
Abstract
Background
Biological functions of biomolecules rely on the cellular compartments where they are located in cells. Importantly, RNAs are assigned in specific locations of a cell, enabling the cell to implement diverse biochemical processes in the way of concurrency. However, lots of existing RNA subcellular localization classifiers only solve the problem of single-label classification. It is of great practical significance to expand RNA subcellular localization into multi-label classification problem.
Results
In this study, we extract multi-label classification datasets about RNA-associated subcellular localizations on various types of RNAs, and then construct subcellular localization datasets on four RNA categories. In order to study Homo sapiens, we further establish human RNA subcellular localization datasets. Furthermore, we utilize different nucleotide property composition models to extract effective features to adequately represent the important information of nucleotide sequences. In the most critical part, we achieve a major challenge that is to fuse the multivariate information through multiple kernel learning based on Hilbert-Schmidt independence criterion. The optimal combined kernel can be put into an integration support vector machine model for identifying multi-label RNA subcellular localizations. Our method obtained excellent results of 0.703, 0.757, 0.787, and 0.800, respectively on four RNA data sets on average precision.
Conclusion
To be specific, our novel method performs outstanding rather than other prediction tools on novel benchmark datasets. Moreover, we establish user-friendly web server with the implementation of our method.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Background
Biological functions of biomolecules rely on various cellular compartments. One cell can be divided into different compartments that are related to different biological processes. Thus, the cellular role of one RNA molecular could be inferred from its localization information. What’s more, there has been a great deal of research on the protein subcellular localization [1–6]. Currently, the biological technology capable of whole-genome that subcellular localization has been indicated to be a fundamental regulation mode in biological cells [7].
With the explosive growth of biological sequences in the post-genomic era, one of the most important but also most difficult problems in computational biology is how to express a biological sequence with a discrete model or a vector, yet still keep considerable sequence-order information or key pattern characteristic. This is because all the existing machine-learning algorithms, such as Optimization algorithm [8], Covariance Discriminant algorithm [9, 10], Nearest Neighbor algorithm [11], and Support Vector Machine algorithm [11, 12]) can only handle vectors as elaborated in a comprehensive review [13]. However, a vector defined in a discrete model may completely lose all the sequence-pattern information. To avoid completely losing the sequence-pattern information for proteins, the pseudo amino acid composition [14] or PseAAC [15] was proposed. Ever since the concept of Chou’s PseAAC was proposed, it has been widely used in nearly all the areas of computational proteomics [16–18]. Because it has been widely and increasingly used, four powerful open access soft-wares, called ‘PseAAC’ [19], ‘PseAAC-Builder’ [20], ‘propy’ [21], and ‘PseAAC-General’ [22], were established: the former three are for generating various modes of Chou’s special PseAAC [23]; while the 4th one for those of Chou’s general PseAAC[24], including not only all the special modes of feature vectors for proteins but also the higher level feature vectors such as Functional Domain mode, Gene Ontology mode, and Sequential Evolution or Position-Specific Score Matrix(PSSM) mode. Encouraged by the successes of using PseAAC to deal with protein/peptide sequences, the concept of PseKNC (Pseudo K-tuple Nucleotide Composition) [25] was developed for generating various feature vectors for DNA/RNA sequences [26–28] that have proved very useful as well. Particularly, in 2015 a very powerful web-server called Pse-in-One [29] and its updated version Pse-in-One2.0 [30] have been established that can be used to generate any desired feature vectors for protein/peptide and DNA/RNA sequences according to the need of users’ studies. Inspired by the Chou’s method[31, 32], we mainly extract the frequency information of the sequence.
Currently, the biological technology capable of whole-genome localization is the subcellular RNA sequencing, called SubcRNAseq, which yields high-throughput and quantitative data. Large amounts of raw subcRNAseq data have recently become available, most notably from the ENCODE consortium. A lot of research work has established the resource to make RNA localization data available to the broader scientific community. Firstly, Zhang et al. [33] built a database called RNALocate, which collected more than 42,000 manually engineered RNA subcellular localization entries. Subsequently, Mas-Ponte et al. [34] constructed a database named LncATLAS to store the subcellular localization of lncRNA. ViRBase[35] is a resource for studying ncRNA-associated interactions between virus and host. Now, Huang et al.[36] have built a manually curated resource of experimentally supported RNAs with both protein-coding and noncoding function.
Considering expensive and inconvenient biological experiments [37], automatic computational tools are the highly relevant measure to speed up RNA-related studies. The computational identification of subcellular localization has been a hot topic for the last decade. In the early days, Cheng et al. [38] systematically studied the distribution of lncRNA localization in gastric cancer and revealed its relationship with gastric cancer. As a pioneer work, Feng et al. [39] developed a computational method to predict the organelle positions of non-coding RNA (ncRNAs) by collecting ncRNAs from centroids, mitochondria, and chloroplast genomes. Subsequently, Zhen et al. [40] developed lncLocator to predict the subcellular localization of long-stranded non-coding RNA. Xiao et al. [41] proposed a novel method used the sequence-to-sequence model to predict microRNA subcellular localization. Besides, Yang et al. [42] developed MiRGOFS being a GO-based functional similarity measurement for miRNA subcellular localization. Then, iLoc-mRNA [43] used binomial distribution and one-way analysis of variance to obtain the optimal nonamer composition of mRNA sequences, and applies a predictor to identify human mRNA subcellular localization. Recently, deep learning methods [44–47] have been used to predict subcellular localization with good results.
However, most existing RNA subcellular localization classifiers only solve the problem of single-label classification. In fact, a single primary RNA transcript is used to make multiple proteins [48–50]. Therefore, it is of great practical significance to expand RNA subcellular localization into multi-label classification problem. In view of the above research, there is no multi-label RNA subcellular localization dataset available for this task. According to RNALocate database, we extract multi-label classification datasets about RNA-associated subcellular localizations on various types of RNAs, and then construct subcellular localization datasets on four RNA categories (mRNAs, lncRNAs, miRNAs and snoRNAs).
In this study, we utilize different nucleotide property composition models to adequately represent important information of nucleotide sequences. In the most critical part, we achieve a major challenge is to fuse the multivariate information through multiple kernel learning[51–58], based on Hilbert-Schmidt independence criterion. The optimal combined kernel can be put into an integration support vector machine model for training a multi-label RNA subcellular localization classifier. We follow Chou’s 5-steps rule [24] to go through the following five steps: (1) construct a valid benchmark dataset to train and test the predictor; (2) utilize different nucleotide property composition models to adequately represent important information of nucleotide sequences; (3) achieve a major challenge is to fuse the multivariate information through multiple kernel learning based on Hilbert-Schmidt independence criterion, and the optimal combined kernel can be put into an integration support vector machine model for training a multi-label RNA subcellular localization classifier; (4) properly perform cross-validation tests to objectively evaluate the anticipated prediction accuracy; (5) establish multiple user-friendly web-servers for different datasets.
Results
In this section, we compare various nucleotide representations, integration strategies and classification tools on our novel benchmark datasets.
Evaluation measurements
Ten-fold cross-validation is a statistical technique to evaluate the performance of models in turn. Six parameters are used to analyze the performance of model [59], including Average Precision (AP), Accuracy (Acc), Coverage (Cov), Ranking Loss (Lr), Hamming Loss (Lh) and One-error (Eone).
where |D| represents the number of samples, |L| represents the number of labels, \(\hat {r}(y)\) indicates the rank of y in Y on the descending order, \(\hat {f}(y)\) represents the score of y predicted by the classifier, Y represents the real label set, \(\hat {Y}\) represents the prediction label set, \(\bar {Y}\) denotes the complementary set of Y, Δ stands for the symmetric difference between two label sets.
For Coverage, Ranking Loss, Hamming Loss and One-error, the model can achieve the best performance with the smallest value. For Average Precision and Accuracy, the model can achieve the best performance with the largest value.
Performance of different nucleotide representations
We analyze seven different nucleotide property composition representations via 10-fold cross validation. Here, we compare single-kernel feature models on four RNA subcellular localization datasets, as shown in Table 1. It can be observed that kmer achieves best performance on mRNAs (AP:0.688) and lncRNAs (AP:0.745), NAC obtains best performance on miRNAs (AP:0.785), and DNC gains best performance on snoRNAs (AP:0.793). Details are shown in Additional file 1: Table S5. Also, we compare single-kernel feature models on four human RNA subcellular localization datasets, as shown in Table 2. It can be noticed that kmer achieves best performance on mRNAs (AP:0.750), lncRNAs (AP:0.753), and snoRNAs (AP:0.817), CKSNAP obtains best performance on miRNAs (AP:0.784). Details are shown in Additional file 1: Table S6.
In order to further analyze characteristics, we make use of random forest (RF) to calculate the importantce score of each feature dimension. On four RNA datasets, feature scores of mRNAs have more balanced overall distribution, but feature scores of miRNAs and snoRNAs have irregular distributions, as shown in Fig. 1. This phenomena is also reflected on four human RNA dataset, as shown in Fig. 2. It indicates that miRNAs and snoRNAs have shorter sequences with less regular nucleotide property composition information.

Feature importantce scores of seven characteristics on four RNA datasets

Feature importantce scores of seven characteristics on four human RNA datasets
Performance of different integration strategies
We study five different integration strategies with SVM model as base classifier via 10-fold cross validation, including binary relevance (BR) [59], ensemble classifier chain (ECC) [60], label powerest (LP) [59], multiple kernel learning with average weights (MK-AW), multiple kernel learning with Hilbert-Schmidt independence criterion (MK-HSIC).
Here, we compare five integrated SVM strategies on four RNA subcellular localization datasets, as shown in Table 3. It can be observed that MKSVM-HSIC achieves best performance on mRNAs (AP:0.703), lncRNAs (AP:0.757), miRNAs (AP:0.787), and snoRNAs (AP:0.800). Details are shown in Additional file 1: Table S7. Also, we compare five integrated SVM strategies on four human RNA subcellular localization datasets, as shown in Table 4. It can be observed that MK-HSIC achieves best performance on mRNAs (AP:0.755), lncRNAs (AP:0.754), miRNAs (AP:0.791), and snoRNAs (AP:0.816). Details are shown in Additional file 1: Table S8. Overall accuracy of our integration strategy is significantly higher than that of other four strategies. It can be found that multiple kernel learning has an obvious advantage over other general integration strategies in dealing with classification problems.
According to MK-HSIC strategy, we optimize all weights of effective kernels, in order to improve the correlation between optimal combined kernel and ideal kernel. All weights for seven kernels are shown in Fig. 3. Details are shown in Additional file 1: Table S9. On miRNAs dataset, KKmer1234 has highest kernel weight, and KNAC has second highest kernel weight. On human miRNAs dataset, KNAC has highest kernel weight. On other six dataset, KDNC similarly has highest kernel weights.

Weights for seven different kernels on various RNA datasets
Comparison with existing classification tools
We compare the performance of different classifiers for solving multi-label classification problem via 10-fold cross validation. We use all feature sets for training SVM [61], RF [40], ML-KNN [59], extreme gradient boosting (XGBT) [62], multi-layer perceptron (MLP) [63].
Here, we compare six classification methods on four RNA subcellular localization datasets, as shown in Table 5. It can be observed that MKSVM-HSIC achieves best performance on mRNAs (AP:0.703), lncRNAs (AP:0.757) and miRNAs (AP:0.787), and XGBT obtains best performance on snoRNAs (AP:0.806). Details are shown in Additional file 1: Table S10. Also, we compare six classification methods on four human RNA subcellular localization datasets, as shown in Table 6. It can be noticed that MKSVM-HSIC achieves best performance on mRNAs (AP:0.755), lncRNAs (AP:0.754), miRNAs (AP:0.791), and snoRNAs (AP:0.816). Details are shown in Additional file 1: Table S11. As is clearly reflected by the chart, MKSVM-HSIC achieved best performance on different RNA datasets, and XGBT and RF also have good prediction results. It proves that our novel method is valid, and our new benchmark dataset is correct and meaningful.
In order to analyze the stability, we perform T-check on MKSVM-HSIC via 10-fold cross validation. We calculate mean value and standard deviation of Average Precision, Accuracy, Coverage, Ranking Loss, Hamming Loss and One-error, as shown in Fig. 4 on RNA dataset and Fig. 5 on human RNA dataset. It can be seen that the variance of MKSVM-HSIC is small, so the stability and robustness of our method is very excellent. Details are shown in Additional file 1: Table S12.

The robustness of our novel method on four RNA datasets

The robustness of our novel method on four human RNA datasets
Importantly, RNAs are assigned in specific locations of a cell, enabling the cell to implement diverse biochemical processes in the way of concurrency. To be specific, our novel method performs outstanding rather than other prediction tools on our novel benchmark datasets. Moreover, we establish user-friendly web server with the implementation of our method.
Web server
A web server is built for the new proposed method in this paper, the URL is http://lbci.tju.edu.cn/Online_services.htm, including four servers: LocmRNA, LocmiRNA, LocmiRNA and LocsnoRNA. Each one supports two prediction formats, an on-line input single sequence or an entire multiple sequence upload file. The sequence format must be.fasta. It will return the possibility of each label for RNA subcellular localization, and also give the suggested labels as final prediction result.
Conclusion
In this paper, we establish multi-label benchmark data sets for various RNA subcellular localizations to verify prediction tools. Furthermore, we design an integration SVM prediction model with one-vs-rest strategy to fuse a variety of nucleic acid sequence to identify RNA subcellular localization. Finally, we propose user-friendly web server with the implementation of our method, which is a useful platform for research community. However, we only consider the frequency information of the sequence, and more characteristic information can be added in the future.In addition, deep learning can be introduced to solve the problem of multiple tags and multiple classifications, which may have good results.
Methods
In this study, we establish RNA subcellular localization datasets, and then propose an integration learning model for multi-label classification. The flowchart of our method is show in Figure S1.
Benchmark dataset
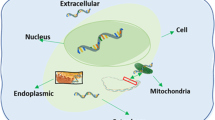
RNAs are generally divided into two categories. One is encoding RNAs, such as messenger RNAs (mRNAs), which play a very important role in transcription. Other is non-coding RNAs, including long non-coding RNA (lncRNA), microRNA (miRNA), small nucleolar RNA (snoRNA), which play an irreplaceable regulatory role in life. In order to study subcellular localization for Homo sapiens, we further establish human RNA subcellular localization datasets. Subcellular localizations of various RNAs in cells are shown in Fig. 6.

Schematic diagram of RNA subcellular localizations in cells
We use the database of RNA subcellular localization in order to integrate, analyze and identify RNA subcellular localization for speeding up RNA structural and functional researches. The first release of RNALocate (http://www.rna-society.org/rnalocate/) contains more than 42,000 manually engineered RNA-associated subcellular locali- zation and experimental evidence entries in more than 23100 RNA sequences, 65 organisms (e.g., homo sapiens, mus musculus, saccharomyces cerevisiae), localization of 42 subcells (e.g., cytoplasm, nucleus, endoplasmic reticulum, ribosomes), and 9 RNA categories (e.g., mRNA, microRNA, lncRNA, snoRNA). Thus, RNALocate provides a comprehensive source of subcellular localization and even insight into the function of hypothetical or new RNAs. We extract multi-label classification datasets about RNA-associated subcellular localizations on four RNA categories (mRNAs, lncRNAs, miRNAs and snoRNAs). The flowchart of mRNA subcellular localization dataset construction framework is shown in Fig. 7.

The flowchart of mRNA subcellular localization dataset construction framework
RNA subcellular localization datasets
We extract four RNA subcellular localization datasets, including mRNAs, lncRNAs, miRNA and snoRNAs. The procedure for constructing RNA datasets is listed as follows.
-
We download total RNA entries with curated subcellular localizations from RNAlocate, and use CD-HIT [64] to remove redundant samples with a cutoff of 80%.
-
We delete samples with duplicate Gene ID and remove samples without corresponding subcellular localization labels, and then construct four RNA subcellular localization datasets.
-
We count the number of samples for each category of subcellular localization labels, and then select some categories with the sample size greater than a reasonable threshold (N/Nmax>1/30).
The statistical distributions of these four RNA datasets are shown in Fig. 8. Details are shown in Additional file 1: Table S1-S2.

The statistical distributions of four RNA subcellular localization datasets
Human RNA subcellular localization datasets
We also extract four Homo sapiens RNA subcellular localization datasets, including H_mRNAs, H_lncRNAs, H_miRNA and H_snoRNAs. The procedure for constructing human RNA datasets is listed as follows.
-
We screen out samples of homo sapiens on above four RNA datasets, and construct four human RNA subcellular localization datasets.
-
We count the number of samples for each category, and then select some categories with the sample size greater than a reasonable threshold (N/Nmax>1/12).
The statistical distributions of these four human RNA datasets are shown in Fig. 9. Details are shown in Additional file 1: Table S3-S4.

The statistical distributions of four human RNA subcellular localization datasets
Nucleotide property composition representation
RNA sequence can be represented as follow: S=(s1,⋯,sl,⋯,sL), where sl denotes the l-th ribonucleic acid and L denotes the length of S. How to formulate varied length RNA sequences as fixed length features, is the key point to effective operational problem-solving. Many studies have shown that the RNA sequence can be encoded by nucleotide property composition representation [65], which can profoundly affect the way of body behaves. Here, we encode the RNA sequence in order to better mine and explore information patterns.
k-mer nucleotide composition
For k-mer descriptor, RNAs are represented as occurrence frequencies of k neighboring nucleic acids, which has been successfully applied to human gene regulatory sequence prediction and enhancer identification. The k-mer (e.g. k=2) descriptor can be calculated as follows.
where N(t) is the number of k-mer type t, while N is the length of a nucleotide sequence.
For k=1,2,3,4, there are four combinations together, each of which has 4k distinct types of nucleotide characteristics. Therefore, we extract 340-dimensional feature vector Fkmer1234.
Only remaining 4-mer, there are 44 types of nucleotide characteristics. Therefore, we extract 256-dimensional feature vector Fkmer4.
Reverse compliment k-mer
The reverse compliment k-mer (RCKmer) is a variant of k-mer descriptor, which is not expected to be strand-specific. For instance, there are 16 types of 2-mer (‘AA’, ‘AC’, ‘AG’, ‘AT’, ‘CA’, ‘CC’, ‘CG’, ‘CT’, ‘GA’, ‘GC’, ‘GG’, ‘GT’, ‘TA’, ‘TC’, ‘TG’, ‘TT’), ‘TT’ is reverse compliment with ‘AA’. After removing the reverse compliment k-mer, there are only 10 distinct types of k-mer in the reverse compliment k-mer approach (‘AA’, ‘AC’, ‘AG’, ‘AT’, ‘CA’, ‘CC’, ‘CG’,‘GA’, ‘GC’, ‘TA’).
For 4-mer with 256 types, after removing reverse compliment 4-mer, there are 136 distinct types in the reverse compliment k-mer approach. Therefore, we extract 136-dimensional feature vector FRCKmer.
Nucleic acid composition
The nucleic acid composition (NAC) encodes the frequency of each nucleic acid type in a nucleotide sequence, which is similar to 1-mer. The frequency of each natural nucleic acid (‘A’, ‘C’, ‘G’, ‘T’ or ‘U’) can be calculated as follows.
where N(t) is the number of nucleic acid type t, while N is the length of a nucleotide sequence.
Therefore, we extract 4-dimensional feature vector FNAC.
Di-nucleotide composition
The di-nucleotide composition (DNC) encodes the frequency of each 2-tuple of nucleic acid type in a nucleotide sequence, which is similar to 2-mer. The frequency of each 2-tuple of natural nucleic acid can be calculated as follows.
where Nij is the number of di-nucleotide type represented by nucleic acid types i and j.
Therefore, we extract 16-dimensional feature vector FDNC.
Tri-nucleotide composition
The tri-nucleotide composition (TNC) encodes the frequency of each 3-tuple of nucleic acid type in a nucleotide sequence, which is similar to 3-mer. The frequency of each 3-tuple of natural nucleic acid can be calculated as follows.
where Nijk is the number of di-nucleotide type represented by nucleic acid types i, j and k.
Therefore, we extract 64-dimensional feature vector FTNC.
Composition of k-spaced nucleic acid pair
The composition of k-spaced nucleic acid pair (CKSNAP) is used to calculate the frequency of nucleic acid pairs separated by any k nucleic acids (k=0,1,2,…). For each k-space, there are 16 types of nucleic acid pair composition (‘A...A’, ‘A...C’, ‘A...G’, ‘A...T’, ‘C...A’, ‘C...C’, ‘C...G’, ‘C...T’, ‘G...A’, ‘G...C’, ‘G...G’, ‘G...T’, ‘T...A’, ‘T...C’, ‘T...G’, ‘T...T’).
For k=0,1,2,3,4,5, there are six different combinations, each of which has 16 distinct types of nucleic acid pair composition. Therefore, we extract 96-dimensional feature vector FCKSNAP.
Multiple kernel support vector machine classifier
We apply radial basis function (RBF) on above feature sets to construct corresponding kernels, respectively. The RBF kernel is defined as follows.
where xi and xj are the feature vectors of samples i and j, N denotes the number of samples, and γ is the bandwidth of Gaussian kernel.
The kernel set with seven distinct kernels is denoted as follows.
Hilbert-Schmidt Independence criterion-multiple kernel learning
We use multiple kernel learning (MKL) to figure out weights of above kernels, and then integrate them together “Multiple kernel support vector machine classifier”, “??”, and “??” sections. The optimal combinatorial kernel can be calculated as follows.
The main purpose of hilbert-schmidt independence criterion (HSIC) [66] is to measure a difference in the distribution of two variables, which is similar to the covariance and is itself constructed according to the covariance. Let X∈RN×d and Y∈RN×1 be two variables from a data set of \(\mathbf {Z}=\left \{(\mathbf {x}_{i},y_{i})\right \}^{N}_{i=1}\), which is jointly from some probability distribution Prxy. HSIC measures the independence between x and y by calculating the norm of cross-covariance operator over domain X×Y.
Hilbert-Schmidt operator norm “Hilbert-Schmidt Independence criterion-multiple kernel learning” section of Cxy is defined as follows.
Given set Z, empirical estimate of HSIC is computed as follows.
where F is the RKHS of feature set X,G is the RKHS of label set Y,e=(1,...,1)T∈RN×1,H=I−eeT/N∈RN×N (centering matrix), K,U∈RN×N are kernel matrices with Kij=k(xi,xj) and Uij=l(yi,yj),I∈RN×N is the identity matrix. The stronger the dependence between K and U, the larger the value. K and U are independent between each other, when HSIC(K,U)=0.
Enligthened by HSIC [67], we define optimization function as follows.
where K∗∈RN×N is the optimal kernel of feature space, and \(\mathbf {U} = \mathbf {y}_{train}\mathbf {y}_{train}^{T} \in \mathbf {R}^{N \times N}\) is ideal kernel matrix (label kernel), β∈RP×1 is the kernel weight vector. We aim to maximize HSIC between K∗ and U.
Convex quadratic programming problem can be solved as follows.
where ν1 is L2 norm regularization term. The final training and testing kernels are linearly weighted by β, respectively.
Support vector machine
Support vector Machine [68] was first proposed by Cortes and Vapnik [69]. It deals primarily with dichotomies. Given a dataset of instance-label pairs {xi,yi},i=1,2,...,N, the classification decision function realized by SVM is expressed as follows.
where xi∈R1×d and yi∈{+1,−1}.
Solving the following convex Quadratic Programming (QP) problem can obtain the coefficient αi.
where C is a regularization parameter that controls the balance between boundary and misclassification errors, and when the corresponding αj>0,xj is called support vector.
One-vs-rest strategy
We use an indirect strategy to solve multi-label classification problem, which can be solved by converting multi-label problem into multiple binary classification problems. The one-vs-rest strategy is to treat one class as positive samples and the rest classes as negative samples. We can build a binary classifier for each class label, thus construct a total of k binary classifiers.
Availability of data and materials
A web server is built for the new proposed method in this paper, the URL is http://lbci.tju.edu.cn/Online_services.htm, including four servers: LocmRNA, LocmiRNA, LocmiRNA and LocsnoRNA.
Abbreviations
- mRNAs:
-
message RNAs
- miRNAs:
-
micro RNAs
- lncRNAs:
-
long non-coding RNAs
- snoRNAs:
-
small nucleolar RNAs
- Ap:
-
Average Precision
- Acc:
-
Accuracy
- Cov:
-
Coverage
- L r :
-
Ranking Loss
- L h :
-
Hamming Loss
- E one :
-
One-error
- BR:
-
Binary Relevance
- ECC:
-
Ensemble Classifier Chain
- LP:
-
Label Powerest
- MK-AW:
-
multiple kernel learning with average weights
- MK-HSIC:
-
multiple kernel learning with Hilbert-Schmidt independence criterion
- RF:
-
Random Forest
- ML-KNN:
-
Mutil-label K-Nearest Neighbors
- XGBT:
-
extreme gradient boosting
- MLP:
-
multi-layer perceptron
- RCKmer:
-
reverse compliment k-mer
- NAC:
-
nucleic acid composition
- DNC:
-
Di-nucleotide composition
- TNC:
-
Tri-nucleotide composition
- CKSNAP:
-
composition of k-spaced nucleic acid pair
- RBF:
-
radial basis function
- MKL:
-
multiple kernel learning
- HSIC:
-
Hilbert-Schmidt independence criterion
- QP:
-
Quadratic Programming
- SVM:
-
Support vector Machine
References
Chou KC, Shen HB. Large-scale plant protein subcellular location prediction. J Cell Biochem. 2006; 100(3):665–78.
Chou KC, Shen HB. Hum-ploc: A novel ensemble classifier for predicting human protein subcellular localization. Biochem Biophys Res Commun. 2006; 347(1):0–157.
Shen H-B, Chou K-C. Nuc-ploc: a new web-server for predicting protein subnuclear localization by fusing pseaa composition and psepssm. Protein Eng Des Sel Peds. 2007; 20(11):561–7.
Shen H-B, Yang J, Chou K-C. Methodology development for predicting subcellular localization and other attributes of proteins. Expert Rev Proteomics. 2007; 4(4):453–63.
Shen HB, Yang J, Chou KC. Euk-ploc: an ensemble classifier for large-scale eukaryotic protein subcellular location prediction. Amino Acids. 2007; 33(1):57–67.
Shen HB, Chou KC. A top-down approach to enhance the power of predicting human protein subcellular localization: Hum-mploc 2.0. Anal Biochem. 2009; 394(2):269–74.
Ayers D. Long non-coding rnas: Novel emergent biomarkers for cancer diagnostics. J Cancer Res Treat. 2013; 1(2):31–5.
Zhang CT, Chou KC. An optimization approach to predicting protein structural class from amino acid composition. Protn ence. 2010; 1(3):401–8.
Chou KC, Elrod DW. Bioinformatical analysis of g-protein-coupled receptors. J Proteome Res. 2002; 1(5):429.
Chou KC, Cai YD. Prediction and classification of protein subcellular location-sequence-order effect and pseudo amino acid composition. (vol 90, pg1250, 2003). J Cell Biochem. 2004; 91(5):1085.
Lele H, Tao H, Xiaohe S, Wen-Cong L, Yu-Dong C, Kuo-Chen C, Christos O. Predicting functions of proteins in mouse based on weighted protein-protein interaction network and protein hybrid properties. PloS ONE. 2011; 6(1):14556.
Cai YD, Feng KY, Lu WC, Chou KC. Using logitboost classifier to predict protein structural classes. J Theor Biol. 2006; 238(1):172–6.
Chou KC. Impacts of bioinformatics to medicinal chemistry. Med Chem. 2015; 11(3):-.
Chou KC. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins-Struct Function Bioinforma. 2010; 43(3):246–55.
Chou K. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005; 21(1):10–9.
Dehzangi A, Heffernan R, Sharma A, Lyons J, Paliwal K, Sattar A. Gram-positive and gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into chou’ s general pseaac. J Theor Biol. 2015; 364:284–94.
Meher PK, Sahu TK, Saini V, Rao AR. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci Rep. 2017; 7(1):1–12.
Chou KC. Progresses in predicting post-translational modification. Int J Pept Res Ther. 2019; 26(2):1–16.
Shen HB, Chou KC. Pseaac: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal Biochem. 2008; 373(2):386–8.
Du P, Wang X, Xu C, Gao Y. Pseaac-builder: a cross-platform stand-alone program for generating various special chou’s pseudo-amino acid compositions. Anal Biochem. 2012; 425(2):117–9.
Cao D-S, Xu Q-S, Liang Y-Z. propy: a tool to generate various modes of chou’s pseaac. Bioinformatics. 2013; 29(7):960–2.
Du P, Gu S, Jiao Y. Pseaac-general: fast building various modes of general form of chou’s pseudo-amino acid composition for large-scale protein datasets. Int J Mol Sci. 2014; 15(3):3495–506.
Chou KC. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr Proteomics. 2009; 6(4):262–74.
Chou K-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol. 2011; 273(1):236–47.
Chen W, Lei TY, Jin DC, Lin H, Chou KC. Pseknc: a flexible web server for generating pseudo k-tuple nucleotide composition. Anal Biochem. 2014; 456:53–60.
Chen W, Lin H, Chou KC. Pseudo nucleotide composition or pseknc: an effective formulation for analyzing genomic sequences. Mol BioSyst. 2015; 11(10):2620–34.
Bin L, Fan Y, De-Shuang H, Kuo-Chen C. ipromoter-2l: a two-layer predictor for identifying promoters and their types by multi-window-based pseknc. Bioinformatics; 34(1):1.
Tahir M, Tayara H, Chong KT. irna-pseknc(2methyl): Identify rna 2’-o-methylation sites by convolution neural network and chou’s pseudo components. J Theor Biol. 2018; 465:1–6.
Liu B, Wang X, Chen J, Fang L, Chou K-C. Pse-in-one: A web server for generating various modes of pseudo components of dna, rna, and protein sequences. Nucleic Acids Res. 2015; 43. https://doi.org/10.1093/nar/gkv458.
Liu B, Wu H, Chou K-C, et al. Pse-in-one 2.0: an improved package of web servers for generating various modes of pseudo components of dna, rna, and protein sequences. Nat Sci. 2017; 9(04):67.
Xiang C, Shu-Guang Z, Wei-Zhong L, Xuan X, Kuo-Chen C. ploc-manimal: predict subcellular localization of animal proteins with both single and multiple sites. Bioinformatics. 2017; 33(22):3524.
Xiao X, Cheng X, Chen G, Mao Q, Chou KC. plocbal-mgpos: Predict subcellular localization of gram-positive bacterial proteins by quasi-balancing training dataset and pseaac. Genomics. 2019; 111(4):886–92.
Zhang T, Tan P, Wang L, Jin N, Li Y, Zhang L, Yang H, Hu Z, Zhang L, Hu C, et al. Rnalocate: a resource for rna subcellular localizations. Nucleic Acids Res. 2016; 45(D1):135–8.
Mas-Ponte D, Carlevaro-Fita J, Palumbo E, Pulido TH, Guigo R, Johnson R. Lncatlas database for subcellular localization of long noncoding rnas. Rna. 2017; 23(7):1080–7.
Li Y, Wang C, Zhengqiang M, Bi X, Wu D, Jin N, Wang L, Wu H, Qian K, Li C, Zhang T, Zhang C, Yi Y, Lai H, Hu Y, Cheng L, Leung K, li X, Zhang F, Wang D. Virbase: A resource for virus-host ncrna-associated interactions. Nucleic Acids Res. 2014; 43. https://doi.org/10.1093/nar/gku903.
Huang Y, Wang J, Zhao Y, Wang H, Liu T, Li Y, Cui T, Li W, Feng Y, Luo J, Gong J, Ning L, Zhang Y, Wang D, Zhang Y. cncRNAdb: a manually curated resource of experimentally supported RNAs with both protein-coding and noncoding function. Nucleic Acids Res. 2020. https://doi.org/10.1093/nar/gkaa791.
Chou KC, Shen HB. Recent progress in protein subcellular location prediction. Anal Biochem. 2007; 370(1):1–16.
Cheng L, Leung K-S. Quantification of non-coding rna target localization diversity and its application in cancers. J Mol Cell Biol. 2018; 10(2):130–8.
Feng P, Zhang J, Tang H, Chen W, Lin H. Predicting the organelle location of noncoding rnas using pseudo nucleotide compositions. Interdiscip Sci Comput Life Sci. 2017; 9(4):540–4.
Cao Z, Pan X, Yang Y, Huang Y, Shen H-B. The lncLocator: a subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics. 2018; 34(13):2185–94. https://doi.org/10.1093/bioinformatics/bty085.
Xiao Y, Cai J, Yang Y, Zhao H, Shen H. Prediction of microrna subcellular localization by using a sequence-to-sequence model. In: 2018 IEEE International Conference on Data Mining (ICDM). IEEE: 2018. p. 1332–7.
Yang Y, Fu X, Qu W, Xiao Y, Shen H-B. Mirgofs: a go-based functional similarity measurement for mirnas, with applications to the prediction of mirna subcellular localization and mirna–disease association. Bioinformatics. 2018; 34(20):3547–56.
Zhang Z-Y, Yang Y-H, Ding H, Wang D, Chen W, Lin H. Design powerful predictor for mRNA subcellular location prediction in Homo sapiens. Brief Bioinform. 2020. https://doi.org/10.1093/bib/bbz177.
Chou KC, Cheng X, Xiao X. Med Chem. 2018; 15(5):472–85.
Shao Y-T, Liu X-X, Lu Z, Chou K-C. plocdeep-mhum: Predict subcellular localization of human proteins by deep learning. Nat Sci. 2020; 12(7):526–51.
Shao Y-T, Liu X-X, Lu Z, Chou K-C. plocdeep-mplant: Predict subcellular localization of plant proteins by deep learning. Nat Sci. 2020; 12(5):237–47.
Shao Y, Chou K-C. plocdeep-mvirus: A cnn model for predicting subcellular localization of virus proteins by deep learning. Nat Sci. 2020; 12(6):388–99.
Shen HB, Chou KC. Virus-mploc: A fusion classifier for viral protein subcellular location prediction by incorporating multiple sites. J Biomol Struct Dyn. 2010; 28(2):175–86.
Shen HB, Chou KC. Hum-mploc: An ensemble classifier for large-scale human protein subcellular location prediction by incorporating samples with multiple sites. Biochem Biophys Res Commun. 2007; 355(4):0–1011.
Ying-Ying X, Fan Y, Hong-Bin S. Incorporating organelle correlations into semi-supervised learning for protein subcellular localization prediction. Bioinformatics. 2016; 32(14):14.
Wang H, Ding Y, Tang J, Guo F. Identification of membrane protein types via multivariate information fusion with hilbert–schmidt independence criterion. Neurocomputing. 2020; 383:257–69. https://doi.org/10.1016/j.neucom.2019.11.103.
Ding Y, Tang J, Guo F. Human protein subcellular localization identification via fuzzy model on kernelized neighborhood representation. Appl Soft Comput. 2020:106596. https://doi.org/10.1016/j.asoc.2020.106596.
Shen Y, Tang J, Guo F. Identification of protein subcellular localization via integrating evolutionary and physicochemical information into chou’s general pseaac. J Theor Biol. 2018; 462. https://doi.org/10.1016/j.jtbi.2018.11.012.
Ding Y, Tang J, Guo F. Identification of drug-target interactions via dual laplacian regularized least squares with multiple kernel fusion. Knowl-Based Syst. 2020; 204:106254.
Zou Y, Wu H, Guo X, Peng L, Ding Y, Tang J, Guo F. Mk-fsvm-svdd: A multiple kernel-based fuzzy svm model for predicting dna-binding proteins via support vector data description. Curr Bioinforma. 2020. https://doi.org/10.2174/1574893615999200607173829.
Ding Y, Tang J, Guo F. Identification of drug-side effect association via semisupervised model and multiple kernel learning. IEEE J Biomed Health Inf. 2019; 23(6):2619–32.
Ding Y, Tang J, Guo F. Identification of drug-side effect association via multiple information integration with centered kernel alignment. Neurocomputing. 2019; 325:211–24.
Ding Y, Tang J, Guo F. Identification of drug-target interactions via multiple information integration. Inf Sci. 2017; 418:546–60.
Zhang M-L, Zhou Z-H. A review on multi-label learning algorithms. IEEE Trans Knowl Data Eng. 2013; 26(8):1819–37.
Read J, Pfahringer B, Holmes G, Frank E. Classifier chains for multi-label classification. Mach Learn. 2011; 85(3):333.
Su Z-D, Huang Y, Zhang Z-Y, Zhao Y-W, Wang D, Chen W, Chou K-C, Lin H. iLoc-lncRNA: predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics. 2018; 34(24):4196–204. https://doi.org/10.1093/bioinformatics/bty508.
Chen T, He T, Benesty M, Khotilovich V, Tang Y. Xgboost: extreme gradient boosting. R package version 0.4-2. 2015:1–4.
Oh C, Zak SH, Mirzaei H, Buck C, Regnier FE, Zhang X. Neural network prediction of peptide separation in strong anion exchange chromatography. Bioinformatics. 2007; 23(1):114–8.
Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010; 26(5):680–2. https://doi.org/10.1093/bioinformatics/btq003.
Chen Z, Zhao P, Li F, Marquez-Lago TT, Leier A, Revote J, Zhu Y, Powell DR, Akutsu T, Webb GI, et al.ilearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of dna, rna and protein sequence data. Brief Bioinform. 2019; 10:1047–57.
Gretton A, Bousquet O, Smola A, Schölkopf B. Measuring statistical dependence with hilbert-schmidt norms: 2005. https://doi.org/10.1007/11564089_7.
Yamada M, Jitkrittum W, Sigal L, et al.High-dimensional feature selection by feature-wise kernelized lasso. Neural Comput. 2013; 26(1):185–207.
Ding Y, Tang J, Guo F. Identification of drug-target interactions via multiple information integration. Inf Sci. 2017; 418-419:546–60.
Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995; 20(3):273–97.
Acknowledgements
This work is supported by a grant from National Key R&D Program of China (2020YFA0908400, 2020YFA0908401, 2018YFC0910405, 2017YFC0908400) and National Natural Science Foundation of China (NSFC 61772362, 61902271 and 61972280).
Funding
This work is supported by a grant from the National Natural Science Foundation of China (NSFC 61772362, 61902271 and 61972280) and National Key R&D Program of China (2018YFC0910405, 2017YFC0908400, 2020YFA0908400, 2020YFA0908401).
Author information
Authors and Affiliations
Contributions
HW conceived and designed the experiments; YD performed the experiments and analyzed the data; FG wrote the paper; JT and QZ reviewed the manuscript. All authors have read and approved the whole manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No application.
Consent for publication
No application.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
Supplemental charts for the article are in the supplemental data file and include 12 more comprehensive tables and a flowchart.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, H., Ding, Y., Tang, J. et al. Identify RNA-associated subcellular localizations based on multi-label learning using Chou’s 5-steps rule. BMC Genomics 22, 56 (2021). https://doi.org/10.1186/s12864-020-07347-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-07347-7