, Elmore DE. Investigating the nucleic acid interactions and antimicrobial mechanism of buforin II. FEBS Lett. 2008;582(12):1715–8.
Bandyopadhyay S, Lee M, Sivaraman J, Chatterjee C. Model membrane interaction and DNA-binding of antimicrobial peptide Lasioglossin II derived from bee venom. Biochem Biophys Res Commun. 2013;430(1):1–6.
Pandey B, Grover A, Sharma P. Molecular dynamics simulations revealed structural differences among WRKY domain-DNA interaction in barley (Hordeum vulgare). BMC Genomics. 2018;19(1):132.
Eswar N, Webb B, Marti-Renom MA, Madhusudhan M, Eramian D, My S, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006;15(1):5.6. 1–5.6. 30.
Maupetit J, Derreumaux P, Tuffery P. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009;37(suppl_2):W498–503.
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–91.
Berendsen HJ, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1–3):43–56.
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–18.
Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25.
MacKerell AD Jr, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56(4):257–65.
Berendsen HJ, Postma JP, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. In: Intermolecular forces. Dordrecht: Springer; 1981. p. 331–42.
Nosé S, Klein M. Constant pressure molecular dynamics for molecular systems. Mol Phys. 1983;50(5):1055–76.
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–93.
Evans DJ, BLJTJocp H. The nose–hoover thermostat. J Chem Phys. 1985;83(8):4069–74.
Kumari R, Kumar R, Consortium OSDD, Lynn A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J Chem Inf Model. 2014;54(7):1951–62.
Cuervo A, Dans PD, Carrascosa JL, Orozco M, Gomila G, Fumagalli L. Direct measurement of the dielectric polarization properties of DNA. Proc Natl Acad Sci. 2014;111(35):E3624–30.
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33(12):889–97.
Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics Poisson− Boltzmann surface area method. Mol Inform. 2012;31(2):114–22.
Xie N-Z, Du Q-S, Li J-X, Huang R-B. Exploring strong interactions in proteins with quantum chemistry and examples of their applications in drug design. PLoS One. 2015;10(9):e0137113.
Bosshard HR, Marti DN, Jelesarov I. Protein stabilization by salt bridges: concepts, experimental approaches and clarification of some misunderstandings. J Mol Recognit. 2004;17(1):1–16.
Kumar S, Nussinov R. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys J. 2002;83(3):1595–612.
Barlow DJ, Thornton J. Ion-pairs in proteins. J Mol Biol. 1983;168(4):867–85.
Gromiha MM. Protein bioinformatics: from sequence to function: academic press; 2010.
Zeng S, Zhou G, Guo J, Zhou F, Chen J. Molecular simulations of conformation change and aggregation of HIV-1 Vpr13-33 on graphene oxide. Sci Rep. 2016;6:24906.
van der Kraan MI, van der Made C, Nazmi K, van’t Hof W, Groenink J, Veerman EC, Bolscher JG, Amerongen AVN: Effect of amino acid substitutions on the candidacidal activity of LFampin 265–284. Peptides 2005, 26(11):2093–2097.
van der Kraan MI, Nazmi K, Teeken A, Groenink J, van't Hof W, Veerman EC, Bolscher JG, Amerongen AVN. Lactoferrampin, an antimicrobial peptide of bovine lactoferrin, exerts its candidacidal activity by a cluster of positively charged residues at the C-terminus in combination with a helix-facilitating N-terminal part. Biol Chem. 2005;386(2):137–42.
Chan DI, Prenner EJ, Vogel HJ. Tryptophan-and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim Biophys Acta. 2006;1758(9):1184–202.
Sim S, Wang P, Beyer BN, Cutrona KJ, Radhakrishnan ML, Elmore DE. Investigating the nucleic acid interactions of histone-derived antimicrobial peptides. FEBS Lett. 2017;591(5):706–17.
Hsu C-H, Chen C, Jou M-L, Lee AY-L, Lin Y-C, Yu Y-P, Huang W-T, Wu S-H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005;33(13):4053–64.
Khabiri M, Freddolino PL. Deficiencies in molecular dynamics simulation-based prediction of protein–DNA binding free energy landscapes. J Phys Chem B. 2017;121(20):5151–61.
Acknowledgments
We are grateful to Faezeh Fatemizadeh and Oscar Conchillo Solé for their contribution to manuscript preparation.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
ZP carried out the project, performed bioinformatics analyses and wrote the manuscript. MT was the corresponding author and contributed to manuscript preparation. XD designed the experiments and revised the final draft. HM conceived the study and commented on the manuscript. MHS was the corresponding author and contributed to manuscript preparation. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Second replicate: Structural fluctuation analysis. (A) RMSD as function of time; (B) RMSF per residue.
Additional file 2: Figure S2.
Third replicate: Structural fluctuation analysis. (A) RMSD as function of time; (B) RMSF per residue.
Additional file 3: Figure S3.
Second replicate: COM distance analysis. (A) COM distances between CLFcin, CLFampin and CLFchimera and DNA along 200 ns. (B) Structures at times t = 0 (cyan) and t = 200 ns (purple): (B1) CLFcin-DNA, (B2) CLampin-DNA, and (B3) CLFchimera-DNA.
Additional file 4: Figure S4.
Third replicate: COM distance analysis. (A) COM distances between CLFcin, CLFampin and CLFchimera and DNA along 200 ns. (B) Structures at times t = 0 (cyan) and t = 200 ns (purple): (B1) CLFcin-DNA, (B2) CLampin-DNA, and (B3) CLFchimera-DNA.
Additional file 5: Figure S5.
second replicate: Number of hydrogen bonds with DNA as function of simulation time (200 ns). (A) CLFampin, (B) CLFcin, (C) CLFchimera.
Additional file 6: Figure S6.
Third replicate: Number of hydrogen bonds with DNA as function of simulation time (200 ns). (A) CLFampin, (B) CLFcin, (C) CLFchimera.
Additional file 7: Table S1.
The value of hydrogen bond, salt bridge and contacting surface area for three replicates.
Additional file 8: Figure S7.
Second and Third replicates: Contacting surface area between peptide and DNA along a 200 ns MD simulation.
Additional file 9: Figure S8
. Second and Third replicates: Estimated binding free energy for the peptide-DNA systems. Calculated with the MM/PBSA method on the 100–200 ns period of one of the simulation replicates.
Additional file 10: Figure S9
. Second and Third Replicates: Contribution to DNA binding free energies of amino-acid residues in CLFchimera.
Additional file 11: Figure S10.
Minimum distances during 200 ns simulation.
Additional file 12: Figure S11.
Second replicate: Number of hydrogen bonds with DNA at different concentrations of CLFchimera, (A) CLFchimera, (B) 2-CLFchimera, (C) 3-CLFchimera, (D) 4-CLFchimera.
Additional file 13: Figure S12.
Third replicate: Number of hydrogen bonds with DNA at different concentrations of CLFchimera, (A) CLFchimera, (B) 2-CLFchimera, (C) 3-CLFchimera, (D) 4-CLFchimera.
Additional file 14: Table S2.
Interaction results from MD simulations of CLF-chimera concentration with DNA.
Additional file 15: Figure S13.
Second and Third replicates: Contacting surface area at different concentrations of CLFchimera.
Additional file 16: Figure S14.
Second and Third replicates: Contribution of amino-acid residues to DNA-binding energy at different concentrations of CLFchimera.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pirkhezranian, Z., Tahmoorespur, M., Daura, X. et al. Interaction of camel Lactoferrin derived peptides with DNA: a molecular dynamics study. BMC Genomics 21, 60 (2020). https://doi.org/10.1186/s12864-020-6458-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-6458-7