
Abstract
Background
The gut microbiome can influence life history traits associated with host fitness such as fecundity and longevity. In most organisms, these two life history traits are traded-off, while they are positively linked in social insects. In ants, highly fecund queens can live for decades, while their non-reproducing workers exhibit much shorter lifespans. Yet, when fertility is induced in workers by death or removal of the queen, worker lifespan can increase. It is unclear how this positive link between fecundity and longevity is achieved and what role the gut microbiome and the immune system play in this. To gain insights into the molecular regulation of lifespan in social insects, we investigated fat body gene expression and gut microbiome composition in workers of the ant Temnothorax rugatulus in response to an experimental induction of fertility and an immune challenge.
Results
Fertile workers upregulated several molecular repair mechanisms, which could explain their extended lifespan. The immune challenge altered the expression of several thousand genes in the fat body, including many immune genes, and, interestingly, this transcriptomic response depended on worker fertility. For example, only fertile, immune-challenged workers upregulated genes involved in the synthesis of alpha-ketoglutarate, an immune system regulator, which extends the lifespan in Caenorhabditis elegans by down-regulating the TOR pathway and reducing oxidant production. Additionally, we observed a dramatic loss in bacterial diversity in the guts of the ants within a day of the immune challenge. Yet, bacterial density did not change, so that the gut microbiomes of many immune challenged workers consisted of only a single or a few bacterial strains. Moreover, the expression of immune genes was linked to the gut microbiome composition, suggesting that the ant host can regulate the microbiome in its gut.
Conclusions
Immune system flare-ups can have negative consequence on gut microbiome diversity, pointing to a previously underrated cost of immunity. Moreover, our results provide important insights into shifts in the molecular regulation of fertility and longevity associated with insect sociality.
Similar content being viewed by others

Background
Why and how organisms age are long-standing scientific questions [1,2,3,4]. Senescence is often considered a consequence of the progressive accumulation of molecular damage due to incomplete somatic repair [5], which results in a decline in physiological function including immunity and homeostasis [6]. Endogenous causes of molecular damage are diverse, and include the production of reactive oxygen species (ROS) through metabolic activity, shortening of telomeres and spontaneous errors in replication, transcription and translation [6,7,8,9,10]. Hence, in an environment free of extrinsic risks (e.g., starvation, predation, pathogens), lifespan ultimately depends on two antagonist processes that determine the ability to maintain the soma: the rate of molecular damage and the efficiency at which such molecular damage is repaired [4, 6, 11]. The well-conserved insulin/insulin-like growth factor (IIS) and the target of rapamycin (TOR) signalling pathways are important regulators of longevity that underlie the negative association between somatic maintenance and reproduction observed in most organisms [12, 13]. While some molecular processes underlying lifespan are well understood, the importance of other factors may have been underestimated. For instance, the influence of diet on lifespan via the ISS and TOR pathways is well studied [14], whereas less attention has been paid to the gut microbiome [15] and the immune system. Yet, recent studies suggest that both play an important role. In Drosophila, host fitness is tightly linked to the microbiome and fly guts promote a high microbial diversity. Interactions between gut bacteria affect both fecundity and longevity of Drosophila flies, but in opposite directions [16, 17]. Also in social bees the composition of the gut microbiome community results from a balance between the immune system of the host and bacterial proliferation [18]. The role of immunity alone or in combination with microbial partners are likely important, by rarely considered regulators of lifespan and fecundity [19, 20].
In most organisms, longevity is traded-off with reproduction, but in social insects, these two life-history traits are positively associated [21]: the most fertile individuals in insect societies, the queens, live the longest [22, 23]. Longevity is a phenotypic plastic trait, as the extreme variation in lifespan between the reproductive queen and the non-reproductive worker castes likely arise from the same genetic background [24]. The positive association between fecundity and longevity does not only hold when queens and workers are compared, but is also observed within a single caste. Indeed, differences in lifespan can not only be set during development, but also during the adult life of a social insect. For example, in Temnothorax ants, despite the fact that workers cannot mate, queen removal induces ovary development and the laying of haploid, male-destined eggs in workers, which extends their lifespan by 13% [23]. Moreover, this impact of queen absence on fecundity and survival does not only affect the few fertile workers in the colony, but to some degree all workers, as dominant workers are less able to inhibit ovary development in their nestmates [23, 25]. What remains unclear is, how this lifespan extension, as well as the general positive link between fecundity and longevity, is achieved in social insects on a molecular and physiological level. Social insects provide thus a unique opportunity to investigate intrinsic and extrinsic regulators of lifespan [26].
Here we propose three non-mutually exclusive hypotheses to explain the increase in worker lifespan with fertility in response to queen removal. First, the lifespan extension of fertile workers could be explained by a higher investment into body maintenance. Possibly, fertile workers extend their lifespan by investing more energy into i) a reduction of ROS production, ii) a better resistance to oxidative stress (e.g., via production of antioxidants) [27, 28], and/or iii) better repair mechanisms. The uncoupling or the positive link between longevity and fecundity in social insects suggests a reshaping of those pathways and/or modifications in their regulation compared to solitary organisms [26].
Second, the increase in worker longevity could stem from changes in the microbial community of the gut [29]. IIS and TOR signalling pathways are nutrient sensitive, thus the gut bacteria could affect the regulation of longevity pathways via the digestion and absorption of specific nutrients by the host [30]. In line with this hypothesis, the microbiome diversity changes with biological age, and in Drosophila gut microbiome composition has been causally linked to aging and lifespan [17, 31]. Moreover, gut bacteria can also be beneficial for immune defence. In Caenorhabditis elegans, inoculation through feeding on non-pathogenic bacteria Lactobacilli and Bifidobacteria not only extends lifespan but also improves pathogen resistance [32, 33]. The positive effect of gut bacteria on growth and pathogen resistance in honey bees is related to improved digestion and absorption of nutrients as well as to higher production of antimicrobial substances [30].
Finally, fertility could be linked to a stimulation of the immune system, so that increased pathogen resistance could extend lifespan. In Lasius niger mating activate the immune system of the queen, but in Temnothorax ants, workers that become fertile lay haploid egg without mating. Immunity is typically costly for body maintenance and impairs survival [34]. For instance, a non-lethal immune challenge reduces lifespan in bumblebee workers [20], likely through the production and activity of radical oxygen species (ROS). If the lifespan extension in ant workers would involve molecular changes in immunity, fertile workers should alter expression of immune genes, an effect that might be more detectable when the immune system is provoked.
In this study, we investigated these three hypotheses by studying the molecular regulation of lifespan, and it’s link to immunity and gut microbiome composition in workers of the ant Temnothorax rugatulus [35]. First, we showed in a long-term survival experiment that worker fertility induced by queen removal extends worker lifespan also in our focal species. Then we subjected these fertile and non-fertile workers to a non-lethal immune challenge and compared gut bacterial diversity as well as their transcriptomic responses in the fat body (Fig. 1), the primary organ of systemic immunity in insects [36]. If fertile workers live longer because of an increased investment in body maintenance, we would predict an upregulation of genes involved in molecular repair mechanisms or antioxidant production. If changes in microbial composition of the gut are contributing to the increased longevity of fertile ant workers, we would expect an impact of worker fertility on the composition of the gut microbiome. Finally, the extension in worker lifespan may be explained by an improved immuno-competence. If so, we envisaged that i) the baseline expression of immune genes differs between fertile and non-fertile workers and ii) the immune response following an immune challenge depends on worker fertility. We show below that fertile workers invest more into repair mechanisms and other somatic maintenance functions, especially when their immune system is challenged. This might explain their lifespan extension, particularly when exposed to pathogens. The microbial composition of the gut is not involved in the lifespan shifts following fertility induction. However, the immune challenge had a severe impact on the gut microbiome by slashing microbial diversity, and these changes in the composition of the gut microbiome were linked to the expression of important immune genes.

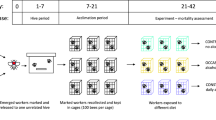
Experimental design. Eight monogynous mother colonies were equally split in two fragments: one containing the queen (queenright: QR) and the other being queenless (QL). After 9 weeks, two workers were sampled from each nest. One was immune-challenged (IC) by pricking with a needle dropped into a 10% lipo-polysaccharide solution (LPS-PGN), while the other non-immune-challenged worker (NIC) received the same manipulation, but the needle only touched the cuticle. Twenty-four hours after the manipulation, the fat body was dissected for RNA extraction, the gut for 16S microbiome sequencing and the ovaries for size measurements. QL workers had longer ovaries than workers from the QR nests (fertile workers F; non-fertile workers NF). The figure was designed by Matteo Negroni using photos of T. rugatulus taken by M. Negroni and S. Foitzik
Results
Influence of queen removal on worker survival and fertility
Queen removal led to an increase in worker survival by 12.5% in queenless colony fragments over 26 weeks (cox survival mixed-effects model: X2 = 18.9; df = 1; P < 0.0001; Fig. 2). Furthermore, T. rugatulus workers in queenless colony fragments showed a 28.6% growth in ovary length within 9 weeks (mean ovariole length ± SD; queenright: 0.49 ± 0.14 mm; queenless: 0.63 ± 0.19 mm; F1 = 5.55, p = 0.027).

Worker survival over time under the presence of the queen (dashed lines) or her absence (solid line). Survival analysis revealed a higher survival in the absence than in the presence of the queen (Cox survival mixed-effects model; x2 = 18.9; df = 1; P < 0.0001)
Influence of fertility and immune challenge on fat body gene expression
We conducted a full-factorial experimental design manipulating the immune system and worker fertility. We challenged the immune system of workers from queenright and queenless experimental colonies by pricking them between the 2nd and 3rd gaster segment with a needle dipped in a 10% lipo-polysaccharide solution. Control workers received the same manipulation, but the needle only touched the cuticle, not penetrating it. After 24 h, we removed the fat body and gut of the focal workers (N = 32) to analyse gene expression via RNAseq and the gut microbiome community by 16S sequencing.
Fertility induction and the immune system provocation interactively altered the expression of 2387 genes (Table S8). More than (55%) half of them displayed opposing expression patterns in response to the queen’s presence and immune challenge (Fig. S1). We consequently compared i) fertile vs. non-fertile workers, separately for the immune and non-immune-challenged workers, and ii) immune-challenged vs. non-challenged workers, separately for fertile and infertile workers [37]. Fertility induction was associated with differential expression of 1466 genes in the immune-challenged workers and 1101 genes in the non-challenged workers (Fig. 3a; Table S9). Conversely, the immune challenge resulted in an altered expression of 2422 genes in non-fertile workers, whereas 1518 genes changed their expression in fertile workers (Fig. 3b; Table S10).

Number of differentially expressed genes and their functional enrichments influenced by the interaction between the immune challenge and the fertility induction: a comparison of upregulated genes in fertile (grey bars) and infertile (white bars) workers within the non-challenged and the immune-challenged treatment groups and irrespective of the immune challenge (non-immune-challenged + immune-challenged) b) comparison of upregulated genes in non-challenged (blue bars) and immune-challenged (red bars) workers, within the non-fertile and fertile treatment groups as well as irrespective from the fertility induction
Both fertility induction and immune challenge led to a changed expression of genes involved in various molecular repair mechanisms (Fig. 3b; Table S12). Fertile workers activated genes of the glutamate degradation into alpha-ketoglutarate pathway in response to the immune challenge (Fig. 3a, b, Table S11, S12). Non-immune-challenged workers stimulated processes involved in oxidative stress reduction (response to oxidative stress, H2O2 catabolism) in response to the fertility induction (Fig. 3a, Table S11), whereas infertile workers upregulated these functions following the immune challenge (Fig. 3b, Table S12).
Transcriptomes clustered mainly by whether or not the immune system of the workers was challenged (Fig. 4), which was reflected in the main effects of the expression analysis. Fertility induction only changed the expression of 4268 genes independently of the immune challenge. Three thousand one hundred twenty-nine genes were upregulated in infertile workers and 1139 in fertile ones, among the latter the vitellogenin receptor (complete list, Table S13). Fertile workers upregulated genes with the function S-adenosylmethionin biosynthetic process (Table S14a, b) This process is not only important for protein synthesis, but also plays a role in the molecular regulation of longevity, as a knockdown of S-adenosylmethionin synthase extends lifespan in Caenorhabditis elegans [38].

Dendrogram showing clustering of samples according to fertility (non-fertile: NF and green; or fertile: F and red), and treatment (non-immune-challenged: NIC and light grey; or immune-challenged: IC and dark grey)
More than twice as many genes (10,717) altered their expression in response to the immune challenge (independently from fertility) than did the fertility induction following the removal of the queen (independently from the immune challenge). Six thousand one hundred thirty-six genes were upregulated in the immune-challenged workers, while 4581 were more highly expressed in non-immune-challenged workers (Table S15). The enrichment analysis of the overexpressed genes in the immune-challenged workers revealed functions related to immunity, such as peptidoglycan catabolic process, defence response, de novo IMP biosynthetic process and innate immune response, or stress reactions such as response to oxidative stress (Fig. 5, Table S14c, d). Immune response was only marginally enriched in immune-challenged workers (P = 0.056). Interestingly, non-immune-challenged workers upregulated genes with enriched functions such as innate immune response and social behaviour.

Gene ontology term (GO) representation of relevant functions from the functional enrichment analysis (significance code: *: < 0.05; **: < 0.01; ***: < 0.001; the dot represents marginal enrichment for a p = 0.056). The number of times a function occurred in the list of upregulated genes for the non-immune-challenged (NIC) workers, immune-challenged (IC) workers, and the expected number (following a random distribution) are coloured blue, red and grey, respectively
Influence of fertility and immune challenge on the gut microbiome
The analysis of pairwise Bray-Curtis dissimilarities between samples revealed no clustering of gut communities based on worker fertility (Fig. 6). Moreover, both the Shannon’s diversity and Simpson’s diversity index did not differ between the gut communities of fertile and infertile ants (Linear mixed-effects model, Fertility: F = 1.75, P = 0.23; Fertility × Immune challenge: F = 1.72, P = 0.21 and Fertility: F = 2.20, P = 0.18; Fertility × Immune challenge: F = 1.82, P = 0.20, respectively). In contrast, most of the gut communities from immune-challenged ants clustered separately from the ants that did not undergo this treatment (Fig. 6). In agreement with these results, the constrained ordination analysis indicated that only the immune challenge and not the fertility induction explained gut community variance (Immune challenge: F = 4.64, P = 0.01; Fertility: F = 0.72, P = 0.71; Fertility × Immune challenge: F = 0.78, P = 0.63). The immune challenge greatly reduced diversity in the microbial communities in the gut as evidence by both the Shannon’s and Simpson’s diversity indices (Linear mixed-effects models, F = 30.12, P < 0.001 and F = 28.18, P < 0.001, respectively; Fig. 7a and b).Three immune-challenged ants showed gut communities composed mainly of a single OTU assigned to the bacterial family Entomoplasmataceae and the intracellular genus Entomoplasma, while the gut communities of seven ants were dominated by Pseudomonaceae, mainly belonging to a single OTU from the genus Pseudomonas (Fig. 6).

Clustering of samples based on the Bray-Curtis dissimilarities using the average linkage method. Clusters with AU values > 98.5 are highly supported clusters and are indicated on the tree with their respective AU values. Red and blue symbols at the tree tips represent the samples from immune-challenged and non-immune-challenged ants, respectively. Fertile and non-fertile workers are indicated by, respectively, circles and triangles. Samples that share the same letter (a to h) originate from ants belonging to the same colony. The relative abundances (≥0.05) of microbial families are plotted next to the samples. The OTUs which did not have a family assigned were binned in the category “na”

Diversity indexes grouped by treatment. The horizontal bars in the boxplots represent the medians and the boxes indicate the first and third quartile. Immune-challenged and non-immune-challenged ants harboured gut communities that differ significantly in a) Shannon’s diversity and b) Simpson diversity index as indicated by the asterisks on the plot. The gut communities of immune-challenged ants had a lower diversity than those of non-immune-challenged ants. c) The number of 16S rRNA copies per ng of total DNA grouped by immunity treatment Samples originating from immune-challenged ants tended to have more copies of the 16S rRNA gene. The samples outside the third quartile are represented by dots and their names, corresponding to those used in Fig. 6, are written on the plot. d) Constrained ordination biplot of the T. rugatulus gut communities showing the association between the expression of the antimicrobial effector genes hymenoptaecin and defensin (TPM) with the bacterial community variance. The length of the arrows reflects the strength of the correlation of each gene with the gut communities
Worker fertility did not affect the relative abundance of microbial families (Table S16), suggesting that the fertility induction did not alter the community composition of the gut. In contrast, 20 of the 64 examined bacterial families contributed less to the gut communities of immune-challenged workers compared to the non-immune-challenged ones (Table S16). The two families that made up the largest parts of the gut community of immune-challenged individuals (Fig. 6), Entomplasmataceae and Pseudomonaceae, are among the few families with a positive log-fold change (Table S16). However, only the increase in the Pseudomonaceae approached significance (adj. P = 0.09). Gram-negative and gram-positive bacteria did not differ in their response to the immune challenge (Pearson’s Chi-squared test, χ2 = 0.16, P = 0.66; Table S17).
The relative abundance of the Aerococcaceae was very similar in the immune-challenged and non-immune-challenged workers, pointing to a possible coevolution between these bacteria and their ant host. Four OTUs were assigned to the Aerococcaceae, but only one was common. An online BLAST search of this OTU sequence against the 16S bacterial/archaeal NCBI database gave the highest identity (100%) with a lactic acid bacterium, Dolosigranulum pigrum; so it is possibly this bacterium or a related species. Lactic acid bacteria are regularly found in insect guts, though most often from the family Lactobacillaceae [39]. One operational taxonomic unit (OTU) of the genus Pseudomonas (Pseudomonaceae) was present in every single gut community. The genus Pseudomonas is known for its metabolic diversity and ability to colonize many niches, including guts. The omnipresence of this OTU makes it also likely to be unintentionally sequenced, but this OTU was on average 23 times more abundant in our experimental samples than in our controls and thus it is unlikely to be a contaminant. A BLAST search of its 16S rRNA sequence gave hits with a variety of Pseudomonas species, but no other known gut inhabitants.
Bacteria belonging to the order Rhizobiales are commonly found in guts of Hymenopterans and have been hypothesized to recycle nitrogen for their host [40, 41]. Among the T. rugatulus gut communities, 12 rhizobial OTUs and five rhizobial families were included in our differential relative abundance tests (Bartonellaceae, Brucellaceae, Methylobacteriaceae, Methylocystaceae and Rhizobiaceae), but only the Methylocystaceae were significantly less abundant in immune-challenged workers (Table S17). The Bartonellaceae and Brucellaceae had a positive log-fold change. Only one OTU belonged to the Bartonellaceae and this OTU was not determined to the genus level. A BLAST search gave no identical hits, but a close hit was found in the NCBI Nucleotide collection. The Bartonellaceae OTU shared most 16S rRNA sequence similarity with a bacterium found in another myrmecine ant [42]. The family Brucellaceae was solely represented by an OTU assigned to the highly diverse genus Ochrobactrum, which encompasses mammalian pathogens, plant and soil associated bacteria, but is also regularly found as part of insect gut communities (e.g. [43, 44]). The immune challenge had no significant effect on the number of 16S rRNA gene copies, although there was a trend that immune-challenged ants harboured more rather than less bacteria (linear mixed-effects model, N = 24, LR = 2.87, P = 0.09; Fig. 7c). The three samples that had the most 16S rRNA copies were from three immune-challenged ants that had a high relative abundance of Entomoplasma in their gut community (Fig. 6).
Interaction between immune gene expression and gut microbiome
Among the genes with an antimicrobial effector functionality (Table S18), seven were significantly upregulated in immune-challenged individuals and three, abaecin, hymenoptaecin and a defensin were associated with the Bray-Curtis distances between gut communities (Table 1). A constrained ordination analysis including only the immune challenge, and the expression (TPM) of abaecin, hymenoptaecin and defensin, explained 31.19% of the variation in gut communities, while hymenoptaecin and defensin remained significant (CAP, hymenoptaecin: F = 6.21, P = 0.01; defensin: F = 2.79, P = 0.01; Fig. 7d), immune challenge and abaecin were no longer significant in this model (backwards elimination, F = 1.59, P = 0.13 and F = 1. 25, P = 0.18, respectively).
Discussion
In Temnothorax ants, as in many other social insects, queen removal induces worker fertility, which can cause an extension in lifespan [23]. In this study, we investigated the independent and combined effects of immune challenge and fertility induction on gut microbiome composition and fat body gene expression. Our gene expression and gut microbiome analyses showed that an immune challenge induced by an injury from a lipopolysaccharide coated needle had a drastic impact on gene expression in the fat body of workers of the ant T. rugatulus, while fertility induction by queen removal had a much smaller effect. Functional enrichment analyses revealed that fertile workers invest more into body maintenance, in particular in molecular repair mechanisms. Transcriptomic responses to the immune challenge depended on worker fertility, though many immune genes were activated in immune-challenged fertile and infertile workers. The immune challenge caused a dramatic drop in gut microbial diversity, while bacterial density remained constant. Microbiome changes were closely linked to changes in the expression of immune genes. Fertility induction did not affect the microbiome of ant workers and thus cannot explain their extended lifespan.
Influence of fertility and immune challenge on fat body gene expression
Many genes altered their expression in response to an interaction between the fertility induction and immune challenge. In the absence of an immune system provocation, fertile workers focussed on degrading reactive oxygen species as indicated by the enriched function hydrogen peroxide catabolism. Yet, immune-challenged fertile workers activated genes of the alpha-ketoglutarate (alpha-KG) pathway. In C. elegans, the immune-enhancing molecule alpha-KG reduces the accumulation of reactive oxygen species in the mitochondria by inhibiting the ATP-synthase (sub-unit V) and reducing energy consumption [38, 45, 46]. Moreover, alpha-KG has been suggested to be an endogenous tumour-suppressor, to downregulate TOR and to extend lifespan in C. elegans [38, 46]. In insects, alpha-KG was not yet found to be involved in aging, but there is evidence that alpha-KG food supplementation enhances antioxidant capacities of female D. melanogaster flies [47]. In addition, in response to the immune challenge, fertile workers upregulate genes with degradation of glutamate for alpha-KG functionalities, while infertile workers activate stress response mechanisms, such as degradation of reactive oxygen species, but also downregulate TOR. TOR signalling has evolved in response to temporal variation in the environment by facilitating plasticity in life history traits, in particular by varying investment into somatic maintenance (somatic maintenance polyphenism [13];). Oxidative stress causes a downregulation of TOR signalling in Drosophila, which in turn increases survival and stress resistance [13, 48]. Thus, a TOR downregulation in response to an immune challenge could increase stress resistance in T. rugatulus.
The transcriptomic changes following fertility induction yielded few candidate genes or enriched functions. The upregulation of a vitellogenin receptor in fertile workers can be explained by the function of these receptors to facilitate vitellogenin up-take of eggs during maturation. Similarly, highly fecund queens of the fire ant Solenopsis invicta overexpress vitellogenin receptors in their ovaries [49]. Also in the ant T. longispinosus the expression of this vitellogenin receptor differs between queen and workers and nurses and foragers [50]. Yet in contrast to our results, vitellogenin receptor expression was independent of worker fertility in this congeneric species. Fertile T. rugatulus workers upregulate genes with the enriched function S-adenosylmethionine biosynthetic processes. S-adenosylmethionin (SAM) accumulates during aging in Drosophila and lifespan is extended by blocking SAM synthesis in the nematode Caenorhabditis or by increasing SAM catabolism in Drosophila flies [51, 52]. The analysis of the interaction between the immune challenge and fertility revealed that fertile workers invest more into molecular repair mechanisms such as telomere maintenance, base-excision repair and nucleotide-excision repair than infertile workers. Since telomere maintenance and DNA damage repair systems are strong predictors of lifespan [8, 53, 54], our findings indicate that fertile workers invest more into somatic maintenance, which could well explain their extended lifespan. Our results show weak changes in the expression of fertility genes in the fat body, indicating that this tissue plays a lesser role in the molecular regulation of fertility. In the absence of immune challenge, we found no difference in immune activity between fertile and non-fertile workers, indicating that the increased survival of fertile workers is not due to a higher basal immune activity, but rather due to an upregulation of DNA damage repair systems and possibly an alteration of the S-adenosylmethionine pathway.
The immune challenge resulted in a stronger transcriptomic response than the fertility indication, as the expression of more than twice as many genes was altered. Not surprising, peptidoglycan catabolism, defence response and immune defence were enriched functions in the genes upregulated in immune-challenged workers. Peptidoglycan is a main component of bacteria cell membrane and it’s catabolism represents a major step in the defence against pathogenic bacteria [55]. Unexpectedly, activation of innate immune response, and inosine monophosphate (IMP) synthesis was enriched among the upregulated genes of non-immune-challenged workers. Supplementation of IMP, a ribonucleotide monophosphate, positively affects survival after an immune challenge in fish [56]. Overall, our data reveal a downregulation of some aspects of the innate immune system in response to a severe immune challenge. This challenge seems to have induced oxidative stress or increased the production of radical oxygen species. We observed a similar pattern in the interaction analysis, as immune-challenged workers activated molecular repair mechanisms (base-excision repair, nucleotide-excision repair, telomere maintenance), which indicate a physiological cost of the immune response. Indeed, an immune reaction is costly, generating reactive oxygen species and molecular damage that have to be compensated [34, 57]. Moreover, an immune response is often accompanied by a proliferation of immune cells - haemocytes in insects [58, 59] - which can shorten telomeres [60], and thus explain the activation of telomere maintenance. All of these observations were made 24 h after the immune challenge. Thus, aspects of an ants’ immune response happening earlier or later would escape our analysis. Analysing the transcriptome of workers sampled at additional time points before and after 24 h would tell us more about the temporal aspect of the transcriptomic response following the immune challenge.
Influence of fertility and immune challenge on the gut microbiome
Animals generally have intimate relationships with their gut microbes. Yet, the physiological, behavioural and hormonal changes that come with an increase in fecundity in worker ants, as evident also in the transcriptomic changes, did not affect the gut microbiome of T. rugatulus workers. Although gene expression of fertile and infertile immune-challenged ants differed markedly, this did not seem to result in differences in antimicrobial peptide production, as the microbiomes in the gut of the immune-challenged ants did not shift based on fertility. Thus, our study did not point to a role of the gut microbiome for the observed lifespan extension with worker fertility. This is surprising, as the experimental inoculation with specific gut microbes increased survival and pathogen resistance in C. elegans and A. mellifera [18, 32, 33].
However, the injury and LPS-PGN exposure caused a drastic decrease in the diversity of the gut bacterial community. In Drosophila, NF-κB/Relish-driven antimicrobial peptide expression is repressed to maintain a normal gut microbiome [61, 62]. Consistent with this, an immune hyperactivity affects the healthy gut microbiome in Drosophila and humans, resulting in disease and reduced lifespan [62,63,64]. Additionally, parasites seem capable of modulating antimicrobial peptide synthesis in their host to impair the growth of certain gut bacterial species that would otherwise have prevented the parasite’s establishment [65]. Our results demonstrate that a single immune challenge can cause a sharp decline in gut microbiome diversity in ants, suggesting that spill-over effects of unspecific immune flare-ups can have negative consequences for gut homeostasis.
Gut bacteria benefit their insect hosts in various ways, for example, by aiding in digestion [66, 67], protecting against pathogens [68] and increasing fecundity [69]. Diversity of the colony gut microbiome is linked to high colony productivity in another Temnothorax species, T. nylanderi [70]. Thus, a turnover of the normal gut microbiome following a provocation of the immune system somewhere else in the body could present an additional cost to immune responses in insects, next to the direct energetic costs of immune activity. To the best of our knowledge, this is the first study to demonstrate negative effects of an externally induced systemic immune response on gut bacteria in insects. If the individual survives the injury or infection that triggered the immune flare-up, the gut community may be able to re-establish by re-inoculation of bacterial strains through frequent mouth-to-mouth and mouth-to-anus contact (“trophallaxis”) with nestmates (e.g. [71] and by contact with nest material [72]). It is possible that these options to re-establish a diverse microbiome enable ants to respond so drastically to an immune challenge. The recovery time of the normal gut bacterial community after an immune flare-up should be investigated as a start to assess the costs of a loss of gut bacterial diversity after immune activity. Moreover, whether these costs depend on the bacterial species that dominate the gut after the immune flare-up is a question that should be considered, as our results revealed that two different bacterial families survive the treatment and become dominants. Interesting would also be to determine whether the bacterial strains that dominate the gut after the immune response influences the re-establishment of a normal gut community.
Testing for associations between the gut microbiome composition and the expression of several antimicrobial effector genes, allowed us to identify genes that may have killed gut bacteria after the immune challenge. Although we did not measure the expression of hymenoptaecin, defensin and abaecin in the gut tissue directly, antimicrobial peptides were both locally produced by epithelial cells and systemically in the fat body in Drosophila, after which they were secreted into the haemolymph [73]. Thus, the expression of hymenoptaecin, abaecin and defensin in the fat body may reflect systemic concentrations of their respective proteins and have a measurable effect on the gut microbiome. Insect defensins are mainly active against gram-positive rather than against gram-negative bacteria [74], while abaecin and hymenoptaecin are active against both [74, 75]. Possibly, the piercing of the cuticle itself may have kicked off an unspecific immune-flare-up.
The lack of difference in absolute bacterial abundance between the samples of immune- and non-immune-challenged ants suggests, that after the host immune response wipes out most bacterial species in the gut, unaffected bacteria species can rapidly increase in population size due to a decrease in competition. Similarly, inhibition of the intestinal homeobox gene Caudal in Drosophila led to an overexpression of antimicrobial peptide genes, resulting in the disappearance of the normal commensal bacteria population and the subsequent proliferation of a pathogen [68].
An increase in relative abundances of bacterial taxonomic groups in T. rugatulus after an immune flare-up may indicate special relationships between the ant host and these bacteria, not necessarily mutually beneficial ones. Bacteria from the families Aerococcaceae, Bartonellaceae, Brucellaceae, Entomplasmataceae and Pseudomonaceae did not decrease in relative abundance in the gut communities of immune-challenged ants, suggesting that these bacteria can withstand the immune defences of T. rugatulus. How these bacteria escaped the immune onslaught, and whether this is the result of host-symbiont coevolution, are interesting topics for future research.
Conclusions
Our investigation of the proximate mechanisms regulating fertility and longevity supported some of our earlier predictions, as fertile workers invested more in somatic maintenance and responded differently to an immune challenge. Other expectations such as a role of the gut microbiome in the lifespan extension of fertile workers were not met. Instead, we uncovered an unpredicted dramatic reduction in gut microbial diversity caused by the immune challenge and associated with an upregulation of important immune effectors. Indeed, the injection of lipopolysaccharide challenged the immune system of ant workers so severely, that fat body gene expression responded to it much more than to the induction of fertility. The challenge apparently induced physiological stress such as oxidative stress and molecular damage, which was counter-acted by the activation of stress response and molecular repair mechanisms. The expression of additional genes was linked to an interaction between the two factors. Fertility induction only weakly altered immune investment, but fertile workers invested more into somatic repair, possibly explaining their extended lifespans. Finally, worker fertility did not alter the composition of the gut microbiome, suggesting that the latter is not involved in the lifespan extension of fertile workers. Yet the single immune challenge caused a drastic loss in gut microbiome diversity and as evidence from other Temnothorax ants indicate beneficial effects of a high microbiome diversity in the gut, our study revealed another potential intrinsic cost of an immune reaction, which is the loss of beneficial symbionts.
Methods
Ant collection and maintenance
Colonies of the ant species Temnothorax rugatulus were collected in the Chiricahua Mountains, Arizona USA in August–September 2015 (Table S1). A collection permit was granted by the Coronado National Forest. A total of 53 queenright monogynous colonies were used for our experiments. Colony size varied between 85 and 304 workers (mean = 143.5; SD = 68.4). After collection, colonies were kept individually in boxes (9 × 9 cm) with a humid plaster floor containing an artificial nest-site, which consisted of a Plexiglas perimeter (2 mm high) sandwiched between two microscope slides. Colonies were fed twice per week with honey and crickets and had access to a constant water supply. They were maintained in controlled climatic conditions simulating field conditions. Our study using a non-endangered insect is exempt from requiring ethical approval or permission from national and regional law.
Queen removal and worker survival
In order to study the impact of queen removal on worker survival in our focal species, we conducted a long-term survival experiment, for which we used 45 colonies. Each colony was split in two size-matched fragments (worker number: mean = 39.5; SD = 13.8; Table S1). We labelled 2 to 6 nurses from each fragment with two coloured wire loops around the petiole (0.02 mm, Elektrisola, combination of red, green, blue or silver). Colonies were transferred to new artificial nest boxes and kept at 22 °C, 12 h/12 h light/dark. The survival of the labelled workers was recorded every 2 weeks following colony splitting for 26 weeks. The influence of the queen’s presence on survival was analysed using Cox proportional hazards model, specifically the coxme function of the coxme package implemented in R, Version 3.2.1. The survival model included N of days until death per individual as the response variable, queen presence (Yes, No) as explanatory variable and colony identity and fragment identity as random factors.
Fertility induction and immune challenge
To obtain non-fertile and fertile workers for the immune challenge experiments, we used eight colonies, with a colony size varying between 34 and 93 workers (mean = 62.3; SD = 23.7). To induce worker fertility, each colony was split in half (Table S1). In each fragment four nurses, which are workers caring for the brood, were individually labelled with a coloured wire loop around the petiole (0.02 mm, Elektrisola, red or green). Young Temnothorax workers take over brood care and are most likely to become behaviourally dominant and start reproduction following queen removal [23, 76, 77]. Colonies were then transferred to new artificial nest boxes and kept at 25 °C, 12 h/12 h light/dark for 9 weeks. Queen removal led to a 28.6% increase in worker ovariole length (mean ± SD; queenright: 0.49 ± 0.14 mm; queenless: 0.63 ± 0.19 mm; F1 = 5.55, p = 0.027; (Fig. 1). The number of eggs in development in the ovaries did not differ between the two groups (mean ± standard error; queenright: 0.35 ± 0.13; queenless: 0.38 ± 0.14; F1 = 0.03, p = 0.86). Temnothorax rugatulus workers can lay male-destined, haploid eggs following queen removal and we observed worker-laid eggs in all queenless colonies during the 9 weeks following queen removal (Table S2). Due to their enlarged ovaries, we refer to workers from the queenless treatment as fertile (F; Fig. 1), and those from the queenright as infertile (NF; Fig. 1).
Nine weeks after colony separation, each nest was placed at − 20 °C for 10 min to reduce worker activity. Immediately following, the nest site was opened and each labelled worker was removed with forceps and individually isolated in a Petri dish. From each fragment, two of the four labelled workers received an immune-challenge (IC) by pricking them with a needle (diameter = 0.15 mm, length = 40 mm) dipped in a 10% lipo-polysaccharide (LPS) solution (containing lipo-polysaccharide and peptidoglycan [PGN]; provided by Sigma-Aldrich). Workers were carefully pricked through the thin cuticle between the 2nd and 3rd segment of the gaster. The non-immune-challenged workers (NIC) received the same manipulation, but the needle only touched the cuticle, not penetrating it. Thereafter, all labelled workers were returned to their colony.
Fat body gene expression
Twenty-four hours after the immune challenge, all labelled workers (Fig. 1) were removed from their nest with soft forceps, immediately killed by decapitation and dissected on ice in 1% PBS. The fat body attached to the two segments of the gaster was crushed into 50 μL of TRIZOL and then stored at − 80 °C until RNA extraction. Dissection of each worker took less than 5 min. Ovaries were dissected and photographed with a Leica DFC425 camera at 20x magnification under a stereomicroscope. Ovary length was measured and the developing eggs were counted using Leica software (LAS version 4.5). RNA was extracted using the RNAeasy mini extraction kit (Qiagen).
Library preparation and sequencing of 100 bp paired reads was done according to standard protocol on an Illumina HiSeq 4000 (StarSeq, Mainz, Germany). We sequenced the fat body transcriptomes of eight workers from each of our four treatments, resulting in 32 sequenced samples of 45 million read pairs each (Fig. 1). The raw reads were trimmed using Trimmomatic-v0.36 [78] and quality checked using FastQC-v0.11.5. Subsequently, all paired reads were assembled de novo using Trinity (trinityrnaseq-Trinity-v2.4.0) [79]. The final assembly contained 150.423 contigs, a mean length of 1128 bp, and a mean back mapping rate of 70.65% (details in supplement, Table S3a).
We conducted a blastX search against the non-redundant invertebrate protein database (state September 2017) to annotate the contigs. About 50.42% were annotated in Blast with 24.15% having a unique Blast hit (contig list and Blast annotation Table S3b). After translation of nucleotide sequences into amino-acid sequences with Transdecoder-v3.0.1 (https://github.com/TransDecoder), the gene ontology (GO) and the Kyoto encyclopaedia of genes and genomes (KEGG) term annotation were performed using InterProScan-v5.25–64.0 [80]. Read count per contig and sample was obtained by using RSEM-v1.3.0 [81], and we completed a differential gene expression analysis with the R package of Deseq2-v1.2.10 [82]. We separately tested the effects of fertility induction and immune challenge, and their interaction, while controlling for colony identity. Additionally, among the contigs that were significantly influenced by the fertility x immunity interaction, we compared the different subgroups (any combination of two factors): i) fertile versus infertile workers, separately and successively, within the immune-challenged workers, and within the non-immune-challenged ones; and ii) immune-challenged versus non-immune-challenged workers, separately and successively, within the fertile workers, and within the non-fertile ones. Among the list of differentially expressed genes we searched for candidate genes associated with fertility, longevity and immunity with the help of the literature, by searching for gene names and traits. Using the online tool KEGG Mapper (http://www.genome.jp/kegg/tool/map_pathway1.html), we conducted a pathway analysis for the upregulated contigs of interest (Table S4 and S5). In order to visualize gene expression patterns across samples categories, among the contigs, which were significantly influenced by the fertility x immunity interaction, we used a clustering tool (degPatterns from the ‘DEGreport’ R package) and plotted their relative expression per cluster (Fig. S1).
To identify biological processes associated with DEGs, we performed a functional enrichment analysis with all upregulated contigs. The enrichment analysis was done for all contigs affected by the interaction through completing separate tests for each factor level. The contigs that were upregulated in response to one treatment irrespective of the other treatment (e.g. upregulated in fertile workers compared to infertile ones irrespective of the immune challenge). We used the R package TopGo [83] for the enrichment analysis, and the p-values for each GO term were obtained with a Fisher’s exact test. In order to identify gene networks, a gene co-expression analysis was performed with WGCNA (Zhao et al. 2010) on the top 20,000 contigs with the highest across sample variance (details see supplement). Finally, GO terms enrichment analyses were performed on the contig list of each of the co-expression modules, as described above. All the results of the WGCNA and functional enrichment of each module are presented in the supplement (Table S6, S7 Fig. S2).
Gut microbiome analyses
Immediately after removing the fat bodies we separated the guts without the crop from the rest of the abdomen with clean dissection tools. The guts were stored individually at − 80 °C in 10 μl 1% PBS until DNA extraction. To control for bacterial contaminants, we included three samples from the 1% PBS, in which the ants were dissected: one with only the PBS, another one in which we dipped the tools used for the dissections and one in which we dipped several intact ants to control for bacteria present on the cuticle. It was not possible to wash the ants before dissection, since this might have interfered with their gene expression. As a fourth control, we took a sample from the food of the ants. Before DNA extraction, the guts were frozen in liquid nitrogen and subsequently crushed with sterile plastic mortars. We used an industrial kit (MasterPure™, from EpiCentre, Wisconsin, USA) for DNA extraction (details in supplement).
The amplification and sequencing of the 16S V4 region was performed by StarSEQ GmbH, Mainz, Germany. Amplification was done with the 515f-806rB primer pair. Overlapping, paired-end reads of 250 bp were generated with Illumina MiSeq. De-multiplexing, adapter trimming and quality filtering was done following the normal Illumina MiSeq workflow by StarSEQ. The initial data set contained 505.050 read pairs (available online see data accessibility section).
We mainly used the Uparse 9.2 pipeline [84] to determine operational taxonomic units (OTUs). Paired-end reads were merged (on average 86.2% ± 2.7 SD could be merged per sample) and subsequently filtered (maxEE cut-off set to 1). The “-fastx_uniques” step was used to de-replicate filtered reads, after which we used the “-cluster_otus” step to cluster the sequences into OTUs with a threshold of 97%. This latter step also removes chimeras. Next, the merged, unfiltered reads were mapped to the OTUs with a 97% identity cut-off with the “usearch_global” algorithm (on average 97.7% ± 1.1 SD were mapped to the OTUs per sample). Using Qiime 1.9.1 [85], we allocated taxonomic groups to the OTUs, the Greengenes database gg_13_8 as a reference. Unassigned OTUs and OTUs classified as chloroplasts or mitochondria were removed from the OTU table, but archaeal OTUs were left in. If an OTU was not at least five times more abundant in any experimental sample than in a control sample, it was discarded. Additionally, we excluded OTUs that did not make up at least 0.1% of the community in at least one sample to further eliminate contaminations. The resulting OTU table contained 378 taxonomic units.
Total bacterial 16S copy numbers per ng DNA were quantified by quantitative realtime PCR (qrt-PCR) with universal 16S primers (details see supplement). The total DNA concentration of each sample in ng/μl was determined with a Qubit Fluorimeter (Thermofisher). For each replicate, we calculated the amount of bacterial 16S copies per ng of DNA in a sample. The means of the replicates were log transformed and subsequently compared between non-immune-challenged and immune-challenged individuals using linear mixed-effects (LME) models from the R package nlme, including colony as random effect.
The OTU table was rarefied to a depth of 1500 reads. Using the rarefied table, we calculated the pairwise Bray-Curtis dissimilarities between the samples in Qiime. Samples were clustered based on their Bray-Curtis dissimilarities (average linkage method, 1000 bootstraps) with the pvclust package in R [86]. Clusters assigned AU values higher than 98.5 are highly supported. The OTUs were binned according to microbial family and the relative abundance of each microbial family was calculated for each sample. This data was used to generate a stacked bar plot, which was combined with the pvclust tree plotted with the R package ggtree [87], to visualize the differences in community composition between samples.
To compare the bacterial diversity between the treatments for each sample we calculated the Shannon’s diversity index with the alpha_diversity.py script of Qiime using the rarefied table. Additionally, we computed Simpson’s diversity index, which gives more weight to common species than Shannon’s diversity, with the diversity function of the R package vegan. We tested for an effect of fertility, immune challenge and their interaction on both measures of diversity with LME models: The fragment and the colony of the ant were included as a nested random effect term. Simpson’s diversity index was arcsine square root-transformed to improve normality. Model residuals were visually checked for deviation from normality.
To identify taxonomic groups that differed in relative abundance between treatments we binned the OTUs into their respective microbial families. To restrict the analysis to the more important taxa, the microbial families that were present in less than 15% of the samples were removed from the OTU table. For the differential abundance testing we performed permutation tests as implemented in the fitLogNormal function of the metagenomeSeq R package [88], using 100 permutations. The p-values were adjusted with the “FDR” method. Taxonomic features with adjusted p-values smaller than 0.05 were regarded as significantly different in relative abundance between the treatments.
We were interested whether variation in T. rugatulus gut communities could be explained by the expression of effector genes involved in antimicrobial immunity. Therefore we looked for the presence of such genes among the contigs and checked whether these contigs were significantly differentially higher expressed in immune-challenged compared to non-immune-challenged individuals. If multiple contigs were annotated with the same effector gene name, we chose the contig with the highest average expression (TPM). With the resulting set of contigs we completed a constrained ordination analysis with the capscale function of the R package vegan (distance-based redundancy analysis, Bray-Curtis) [89]. To test for an association between the expressions of the antimicrobial effector genes on the gut communities, we compared two models with an ANOVA for each contig separately: one model with only fixed factors and one model with next to the fixed factors the expression of the contig (TPM).
Availability of data and materials
All sequence data for this study were archived at NCBI’s Short Read Archive (SRA) with links to BioProject accession numbers PRJNA509332 and PRJNA513968 (https://www.ncbi.nlm.nih.gov/bioproject/).
Abbreviations
- ROS :
-
Reactive oxygen species
- IIS:
-
Insulin/insulin-like growth factor
- TOR:
-
Target of rapamycin
- OTU:
-
Operational taxonomic unit
- TPM:
-
Highest average expression
- CAP:
-
Capscale
- Alpha-KG:
-
Alpha-ketoglutarate pathway
- SAM :
-
S-adenosylmethionin
- IMP:
-
Inosine monophosphate
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- GO:
-
Gene ontology
- LME:
-
Linear mixed-effects
- WGCNA:
-
Weighted gene co-expression analysis
- FDR:
-
False discovery rate
- IC:
-
Immune-challenged workers
- NIC:
-
Non-immune challenged workers
- F:
-
Fertile workers
- NF:
-
Infertile workers
References
Weismann. Über die Dauer des Lebens. Am J Psychol. 1890;3(1):105.
Medawar PB. An Unsolved Problem of Biology. Evolution in Health and Disease. London: H.K. Lewis; 1951.
Williams GC. Pleiotropy, natural selection, and the evolution of senescence. Evolution. 1957;11:398–411.
Flatt T, Partridge L. Horizons in the evolution of aging. BMC Biol. 2018;16:1–13.
Gaillard JM, Lemaître JF. An integrative view of senescence in nature. Funct Ecol. 2020;34:4–16.
Finkel T, Holbrook NJ. Oxidants, oxidative stress and biology of ageing. Insight Rev Artic. 2000;408:239–47.
Metcalfe NB, Alonso-Alvarez C. Oxidative stress as a life-history constraint: the role of reactive oxygen species in shaping phenotypes from conception to death. Funct Ecol. 2010;24:984–96.
Heidinger BJ, Blount JD, Boner W, Griffiths K, Metcalfe NB, Monaghan P. Telomere length in early life predicts lifespan. Proc Natl Acad Sci U S A. 2012;109:1743–8.
Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013;75:621–44.
Rattan SIS. Theories of biological aging: genes, proteins, and free radicals. Free Radic Res. 2006;40:1230–8.
Partridge L, Gems D. Mechanisms of ageing: public or private? Nat Rev Genet. 2002;3:165–75.
Narasimhan SD, Yen K, Tissenbaum HA. Converging pathways in lifespan regulation. Curr Biol. 2009;19(15):R657–66.
Flatt T, Amdam GV, Kirkwood TBL, Omholt SW. Life-history evolution and the polyphenic regulation of somatic maintenance and survival. Q Rev Biol. 2013;88:185–218.
Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015;161:106–18.
Ottaviani E, Ventura N, Mandrioli M, Candela M, Franchini A, Franceschi C. Gut microbiota as a candidate for lifespan extension: an ecological/evolutionary perspective targeted on living organisms as metaorganisms. Biogerontology. 2011;12:599–609.
Obata F, Fons CO, Gould AP. Early-life exposure to low-dose oxidants can increase longevity via microbiome remodelling in Drosophila. Nat Commun. 2018;9:975.
Gould AL, Zhang V, Lamberti L, Jones EW, Obadia B, Korasidis N, et al. Microbiome interactions shape host fitness. Proc Natl Acad Sci U S A. 2018;115:E11951–60.
Kwong WK, Moran NA. Gut microbial communities of social bees. Nat Rev Microbiol. 2016;14:374–84.
Fabian DK, Garschall K, Klepsatel P, Santos-Matos G, Sucena É, Kapun M, et al. Evolution of longevity improves immunity in Drosophila. Evol Lett. 2018;2:567–79.
Moret Y, Schmid-Hempel P. Survival for immunity: the price of immune system activation for bumblebee workers. Science. 2000;290:1166–8.
Korb J. Why do social insect queens live so long? Approaches to unravel the sociality-aging puzzle. Curr Opin Insect Sci. 2016;16:104–7.
Heinze J, Schrempf A. Aging and reproduction in social insects - a mini-review. Gerontology. 2008;54:160–7.
Kohlmeier P, Negroni MA, Kever M, Emmling S, Stypa H, Feldmeyer B, et al. Intrinsic worker mortality depends on behavioral caste and the queens’ presence in a social insect. Sci Nat. 2017;104:34.
Keller L, Jemielity S. Social insects as a model to study the molecular basis of ageing. Exp Gerontol. 2006;41:553–6.
Konrad M, Pamminger T, Foitzik S. Two pathways ensuring social harmony. Naturwissenschaften. 2012;99:627–36.
Rodrigues MA, Flatt T. Endocrine uncoupling of the trade-off between reproduction and somatic maintenance in eusocial insects. Curr Opin Insect Sci. 2016;16:1–8.
Sun J, Tower J. FLP Recombinase-mediated induction of cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Mol Cell Biol. 1999;19(1):216–28.
Tasaki E, Kobayashi K, Matsuura K, Iuchi Y. An efficient antioxidant system in a long-lived termite queen. PLoS One. 2017;12:e0167412.
O’Toole PW, Jeffery IB. Gut microbiota and aging. Science. 2015;350:1214–5.
Zheng H, Powell JE, Steele MI, Dietrich C, Moran NA. Honeybee gut microbiota promotes host weight gain via bacterial metabolism and hormonal signaling. Proc Natl Acad Sci U S A. 2017;114:4775–80.
Clark RI, Salazar A, Yamada R, Fitz-Gibbon S, Morselli M, Alcaraz J, et al. Distinct shifts in microbiota composition during Drosophila aging impair intestinal function and drive mortality. Cell Rep. 2015;12:1656–67.
Ikeda T, Yasui C, Hoshino K, Arikawa K, Nishikawa Y. Influence of lactic acid bacteria on longevity of Caenorhabditis elegans and host defense against Salmonella enterica serovar Enteritidis. Appl Environ Microbiol. 2007;73:6404–9.
Komura T, Ikeda T, Yasui C, Saeki S, Nishikawa Y. Mechanism underlying prolongevity induced by bifidobacteria in Caenorhabditis elegans. Biogerontology. 2013;14:73–87.
Flatt T, Garschall K. The interplay between immunity and aging in Drosophila. F1000Res. 2018;7:160.
Negroni MA, Foitzik S, Feldmeyer B. Long-lived Temnothorax ant queens switch from investment in immunity to antioxidant production with age. Sci Rep. 2019;9:7270.
Hoffmann JA. The immune response of Drosophila. Nature. 2003;426:33–8.
Palakurty SX, Stinchcombe JR, Afkhami ME. Cooperation and coexpression: how coexpression networks shift in response to multiple mutualists. Mol Ecol. 2018;27:1860–73.
Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;510:397–401.
Engel P, Moran NA. The gut microbiota of insects - diversity in structure and function. FEMS Microbiol Rev. 2013;37:699–735.
Van Borm S, Buschinger A, Boomsma JJ, Billen J. Tetraponera ants have gut symbionts related to nitrogen-fixing root-nodule bacteria. Proc R Soc B Biol Sci. 2008;49:2023–7.
Segers FH, Kešnerová L, Kosoy M, Engel P. Genomic changes associated with the evolutionary transition of an insect gut symbiont into a blood-borne pathogen. ISME J. 2017;11:1232–44.
Liberti J, Sapountzis P, Hansen LH, Sørensen SJ, Adams RMM, Boomsma JJ. Bacterial symbiont sharing in Megalomyrmex social parasites and their fungus-growing ant hosts. Mol Ecol. 2015;24:3151–69.
Mathew GM, Ju YM, Lai CY, Mathew DC, Huang CC. Microbial community analysis in the termite gut and fungus comb of Odontotermes formosanus: the implication of Bacillus as mutualists. FEMS Microbiol Ecol. 2012;79:504–17.
Aksoy E, Telleria EL, Echodu R, Wu Y, Okedi LM, Weiss BL, et al. Analysis of multiple tsetse fly populations in Uganda reveals limited diversity and species-specific gut microbiota. Appl Environ Microbiol. 2014;80:4301–12.
Wu N, Yang M, Gaur U, Xu H, Yao Y, Li D. Alpha-ketoglutarate: physiological functions and applications. Biomol Ther. 2016;24:1–8.
Fu X, Chin RM, Vergnes L, Hwang H, Deng G, Xing Y, et al. 2-hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 2015;22:508–15.
Bayliak MM, Lylyk MP, Shmihel HV, Sorochynska OM, Manyukh OV, Pierzynowski SG, et al. Dietary alpha-ketoglutarate increases cold tolerance in Drosophila melanogaster and enhances protein pool and antioxidant defense in sex-specific manner. J Therm Biol. 2016;60:1–11. https://doi.org/10.1016/j.jtherbio.2016.06.001.
Teleman AA, Chen YW, Cohen SM. 4E-BP functions as a metabolic brake used under stress conditions but not during normal growth. Genes Dev. 2005;19:1844–8.
Lu HL, Vinson SB, Pietrantonio PV. Oocyte membrane localization of vitellogenin receptor coincides with queen flying age, and receptor silencing by RNAi disrupts egg formation in fire ant virgin queens. FEBS J. 2009;276:3110–23.
Feldmeyer B, Elsner D, Foitzik S. Gene expression patterns associated with caste and reproductive status in ants: worker-specific genes are more derived than queen-specific ones. Mol Ecol. 2014;23:151–61.
Lee BC, Kaya A, Ma S, Kim G, Gerashchenko MV, Yim SH, et al. Methionine restriction extends lifespan of Drosophila melanogaster under conditions of low amino-acid status. Nat Commun. 2014;5:3592.
Obata F, Miura M. Enhancing S-adenosyl-methionine catabolism extends Drosophila lifespan. Nat Commun. 2015;6:8332.
Lucas ER, Keller L. The co-evolution of longevity and social life. Funct Ecol. 2020;34:76–87.
Marra NJ, Stanhope MJ, Jue NK, Wang M, Sun Q, Bitar PP, et al. White shark genome reveals ancient elasmobranch adaptations associated with wound healing and the maintenance of genome stability. Proc Natl Acad Sci U S A. 2019;116:4446–55.
Steiner H. Peptidoglycan recognition proteins: on and off switches for innate immunity. Immunol Rev. 2004;198:83–96.
Song JW, Lim SJ, Lee KJ. Effects of dietary supplementation of inosine monophosphate on growth performance, innate immunity and disease resistance of olive flounder (Paralichthys olivaceus). Fish Shellfish Immunol. 2012;33:1050–4.
Navrotskaya V, Oxenkrug G. Effect of kynurenic acid on development and aging in wild type and vermilion mutants of Drosophila melanogaster. Pharmacol Drug Dev Ther. 2016;1:10.
Buchmann K. Evolution of innate immunity: clues from invertebrates via fish to mammals. Front Immunol. 2014;5:459.
Kutsukake M, Moriyama M, Shigenobu S, Meng XY, Nikoh N, Noda C, et al. Exaggeration and cooption of innate immunity for social defense. Proc Natl Acad Sci U S A. 2019;116:8950–9.
Wong JMY, Collins K. Telomere maintenance and disease. Lancet. 2003;362:983–8.
Dantoft W, Davis MM, Lindvall JM, Tang X, Uvell H, Junell A, et al. The Oct1 homolog nubbin is a repressor of NF-κB-dependent immune gene expression that increases the tolerance to gut microbiota. BMC Biol. 2013;11:99.
Dantoft W, Lundin D, Esfahani SS, Engström Y. The POU/Oct transcription factor Pdm1/nub is necessary for a beneficial gut microbiota and Normal lifespan of Drosophila. J Innate Immun. 2016;8:412–26.
Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140:859–70.
Mistry R, Kounatidis I, Ligoxygakis P. Interaction between familial transmission and a constitutively active immune system shapes gut microbiota in Drosophila melanogaster. Genetics. 2017;206:889–904.
Vieira CS, Waniek PJ, Castro DP, Mattos DP, Moreira OC, Azambuja P. Impact of Trypanosoma cruzi on antimicrobial peptide gene expression and activity in the fat body and midgut of Rhodnius prolixus. Parasit Vectors. 2016;9:119.
Hu Y, Sanders JG, Łukasik P, D’Amelio CL, Millar JS, Vann DR, et al. Herbivorous turtle ants obtain essential nutrients from a conserved nitrogen-recycling gut microbiome. Nat Commun. 2018;9:964.
Salem H, Kreutzer E, Sudakaran S, Kaltenpoth M. Actinobacteria as essential symbionts in firebugs and cotton stainers (Hemiptera, Pyrrhocoridae). Environ Microbiol. 2013;15:1956–68.
Caudal HG, Ryu J, Kim S, Lee H, Bai JY, Nam Y, et al. Innate immune homeostasis by the Homeobox gene Caudal and commensal-gut mutualism in Drosophila. Science. 2008;319:777–82.
Rosengaus RB, Zecher CN, Schultheis KF, Brucker RM, Bordenstein SR. Disruption of the termite gut microbiota and its prolonged consequences for fitness. Appl Environ Microbiol. 2011;77:4303–12.
Segers FHID, Kaltenpoth M, Foitzik S. Abdominal microbial communities in ants depend on colony membership rather than caste and are linked to colony productivity. Ecol Evol. 2019;9:13450–67.
Grüter C, Acosta LE, Farina WM. Propagation of olfactory information within the honeybee hive. Behav Ecol Sociobiol. 2006;60:707–15.
Powell JE, Martinson VG, Urban-Mead K, Moran NA. Routes of acquisition of the gut microbiota of the honey bee Apis mellifera. Appl Environ Microbiol. 2014;80:7378–87.
Zhang LJ, Gallo RL. Antimicrobial peptides. Curr Biol. 2016;26:R14–9.
Bulet P, Hetru C, Dimarcq JL, Hoffmann D. Antibacterial peptides in insects; structure and function. Dev Comp Immunol. 1999;23:329–44.
Casteels P, Ampell C, Jacobs F, Tempst P. Functional and chemical characterization of Hymenoptaecin, an antibacterial polypeptide that is infection-inducible in the honeybee. J Biol Chem. 1993;268:7044.
Brunner E, Kroiss J, Trindl A, Heinze J. Queen pheromones in Temnothorax ants: control or honest signal? BMC Evol Biol. 2011;11:55.
Kohlmeier P, Feldmeyer B, Foitzik S. Vitellogenin-like A–associated shifts in social cue responsiveness regulate behavioral task specialization in an ant. PLoS Biol. 2018;16:e2005747.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512.
Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–40.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Anders S. Analysing RNA-Seq data with the DESeq package. 2011. http://www-huber.embl.de/users/.
Alexa A, Maintainer JR. Package “topGO” type package title enrichment analysis for gene ontology. 2020. https://git.bioconductor.org/packages/topGO.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–8.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–2.
Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. Ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 2017;8:28–36.
Paulson N, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10:1200–2.
Oksanen J, Kindt R, Legendre P, O’Hara B, Simpson GL, Solymos P, Wagner H. R package community ecology package Vegan; 2009.
Acknowledgements
Many thanks to Marion Kever for lab support and Barbara Feldmeyer for her help with ant collection and her general contributions to the project. We are greatly indebted to Thomas Onchuru and Martin Kaltenpoth for providing us with the firebug 16S DNA copy mixture and the universal 16S primers. The authors thank the LOEWE-Centre TBG funded by the Hessen State Ministry of Higher Education, Research and the Arts (HMWK).
Funding
This work was supported by the Humboldt foundation (grant to F.V.), JGU internal university grants and the Deutsche Forschungsgemeinschaft (FO 298/19–1 to S.F.). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
All coauthors conceived the study and developed the experimental design. Ants were collected by S.F. and M.N., and the experiments and sample collection was performed by F.V., F.S. and M.N. The analysis of fat body transcriptomic data was done by M.N. with the help of F.V and F.S, and guided by S.F. The analysis of 16S RNAseq data was performed by F.S. A first draft of the manuscript was written by M.N. and F.S. and all coauthors improved the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
There are no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Additional methodological details, Figures S1 and S2 and Table S2. Description of additional supplementary tables (Tables S1, S3-S18) are also provided.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Negroni, M.A., Segers, F.H.I.D., Vogelweith, F. et al. Immune challenge reduces gut microbial diversity and triggers fertility-dependent gene expression changes in a social insect. BMC Genomics 21, 816 (2020). https://doi.org/10.1186/s12864-020-07191-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-07191-9