
Abstract
Background
Burkholderia cenocepacia is a human opportunistic pathogen causing devastating symptoms in patients suffering from immunodeficiency and cystic fibrosis. Out of the 303 B. cenocepacia strains with available genomes, the large majority were isolated from a clinical context. However, several isolates originate from other environmental sources ranging from aerosols to plant endosphere. Plants can represent reservoirs for human infections as some pathogens can survive and sometimes proliferate in the rhizosphere. We therefore investigated if B. cenocepacia had the same potential.
Results
We selected genome sequences from 31 different strains, representative of the diversity of ecological niches of B. cenocepacia, and conducted comparative genomic analyses in the aim of finding specific niche or host-related genetic determinants. Phylogenetic analyses and whole genome average nucleotide identity suggest that strains, registered as B. cenocepacia, belong to at least two different species. Core-genome analyses show that the clade enriched in environmental isolates lacks multiple key virulence factors, which are conserved in the sister clade where most clinical isolates fall, including the highly virulent ET12 lineage. Similarly, several plant associated genes display an opposite distribution between the two clades. Finally, we suggest that B. cenocepacia underwent a host jump from plants/environment to animals, as supported by the phylogenetic analysis. We eventually propose a name for the new species that lacks several genetic traits involved in human virulence.
Conclusion
Regardless of the method used, our studies resulted in a disunited perspective of the B. cenocepacia species. Strains currently affiliated to this taxon belong to at least two distinct species, one having lost several determining animal virulence factors.
Similar content being viewed by others

Background
Over the past years, the genus Burkholderia has been progressively revised, leading to the description of six current genera, Burkholderia sensu stricto, Paraburkholderia, Caballeronia, Trinickia, Mycetohabitans and Robbsia [1, 2]. Burkholderia sensu stricto englobes at least 31 distinct species, including 22 that belong to the Burkholderia cepacia complex (BCC) [1]. The BCC harbors species that are opportunistic human pathogens, causing devastating symptoms in immunocompromised individuals. These pathogens are mainly causing nosocomial infections and severely affect patients suffering from cystic fibrosis (CF). In some cases, the infected patients can develop the fatal “cepacia syndrome” characterized by progressive respiratory failure and necrotizing pneumonia, often resulting in early death [3]. However, some BCC strains seem to be more virulent than others as most infections are caused by either Burkholderia cenocepacia or Burkholderia multivorans [4]. In some regions of Europe as well as Canada, B. cenocepacia infections account for over 80% of bacterial infections in CF patients [5,6,7]. One lineage in particular, ET 12, is highly transmissible and responsible for most B. cenocepacia outbreaks [8]. It is no surprise that this deadly species has received a considerable amount of attention considering its clinical implication in human health [9].
The specific description of B. cenocepacia occurred in 2003. It was originally part of Burkholderia cepacia whose type strain, LMG1222, was isolated from decaying onions and identified as a plant pathogen [10]. B. cepacia later appeared to be recurrently isolated from immunocompromised patients and was recognized as an opportunistic pathogen. However, B. cepacia also proved to be useful as a biocontrol agent against plant pathogens, inhibiting growth of diverse oomycetes, fungi, bacteria and nematodes [11, 12]. With the advancements of genomics, it was demonstrated that the presumed B. cepacia species should be divided in five genetically distinct but phenotypically undistinguishable genomovars [13]. With further studies, the number of B. cepacia genomovars increased and were progressively classified into nine separate taxa, mostly using recA-based identification [14,15,16].
B. cenocepacia (initially genomovar III) was distinguished from B. cepacia by DNA-DNA hybridization studies but recA sequence phylogeny still suggested different subgroups within B. cenocepacia [17]. At least four different recA-lineages (IIIA, IIIB, IIIC and IIID) are observed with lineages IIIA and IIIB being predominant in clinical isolations. The highly virulent strains of the ET 12 lineage belong to group IIIA [17,18,19]. Moreover, using microarray experiments, it was observed that various B. cenocepacia strains reacted differentially to conditions mimicking the human host environment. Out of several hundreds of differentially regulated genes, only nine displayed similar regulations across the different strains, suggesting important differences in infection capacity across strains of B. cenocepacia [20,21,22]. Despite the apparent genetic contrast between B. cenocepacia strains, no large scale comparative genomics study has been conducted on this species yet [23].
Albeit it has received most its attention from clinical studies, it is not uncommon to recover B. cenocepacia isolates from soil samples. Isolates of this species have also been frequently sampled from plant material [19, 24,25,26]. Plants could thus represent alternative hosts and potential reservoirs for BCC strains. Still, their adaptation for plant infection or colonization remains poorly documented. Four studies investigated the biocontrol potential of recognized B. cenocepacia strains that all belong to the IIIB recA-lineage. Altogether, they suggest strong biocontrol potential of plant-associated B. cenocepacia strains against diverse plant-pathogens [26,27,28,29].
Our study aims at clarifying the taxonomic position of B. cenocepacia strains isolated from different sources by investigating the correlation between genomic identity and environmental distribution within the species. We also strive to elucidate if plants may represent a reservoir of human opportunistic B. cenocepacia strains. By using bioinformatics and phylogenetic tools, we compared the whole genome sequences of 31 B. cenocepacia strains isolated from either clinical or environmental sources. We highlight the existence of a new Burkholderia species and describe its reduced adaptation to animal infection and virulence as compared to its closest parent, B. cenocepacia.
Results
Characteristics of selected B. cenocepacia strains selected for comparative analyses
Two hundred forty-six of the 303 genomes (either full or draft) of B. cenocepacia strains available on the NCBI database, at the time of this study, are clinical isolates (Additional file 1: Figure S1). They were sampled from patients suffering from CF, from other pathologies or from healthy patients. The isolates also vary according to the source of biological sample they originate from. Most clinical isolates were obtained from sputum or blood samples, but some were also isolated from hospital equipment [30] as B. cenocepacia is resistant to many common antibiotics as well as several sanitizers [31, 32].
The remaining B. cenocepacia strains with available genomes come from environmental sources. These can be aerosol and water samples but also agricultural soil and plant roots (Additional file 1: Figure S1, Table 1). The recA phylogeny of all genomes available showed that the recA-IIIA lineage includes in a very large majority strains isolated from a clinical context (228 isolates; 94.2%), with only 10 strains obtained from an environmental context and four with an unknown origin (Additional file 1: Figure S1). Conversely the recA-IIIB clade mixed clinical isolates (18; 43.9%), environmental isolates (15; 36.6%), plant isolates (4; 9.8%) and isolates with unknown origin (4; 9.8%). It should however be noted that, among the 228 isolates clinical isolates of the recA-IIIA clade, 188 were isolated from the same place, a hospital in Vancouver, Canada. Similarly, 12 of the 18 clinical isolates of the recA-IIIB clade are from the same hospital. There is thus a strong bias in the sampling, and highly similar strains might coexist in the database.
Core-genome phylogenetic analysis
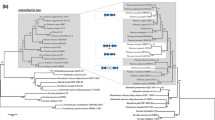
The genomes of 31 B. cenocepacia strains were compared and their core-genome extracted (Additional file 4: Table S1; refer to Methods section for details on strain selection). The resulting 1057 conserved genes were aligned and studied in a phylogenetic analysis using the Maximum Likelihood method (Fig. 1).

Phylogeny and distribution of host-adaptation genes for 31 B. cenocepacia strains. The evolutionary distances were computed using the Maximum Composite Likelihood method. A total of 1057 conserved core-genes, totaling 1,039,265 positions were used in the final dataset. Branch label colors are indicative of the isolation source of the respective strains. These can either be clinic (red), rhizospheric (green) or environmental (grey). The colored shapes indicate the presence of genetic elements in the genomes of the corresponding strains. Squares correspond to genes that were found to be preferably enriched in clinical (vir.) or environmental (env.) species. From left to right: cable pilus (cblA), 22 kDa adhesion (adhA), Burkholderia cenocepacia epidemic strain marker (BCESM), transcriptional regulator kdgR, bile acid 7-alpha dehydratase (baiE), taurine dehydrogenase (tauX), sulfoacetaldehyde acetyltransferase (xsc), tellurite resistance cluster (telA), low oxygen activated locus (lxa), respiratory nitrate reductase cluster (narIJHGK), nitrate sensor and regulation cluster (narLX), lectin like bacteriocin 88 (llpA), nitrile hydratase cluster (nthAB), phenylacetaldoxime dehydratase (oxd), feruloyl-esterase (faeB), pyrrolnitrin biosynthesis cluster (prn), galacturonate metabolism genes (uxaAB). Circles indicate the presence of the pC3 megaplasmid and the afc cluster. This figure was generated using iTOL [48]
This result was validated through a Bayesian prediction using the BEAST software (Additional file 2: Figure S2). Both approaches yielded comparable reconstructions. An additional tree resulting from a Neighbor Joining analysis with 1000 bootstrap repetitions is also available (Additional file 3: Figure S3). Among the 31 strains labelled as B. cenocepacia, three (869 T2, DDS 22E-1 and DWS 37E-2) fall outside the main clade. The 28 other strains fall within three main clades. One clade gathers the strains belonging to the recA-IIIA lineage, including the ET 12 lineage (J2315T, BC-7, K56) plus 11 other strains, among which only one was isolated from the environment (F01, from a soil in Burkina Faso). The sister clade of this latter group is composed of 11 strains which belong to the recA-IIIB lineage. Seven originate from a plant environment and four from a hospital environment.
The closest outgroup of these two clades contains three strains (Bp8974, Bp9038 and CEIB S5–2) isolated from soil in Mexico and Puerto Rico. These three clades are all extremely well supported by bootstrap values (Additional file 3: Figure S3).
Whole-genome comparisons
Based on the ANI analyses and considering the 95% threshold for species delimitation, most input strains cluster in three main species identity groups (Fig. 2a, Additional file 5: Table S2). This distribution is identical to the three clades detected in the previous phylogenetic analyses. The first group includes mainly clinical strains with the exception of strain F01. Consistently, this cluster contains the highly transmissible strains belonging to the ET 12 lineage and can thus be considered as the B. cenocepacia sensu stricto (s.s.) species. Eleven strains belong to the sister clade of B. cenocepacia s.s. and their average nucleotide identity to this latter ranges from 92 to 94% (Additional file 5: Table S2). No closer ANI was found with any of the phylogenetically closest Burkholderia species (data not shown). Similarly, the three strains of the third clade do not display any ANI ≥ 95% with B. cenocepacia. Their closest Burkholderia relative is B. cenocepacia strain FL-5-3-30-S1-D7 with 94% ANI value.

Whole-genome comparisons of 31 B. cenocepacia strains. The calculations were performed using the Python module PYANI [49]. Two major identity clusters are formed. The bottom cluster consists of B. cenocepacia strains and the second cluster consists of Burkholderia sp. nov. strains. One minor identity cluster is formed by the three outlier strains (Bp9038, CEIB_S5–2, Bp8974) and the last three strains are neither genetically related to B. cenocepacia nor to each other. A double entry heatmap was used to depict the ANI results with ANIm as left entry and ANIb as right entry (a). the dDDH results are depicted on a single heatmap (b). The species demarcation threshold is at ≥95% identity on ≥70% aligned genomic sequence for ANI and at ≥70% identity for dDDH. The exact values and sequence cover ratios are available in Additional file 5: Table S2
Finally, three strains that were originally described as B. cenocepacia show closer identity with other species (Additional file 5: Table S2). Strain 869 T2 should be affiliated to the Burkholderia seminalis taxon (98.99% identity with 88.85% cover). Strain DDS 22E-1 shares high ANI scores with Burkholderia pseudomultivorans (97.57% identity with 80.85% cover) while strain DWS 37E-2 is related to Burkholderia latens with 99.01% homology and 89.92% cover.
Two genome alignment methods were used for the ANI analyses, one based on BLAST+ (ANIb) and the other on MUMmer (ANIm). ANIb resulted in a robust species delimitation between B. cenocepacia and Burkholderia sp. nov. as the values between those clusters are below the 95% threshold. ANIm improved the proximity among species within the clusters. The minimal identity value between B. cenocepacia and Burkholderia sp nov. strains respectively increased from 94.86 to 97.57% and 97.97 to 98.92%. However, the maximal identity values between the clusters increased as well going from 94.76 to 95.32% and thus passing, although marginally, the threshold value for species delimitation.
The species delimitation was validated through a digital DNA-DNA hybridization (dDDH) analysis (Fig. 2b, Additional file 5: Table S2). For a pairwise comparison between two genomes, a dDDH value ≤70% indicates that the tested organisms indeed belong to different species. Considering this threshold, the species delimitation between B. cenocepacia and Burkholderia sp. nov. is very well supported with values ranging from 49.9 to 61% .
General genomic features of Burkholderia sp. nov.
In the following parts, we will refer to the clade harboring in majority environmental strains as Burkholderia sp. nov., while isolates that fall together in the same clade as the ET 12 lineage will keep the name B. cenocepacia s.s.. Occasionally, the group formed by those two main clades will be referred to as B. cenocepacia sensu lato (s.l.). The third clade englobes strains with high similarity originating from only two sampling sites and needs to be completed with other isolates from other sites to be confirmed as a new species. As the quality of genome sequences is heterogeneous for the strains used in this study, no comparison of the global genomic architecture was carried out. We screened the strains for the presence of the pC3 megaplasmid containing the virulence associated afc cluster [50, 51]. Although we cannot confirm its megaplasmid structure from the draft genomes, large genetic portions of the pC3 were detected in all strains but FL-5-3-30-S1-D7. Strains 869 T2, DDS 22E-1 and DWS 37E-2 harbor a pC3 lacking the afc cluster (Fig. 1). On average, Burkholderia sp. nov. strains have a slightly, yet significantly (Student’s t-test, p < 5.10− 5), smaller genome than their closest related species, with a median value of 7.51 Mb as compared to 8.03 Mb for B. cenocepacia (Fig. 3). Accordingly, the putative new species has an average of 509 coding sequences less than B. cenocepacia with a mean of 6711 and 7220 CDS respectively. The GC % content of both species is comparable with approximately 67% (Fig. 3, Additional file 6: Table S3). Nevertheless, both species share a relatively large genome in regards to the genus Burkholderia which averages at 7.2 Mb.

Variations in genomic organization between B. cenocepacia and Burkholderia sp. nov.. The data of 304 genomes presented in Additional file 6: Table S3 was used to represent the differences in genomic organization between B. cenocepacia and Burkholderia sp. nov. strains. Significant levels in variations were determined using Student’s t-test (p < 2.10− 4, p < 2.10− 5 for *** and **** respectively)
Analysis of core-genome features involved in host-adaptation
We analyzed the core-genome of B. cenocepacia s.s. and looked for genes that are strictly absent from the core-genome of Burkholderia sp. nov. and reciprocally. This list was further curated from genes with convergent functions when those were successfully annotated. The core-genome of Burkholderia sp. nov. comprises 150 genes that are missing from B. cenocepacia whereas the latter harbors 244 genes which Burkholderia sp. nov. strains do not harbor. These genes sets were further curated from uncharacterized genes which yields 67 core-genes for Burkholderia sp. nov and 37 core-genes for B. cenocepacia (Additional file 7: Table S4). For both groups, we found several antimicrobial-compound coding genes as well as metabolical genes contributing to environmental competitiveness or improved survival inside their respective hosts. Still, many of the conserved genes play an unknown role. Below, we further elaborate on conserved genes that are susceptible to play a role in specific ecological adaptation. The conservation of these genes of interest across the different taxa and 303 genomes of B. cenocepacia s.l. is given in Additional file 8: Table S5.
Distribution of virulence-associated genes
Based on the literature [50,51,52,53,54], all 31 strains were screened for the presence of several genes previously demonstrated to be involved in virulence (Fig. 1, Table 2). Two well described virulence genes have a striking unbalanced repartition between B. cenocepacia and Burkholderia sp. nov.: The cable pilus coding gene, cblA, is only present in the ET 12 lineage strains and strain F01, and its associated 22 kDa adhesin coding gene, adhA, is ubiquitously found among B. cenocepacia strains but strictly absent from Burkholderia sp. nov. We further focused on candidate genes that are potentially involved in human-host adaptation and specific to B. cenocepacia (Table 2). We found a putative bile-acid dehydratase (BCAM1585–86), a taurine dehydrogenase (BCAM1182–83) and a cluster potentially involved in fatty acid degradation (BCAM1620–48).
We also searched for genes putatively involved in defense against the host immune system but also in host specific resilience and virulence. In those categories B. cenocepacia specifically possesses resistance genes towards tellurite (BCAL2268–71) as well as adaptation genes towards anaerobic metabolism. Considering the different pathways and components potentially allowing anaerobic survival of bacteria (Fig. 1, Table 2), only B. cenocepacia s.s. and the third cluster harbor the required operon for respiratory nitrate reduction (narIJHGK). Within B. cenocepacia, this operon is absent from the ET 12 lineage and strain H111 (Fig. 1, Table 2). Still, all B. cenocepacia s.s. strains possess the genes coding for the nitrate/nitrite sensor (narX) and the associated regulator (narL). However, the gene clusters necessary for subsequent respiratory reduction of nitrite, nitric-oxide and nitrous-oxide are missing in every B. cenocepacia s.l. strain. The lxa genomic island spans over 50 genes and is involved in cell viability after oxygen depletion [69]. This cluster was detected in most B. cenocepacia strains (BCAM0275a-323) as well as in those of the third cluster, but was completely lacking or missing vast genetic portions (at least 37% of the total cluster length) in Burkholderia sp. nov. strains.
Distribution of plant-adaptation and environmental-resilience genes
We investigated the presence of five genes or gene clusters which are, according to previous studies, involved in improving the fitness of plant associated bacteria [70, 71]. Two genes involved in defense strategies were detected in Burkholderia sp. nov., the lectin-like bacteriocin LlpA-88 (Bcen_1091) and the antifungal antibiotic pyrrolnitrin (Bcenmc03_6983–86).
Regarding metabolic features, Burkholderia sp. nov. strains were found to possess several enzymes such as a nitrile hydratase (Bcen_4082–85), a phenylacetaldoxime dehydratase (Bcen_4078–81), a feruloyl-esterase (Bcen_1301) and a galacturonate metabolism operon (Bcen_6467–68) allowing these bacteria to catabolize plant derivatives. It is important to point out that numerous additional plant-adaptive genes are present in the genomes of Burkholderia sp. nov. strains. However, these genes are not addressed here as they are shared with B. cenocepacia strains.
Evolutionary history of B. cenocepacia
The clade, formed by the third identity cluster, possesses several plant-adaptive traits which are part of the Burkholderia sp. nov. specific core-genome (i. e. nitrile hydratase, phenylacetaldoxime dehydratase, pyrrolnitrin synthase) (Fig. 1, Table 3). Conversely, these isolates do not possess any of the investigated genes suggested to confer a direct advantage to B. cenocepacia during human infection (i.e. BCESM, cblA, adhA) (Fig. 1, Table 2). This observation can be extended to the outgroup species B. seminalis, B. latens and B. pseudomultivorans.
The phylogenetic reconstructions also support a different pattern of molecular evolution between the two main clades. The reconstruction shows a longer branch leading to B. cenocepacia followed by short inner branches. Burkholderia sp. nov displays an opposite pattern, with a shorter basal branch and longer inner branches (Fig. 1, Additional file 2: Figure S2 and Additional file 3: Figure S3).
Discussion
B. cenocepacia strains have a polyphyletic organization
Regardless of the method used for their genomic comparisons (phylogenetic and ANI analyses), the results yielded a disunited perspective of the B. cenocepacia species (Figs. 1 & 2). The ANIb and dDDH analyzes yielded strong separations of the different clades based on the conventional threshold values for species delimitation. While the ANIm analysis strengthened the proximity within the clusters, it did not provide as clear differences between B. cenocepacia and Burkholderia sp. nov. as the two previous approaches. However, given the results from the remaining whole-genome comparison methods and the MLSA approach, we are confident that our results showed that the B. cenocepacia taxon should be split in two or possibly three distinct species (not considering three strains for which we proved a clear false taxonomic attribution).
Here, we propose to keep the B. cenocepacia name for all strains clustering with the epidemic ET 12 lineage, representing B. cenocepacia in its most studied state, as a potential human opportunistic pathogen. We further propose to reclassify its sister clade, Burkholderia sp. nov., as a new species (see below for a suggested name description). The third cluster, sister clade of the two latter species could also represent a novel Burkholderia species and deserve to be investigated independently. Still, more sampling is needed since the species are very similar to each other and were isolated from only two different geographic areas (Additional file 1: Figure S1).
Unexpectedly, four clinical isolates, namely AU1054, PC184Mulks, VC12802 and VC7848, fall in the Burkholderia sp. nov. clade. As a first hypothesis, these strains could have survived in a clinical environment solely as commensal bacteria, causing no harm to their host. The previous isolation of Burkholderia strains from CF patients or patients suffering from another infectious pathology, without being the apparent causative agent supports this hypothesis [79,80,81]. Although strain AU1054 was shown to cause high mortality rates in diverse plant and animal models [50, 82], this is not sufficient to assess its pathogenesis against humans. Indeed, strain H111, which belongs to the B. cenocepacia s.s. clade, presents the same characteristics as AU1054 on several pathogenesis models [83] and yet does not cause any symptoms in humans. When detected in patients, the bacterial population of H111 decreases over time, unable to maintain itself in a CF context [84]. At this point, we cannot exclude the alternative hypothesis that these clinical strains are in fact human opportunists. However, as discussed in the next section, these isolates lack many key virulence traits that are present in B. cenocepacia s.s.. Still, more virulence and pathogeny tests are needed on strains of this clade to fully rule out their potential human virulence.
B. cenocepacia possess specific key virulence traits compare to Burkholderia sp. nov
Over the past years, many genetic markers have been investigated for their involvement in B. cenocepacia pathogenesis. When mapping some of these virulence factors, the most striking pattern in the distribution between the two clades concerns the adhA and cblA genes. Both cable pilus and the associated 22-kDa adhesin have been shown to be involved and decisive for host cell binding [55, 56]. Their distributions are congruent with previous studies that found adhA to be mandatory and sufficient for cell binding but cblA required for optimal binding [54]. Our data supports that the presence of adhA is essential for the opportunistic potential of B. cenocepacia strains.
Additionally, a genomic island termed B. cenocepacia epidemic strain marker (BCESM) was frequently found in CF isolates [53]. However, it was demonstrated that the BCESM is not an absolute marker for the ability of B. cenocepacia to cause CF infection [57]. In our study, the distribution of this cluster is less clear, as it was detected at a lower frequency in the Burkholderia sp. nov. strains compare to B. cenocepacia (58 and 83% respectively, Additional file 8: Table S5). This difference might just be random, but we believe that it reflects a specialization trend of the BCESM cluster for pathogenicity.
As a counter example, the pC3 megaplasmid was detected in every B. cenocepacia strain and all but one Burkholderia sp. nov. strains. This replicon was shown to play an important role in the pathogenesis of various BCC strains, including B. cenocepacia, in diverse infection models [50]. Located on this pC3, the afc cluster and the adjacent transcriptional regulator shvR were recently demonstrated to be required for acute infection of B. cenocepacia in the zebrafish model [51]. However, the virulence levels of B. cenocepacia strains are strongly depending on the host model. To date, strains of B. cenocepacia s.s. and Burkholderia sp. nov. have been tested on nematodes, wax moth larvae and zebrafish and the strains respective virulence sometimes varied drastically depending on the tested host [51, 85, 86]. Tests that aim to study the human infection route through the lungs have only been performed with ET-12 representatives and we support that clinical research would greatly benefit if a wider diversity of strains including Burkholderia sp. nov. representatives were tested in future studies.
The following genes have not yet been described in B. cenocepacia but their homology to virulence factors of other pathogens might indicate a similar role in our species of interest.
While analyzing the specific core genome of each clade, we detected that Burkholderia sp. nov lacks two B. cenocepacia specific enzymes, a bile-acid dehydratase and a taurine dehydrogenase. Bile acids can be conjugated to taurine before they are secreted inside the digestive track. Furthermore, bile acids are cholesterol-derivatives formed by conversion in the liver [87] and it has been previously demonstrated that several human pathogens could thrive on cholesterol compounds as a carbon source [61, 62]. Mycobacterium tuberculosis is able to maintain itself within host macrophages through this same steroid degradation pathway [63]. Thus, in addition to an improved ability to bind epithelial cells, B. cenocepacia also displays key features that could promote its survival inside host cells as compared to Burkholderia sp. nov..
A resistance gene for tellurite is also present in the core-genome specific to B. cenocepacia. This antimicrobial compound has strong oxidizing abilities but is also believed to be a substitute for sulfur in various cellular functions with drastic outcomes for cell metabolism. Tellurite resistance is widespread in pathogenic bacteria and was taken advantage off for generic screening of pathogens using tellurite in selective media [88]. More recently, the human pathogen Yersinia pestis was shown to express several tellurite resistance genes during macrophage infection and subsequent studies led to speculate that these genes are part of a bacterial adaptive strategy to macrophage associated stress [66, 67].
Finally, in a CF endobronchial context, important amounts of mucus are secreted, leading to partial or total anaerobiosis [89]. Several bacterial species, such as P. aeruginosa or the B. cenocepacia related Burkholderia pseudomallei, can maintain themselves in micro-oxic conditions through denitrification [90, 91]. The complete respiratory nitrate reduction process works sequentially and involves genes responsible for the reduction of nitrate, nitrite, nitric-oxide and nitrous-oxide [92]. B. cenocepacia is usually considered to be an obligate aerobic non-fermenting bacterium, and doesn’t possess all the required genes to perform the complete nitrate reduction reaction. However, most B. cenocepacia s.s. strains have the genetic material to use nitrate as electron acceptor and transfer the resulting nitrite from the cytoplasm to the periplasm. This chain reaction allows the transfer of 2 H+ from the cytoplasm to the periplasm. These protons can be thereafter used for ATP synthesis. An additional specificity of B. cenocepacia over Burkholderia sp. nov. is the presence of the lxa locus. This 50 gene cluster is crucial for the bacterium’s survival in low oxygen conditions [69]. It should be noted that the lack of both respiratory nitrate reduction and the lxa cluster in H111 might explain why this strain cannot maintain itself in CF patients [84]. Overall, Burkholderia sp. nov. lacks several adaptation genes for survival in anaerobic environments.
Taken together, the distribution of these pathogenic or adaptive genes showed a strong tendency in favor of an adaptation of B. cenocepacia strains for human pathogenicity compared to Burkholderia sp. nov. strains.
B. cenocepacia lacks environmental traits present in Burkholderia sp. nov.
Burkholderia sp. nov. encompasses mostly strains that have been retrieved from diverse environmental samples, including healthy plant roots. These bacteria are under the selection of a fluctuant environment and heavy competition from other microorganisms that thrive in these niches. Following this view, several strains of Burkholderia sp. nov. possess the gene cluster coding for the lectin-like bacteriocin LlpA88 and for the antifungal metabolite pyrrolnitrin. Both these metabolites have a very broad-spectrum antibiotic activity [26, 93].
Adaptation to these constraints also requires the ability to exploit various nutriments, including root exudates. When mining in the core-genome of Burkholderia sp. nov., but also the core genome of the third identity clade, we detected two genes supporting a clear adaptation to the plant environment. The first is a nitrile hydratase allowing these bacteria to use nitrile as carbon and nitrogen source. Several natural sources for nitrile are known and many are plant-based [94]. In the same genomic cluster, a gene coding for a phenylacetaldoxime dehydratase was also found. Aldoximes are volatile plant-derived metabolites which can be used as defense mechanisms against insect herbivores and pathogens [74]. Moreover, phenylacetaldoxime dehydratase is very efficient at catalyzing the production of phenylacetonitril which could be further used by the previously described nitrile hydratase [72]. Alternatively, both enzymes can be involved in indole-3-acetic acid (IAA) production, which is one of the traits that is commonly used to screen for endophytic and rhizosphere competent bacteria [73, 95, 96]. One additional advantage of Burkholderia sp. nov. on B. cenocepacia in the effective use of rhizodeposits as carbon source may come from a feruloyl-esterase. This enzyme liberates polysaccharides, such as xylan, from its bond with ferulic acid as it is commonly found in plant cell walls [75]. One other major component of plant cell walls is pectin, a polymer of galacturonic acid. Burkholderia sp. nov. possess the import and catabolism genes to process D-galacturonate and use it as a carbon source [78].
Genes associated with adaptation to the soil environments can be highly multifarious and are more elusive than genes conferring adaptation to a single niche such as the human body. A more profound analysis is required to determine how efficient and how generalized environmental adaptation is for Burkholderia sp. nov. strains.
Figure 4 summarizes the discussed genes and functions conferring Burkholderia sp. nov and B. cenocepacia specific ascendancy in soil environments and human infections respectively.

Summary diagram of differential adaptation of B. cenocepacia and Burkholderia sp. nov. to different environments. Strains of B. cenocepacia and Burkholderia sp. nov. have been isolated from soils (brown), where they compete with other microbes, plants (green) and animals (red). Burkholderia sp. nov. was repeatedly isolated from soil environment (but also plants, water and aerosols) and possesses several genes improving its fitness in those contexts (green factors). While B. cenocepacia can also thrive in soils, it is often found as an opportunistic pathogen of humans and bears several genes improving its virulence (red factors). Burkholderia sp. nov. can use different plant derivatives (galacturonic acid, xylans, pectin) as carbon sources. It is also proposedly able to synthetize the plant hormone auxin (IAA) through a pathway involving Oxd to convert indole-3-acetaldoxime (IAG) to indole-3-acetonitrile (I3A) which is processed to IAA though the action of Nth. It is also able to produce antibiotics with activity against bacteria (Llpa88) and fungi (pyrrolnitrin). B. cenocepacia strains possess a 22 kDa adhesin which improves its binding to target cells and their invasion. Proposedly, they can also metabolize bile acids, derivatives of cholesterol. In anoxic conditions, B. cenocepacia can survive using its low oxygen activated locus (lxa) and the respiratory nitrate reduction pathway (narIJHG). It also possesses the resistance genes against tellurite, for which the exact functions remain elusive. Source: authors’ design
The opportunistic pathogens might have evolved from plant-adapted bacteria
One approach to determine the ancestral state of the two clades of B. cenocepacia and Burkholderia sp. nov. is to focus on the outgroup life style. The conservation of environment and plant adaptive traits and the absence of key virulence genes in strains of the third identity cluster and more distant outgroup species, indicate that B. cenocepacia might have evolved from a plant-associated life style towards a human opportunist state.
The pattern observed using the phylogenetic reconstruction, reflects a significant evolutionary trend. It shows a lower within-species diversity in B. cenocepacia which may have undergone a rapid evolution, by either positive selection and/or population bottleneck, before diversification. Following this view, the core-genome of B. cenocepacia strains is higher than that of Burkholderia sp. nov. (69.9 and 49.7% of the average amount of genes across strains respectively), confirming that B. cenocepacia is less diversified than Burkholderia sp. nov.. This might again be a sign for higher selective pressure applied by its environment on B. cenocepacia during a pathogenic lifestyle, and/or a more recent evolution, leaving less time for genetic divergence. This would implicate that B. cenocepacia has evolved the human-pathogen trait after its ancestors had adapted for interactions with plants.
Proposition of Burkholderia servocepacia sp. nov.
Proposition of name Burkholderia servocepacia sp. nov. (servocepacia: ser.vo.ce.pa’cia. M.L. v. servare, protect / guard; specific epithet, cepacia; N.L. n. servocepacia, protecting cepacia). The prefix “ceno” from the Greek “kainos” was initially chosen to mean “new” since B. cenocepacia was a derivative of the B. cepacia species. Interestingly, the Latin meaning of the same prefix is “to dine”. Given that cepa is Latin for onion, inspired by the isolation source of the first Burkholderia species, the combination ceno-cepacia can be understood as “feasting on onions”. This interpretation also has the benefit of harmonizing the etymological origins of both components. Additionally, this predatorily name is in accordance with the serious harm the bacteria can cause in humans. As a sequel, we propose the name Burkholderia servocepacia sp. nov. coined from the same epithet and using as a prefix the Latin “servo” meaning “to protect/watch over” in regard of the biocontrol potential of this species.
We propose that B. servocepacia Tatl-371 be used as type strain. Its phenotype has been extensively characterized and it was deposited in the BCCM/LMG database under accession number LMG30279 [26]. As a type strain should be representative of its species, it is relevant that Tatl-371 possesses a central position in the phylogeny of its clade (Fig. 1).
Conclusion
The recurrent isolation of B. cenocepacia from plant environments was the premise for the investigation of 31 B. cenocepacia strains using comparative genomics.
Despite the nature of plants as environmental reservoir for diverse human pathogens, our study suggests that multiple B. cenocepacia affiliated strains are instead specialized for an environmental lifestyle and interactions with plants, and potentially unfit to colonize and cause harm to humans. We demonstrate that those strains are phylogenetically distinct from B. cenocepacia s.s. and should be affiliated to a new species. We also suggest that B. cenocepacia has acquired the human opportunist trait from a plant adapted basis. B. cenocepacia is not a united group at the genomic level. This disunity has high chances to impact the metabolism and the virulence of these bacteria, especially considering the specific or over-represented genes we detected in each clade. We hope that these insights will open promising leads in research on virulence promoting factors of B. cenocepacia. It would be most noteworthy if future studies were to consider the large diversity of the cenocepacia group and especially in lung infection models. This study is one of many example that underlines the benefits of interdisciplinary research and in the present case, between environmental microbiology and health sciences. Enhanced communication and collaboration between disciplines is a worthy pursuit which can positively impact the global knowledge.
Methods
Bacterial strains & genome sequencing
For the 303 B. cenocepacia genomes, publicly available at the time of the study, the recA sequences were retrieved (except for VC5279, no recA sequence in genomic data) and used to build a phylogenetic prediction using the Maximum Composite Likelihood method (1000 bootstrap repetitions). The resulting tree is available as Additional file 1: Figure S1 and in interactive form online: https://itol.embl.de/tree/912033414356601565192630#. We chose 31 different strains, representative of the diversity in source of isolation. We prioritized fully annotated genomes and included draft genomes when those increased the global diversity. All genomes from plant- or rhizosphere-isolated strains, available at the time of the study, were included in the analyses. All genomes from environment isolated strains outside the recA-IIIA clade were also included, plus 2 of the 5 genomes that fall in the recA-IIIA clade.
Three hundred one of the bacterial genomes studied here are publicly available on the NCBI database. One strain, CEIB S4–3, is publicly available on the JGI website. The respective strains are all registered as belonging to the B. cenocepacia species and are listed in Additional file 6: Table S3 with additional genomic data.
Strain ABIP444, was isolated during a survey of rice roots endophytes in Cameroon (E. Ngonkeu, unpublished). A single colony was grown in Luria low salt medium (Merck, Inc., Darmstadt, Germany) for 24 h at 28 °C and its DNA was extracted using a modified JGI protocol for bacterial DNA isolation using CTAB. A genomic library with average insert size of 350 bp was prepared for sequencing using a TruSeq Nano DNA library preparation kit (Illumina, Inc., San Diego, CA, USA). Paired-end sequencing was performed by the MGX platform (CNRS, Montpellier, France) using a HiSeq 2500 (Illumina), generating 12,709,495 raw read pairs. Reads with overlapping sequence were assembled using CLC Genomics Workbench version 7.04 resulting in 292 contigs ranging from 586 bp to 20.2 kbp. The genome sequences can be found at the European Nucleotide Archive with accession number PRJEB31911.
Core-genome calculation
The general core-genome of the 31 strains labeled as B. cenocepacia was retrieved from protein sequences using the Roary pipeline [97]. Protein clusters are initially produced with CD-HIT using iterations with similarity thresholds going from 100% down to 98% with 0.5% decrements. An all-against-all comparison is performed on the resulting set of protein sequences with BLASTP at 95% sequence identity. The genome annotations and GFF3 format files were obtained through the Prokka pipeline [98].
Further analysis of the data generated by core-genome calculation (described below), identified two main clades. Their specific (i.e. strictly absent from the neighbor clade) core-genome (gene conservation across ≥80% of strains) were generated using the Roary pangenome output files.
Genomic comparisons
Pairwise genome comparison using Average Nucleotide Identity (ANI) based on BLAST+ (ANIb) or MUMmer (ANIm) was performed using the PYANI software [49]. Two strains were considered co-specific when they shared more than 95% nucleotide identity on at least 70% of their whole genome sequence [99]. For the strains that did not fall within a clear ANI cluster, comparisons to the phylogenetically closest Burkholderia species were carried out [1].
Digital DNA-DNA hybridization was carried out using the GGDC 2.1 web platform [100]. Briefly, this tool uses BLAST+ to align one genome against another and reciprocally and generate high-scoring segment pairs (HSPs). The intergenomic distance is then calculated by dividing the sum of all identities found in HSPs by overall HSP length. A logistic regression is used for reporting the probability that the distance value is above 70%, the similarity threshold to consider two organism as distinct species. Compared to ANI, dDDH can offer more robust estimations of genomic similarities for incomplete genomes as the distance calculation is independent of genome length.
Phylogenetic analysis
The phylogenetic analyses were conducted using the MEGA v7.0.26 software [101]. The core-genome output of 1057 genes (1,039,265 positions) was evaluated using a Maximum Likelihood phylogenetic reconstruction approach with a General Time Reversible model (Gamma distributed rates with invariant sites and 5 discrete gamma categories). We validated the resulting tree through an independent Bayesian analysis using BEAST v1.10.2 [102], with the same substitution model, a strict clock model and the tree prior calculated using the Yule model. The MCMC length of chain was set to 1.107 and the burn-in value was set to 1.106 for analysis. The resulting consensus tree was constructed using a maximum clade credibility prediction.
Host-adaptive gene detection
The specific core genomes of the two main clades, were screened for genes described as involved in host adaptation in the literature [50,51,52,53,54, 70, 71]. The complete sequence of the 875 kbp long virulence plasmid from B. cenocepacia J2315 and the 24 genes of the afc pathogenic cluster were also searched in all 31 genomes by BLAST [50, 51].
Gene clusters involved in anaerobic metabolism, showing potential adaptation to CF lung environments, were identified through similarity with their B. pseudomallei homologues [68]. We also searched for the low-oxygen-activated locus (lxa) which was discovered in B. cenocepacia [69].
The antiSMASH 4.0 software [103] was used to screen for gene clusters allowing the production of secondary metabolites such as antibiotics and siderophores, involved in environmental resilience and competition with other microorganisms.
The specificity to one taxa was validated for each gene through its alignment against a database of the 303 available B. cenocepacia s.l. sequences using BLAST.
At the first occurrence of each genetic element, we provide its gene name in UniProt format. When possible, we use the gene name of the B. cenocepacia type strain J2315. When the gene is absent from the type strain, the next best annotated genome is used.
Availability of data and materials
301 B. cenocepacia genomes analyzed during the current study are available in the NCBI repository (https://www.ncbi.nlm.nih.gov/genome/genomes/475). Strain CEIB S4–3, not currently annotated as B. cenocepacia, is available under RefSeq accession number JSBM00000000.1. The novel sequenced strain ABIP444 is available at the European Nucleotide Archive under study accession number PRJEB31911, http://www.ebi.ac.uk/ena/data/view/PRJEB31911. The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ANI:
-
Average Nucleotide Identity
- BCC:
-
Burkholderia cepacia complex
- BCESM:
-
Burkholderia cepacia epidemic strain marker
- CF:
-
Cystic fibrosis
- dDDH:
-
Digital DNA-DNA hybridization
- IAA:
-
Indole-3-acetic acid
References
Estrada-de los Santos P, Palmer M, Chávez-Ramírez B, Beukes C, Steenkamp E, Briscoe L, et al. Whole genome analyses suggests that burkholderia sensu lato contains two additional novel genera (mycetohabitans gen. nov., and trinickia gen. nov.): implications for the evolution of diazotrophy and nodulation in the Burkholderiaceae. Genes (Basel). 2018;9:389.
Dobritsa AP, Samadpour M. Transfer of eleven species of the genus Burkholderia to the genus Paraburkholderia and proposal of Caballeronia gen. Nov. to accommodate twelve species of the genera Burkholderia and Paraburkholderia. Int J Syst Evol Microbiol. 2016;66:2836–46.
Jones AM, Dodd ME, Webb AK. Burkholderia cepacia: current clinical issues, environmental controversies and ethical dilemmas. Eur Respir J. 2001;17:295–301.
LiPuma JJ. Update on the Burkholderia cepacia complex. Curr Opin Pulm Med. 2005;11:528–33.
Agodi A, Barchitta M, Gianninò V, Collura A, Pensabene T, Garlaschi ML, et al. Burkholderia cepacia complex in cystic fibrosis and non-cystic fibrosis patients: identification of a cluster of epidemic lineages. J Hosp Infect. 2002;50:188–95.
Speert DP. Advances in Burkholderia cepacia complex. Paediatr Respir Rev. 2002;3:230–5.
Mcdowell A, Mahenthiralingam E, Dunbar KEA, Moore JE, Crowe M, Elborn JS. Epidemiology of Burkholderia cepacia complex species recovered from cystic fibrosis patients: issues related to patient segregation. J Med Microbiol. 2004;53:663–8.
Johnson WM, Tyler SD, Rozee KR. Linkage analysis of geographic and clinical clusters in Pseudomonas cepacia infections by multilocus enzyme electrophoresis and ribotyping. J Clin Microbiol. 1994;32(4):924–30.
Scoffone VC, Chiarelli LR, Trespidi G, Mentasti M, Riccardi G, Buroni S. Burkholderia cenocepacia infections in cystic fibrosis patients: drug resistance and therapeutic approaches. Front Microbiol. 2017;8:1592.
Burkholder WH. Sour skin, a bacterial rot. Phytopathology. 1950;40:115–7.
Parke JL. Population dynamics of Pseudomonas cepacia in the pea Spermosphere in relation to biocontrol of Pythium. Phytopathology. 1990;80:1307.
Parke JL, Gurian-Sherman D. Diversity of the Burkholderia cepacia complex and implications for risk assessment of biological control strains. Annu Rev Phytopathol. 2001;39:225–58.
Vandamme P, Holmes B, Vancanneyt M, Coenye T, Hoste B, Coopman R, et al. Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int J Syst Bacteriol. 1997;47(4):1188-200.
Vandamme P, Henry D, Coenye T, Nzula S, Vancanneyt M, LiPuma JJ, et al. Burkholderia anthina sp. nov. and Burkholderia pyrrocinia, two additional Burkholderia cepacia complex bacteria, may confound results of new molecular diagnostic tools. FEMS Immunol Med Microbiol. 2002;33:143–9.
Vermis K, Coenye T, LiPuma JJ, Mahenthiralingam E, Nelis HJ, Vandamme P. Proposal to accommodate Burkholderia cepacia genomovar VI as Burkholderia dolosa sp. nov. Int J Syst Evol Microbiol. 2004;54:689–91.
Coenye T, Laevens S, Gillis M, LiPuma JJ, Henry D, Mahenthiralingam E, et al. Burkholderia ambifaria sp. nov., a novel member of the Burkholderia cepacia complex including biocontrol and cystic fibrosis-related isolates. Int J Syst Evol Microbiol. 2001;51:1481–90.
Vandamme P, Holmes B, Coenye T, Goris J, Mahenthiralingam E, Lipuma JJ, et al. Burkholderia cenocepacia sp. nov. : a new twist to an old story. Res Microbiol. 2003;154:91–6.
Mahenthiralingam E, Bischof J, Byrne SK, Radomski C, Davies JE, Av-Gay Y, et al. DNA-based diagnostic approaches for identification of Burkholderia cepacia complex, Burkholderia vietnamiensis, Burkholderia multivorans, Burkholderia stabilis, and Burkholderia cepacia genomovars I and III. J Clin Microbiol. 2000;38:3165–73.
Jacobs JL, Fasi AC, Ramette A, Smith JJ, Hammerschmidt R, Sundin GW. Identification and onion pathogenicity of Burkholderia cepacia complex isolates from the onion rhizosphere and onion field soil. Appl Environ Microbiol. 2008;74:3121–9.
Drevinek P, Holden MTG, Ge Z, Jones AM, Ketchell I, Gill RT, et al. Gene expression changes linked to antimicrobial resistance, oxidative stress, iron depletion and retained motility are observed when Burkholderia cenocepacia grows in cystic fibrosis sputum. BMC Infect Dis. 2008;8:121.
Yoder-Himes DR, Chain PSG, Zhu Y, Wurtzel O, Rubin EM, Tiedje JM, et al. Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc Natl Acad Sci U S A. 2009;106:3976–81.
Loutet SA, Valvano MA. A decade of Burkholderia cenocepacia virulence determinant research. Infect Immun. 2010;78:4088–100.
Bodilis J, Denet E, Brothier E, Graindorge A, Favre-Bonté S, Nazaret S. Comparative genomics of environmental and clinical Burkholderia cenocepacia strains closely related to the highly transmissible epidemic ET12 lineage. Front Microbiol. 2018;9:383.
Balandreau J, Viallard V, Cournoyer B, Coenye T, Laevens S, Vandamme P. Burkholderia cepacia genomovar III is a common plant-associated bacterium. Appl Environ Microbiol. 2001;67:982–5.
Zekic F, Weselowski B, Yuan Z-C. Complete genome sequence of Burkholderia cenocepacia CR318, a phosphate-solubilizing bacterium isolated from corn root. Genome Announc. 2017;5:e00490–17.
Rojas-Rojas FU, Salazar-Gomez A, Vargas-Díaz ME, Vasquez-Murrieta MS, Hirsch AM, De Mot R, et al. Broad-spectrum antimicrobial activity by burkholderia cenocepacia TATL-371, a strain isolated from the tomato rhizosphere. Microbiology. 2018;164:1072–86.
Bevivino A, Peggion V, Chiarini L, Tabacchioni S, Cantale C, Cl D. Effect of Fusarium verticillioides on maize-root-associated Burkholderia cenocepacia populations. Res Microbiol. 2005;156:974–83.
Ho Y-N, Chiang H-M, Chao C-P, Su C-C, Hsu H-F, Guo C-T, et al. In planta biocontrol of soilborne Fusarium wilt of banana through a plant endophytic bacterium, Burkholderia cenocepacia 869T2. Plant Soil. 2015;387:295–306.
Tagele S, Kim S, Lee H, Lee Y. Potential of novel sequence type of burkholderia cenocepacia for biological control of root rot of maize (zea mays l.) caused by fusarium temperatum. Int J Mol Sci. 2019;20:1005.
Oie S, Kamiya A. Microbial contamination of antiseptics and disinfectants. Am J Infect Control. 1996;24:389–95.
Rhodes KA, Schweizer HP. Antibiotic resistance in Burkholderia species. Drug Resist Updat. 2016;28:82–90.
Ahn Y, Kim JM, Kweon O, Kim S-J, Jones RC, Woodling K, et al. Intrinsic resistance of Burkholderia cepacia complex to Benzalkonium chloride. MBio. 2016;7(6).
Patil PP, Mali S, Midha S, Gautam V, Dash L, Kumar S, et al. Genomics reveals a unique clone of Burkholderia cenocepacia harboring an actively excising novel Genomic Island. Front Microbiol. 2017;8:590.
Sajjan US, Corey M, Karmali MA, Forstner JF. Binding of Pseudomonas cepacia to normal human intestinal mucin and respiratory mucin from patients with cystic fibrosis. J Clin Invest. 1992;89:648–56.
Voronina OL, Kunda MS, Ryzhova NN, Aksenova EI, Sharapova NE, Semenov AN, et al. On Burkholderiales order microorganisms and cystic fibrosis in Russia. BMC Genomics. 2018;19(Suppl 3):74.
Geisenberger O, Givskov M, Riedel K, Hoiby N, Tümmler B, Eberl L. Production of N -acyl-L-homoserine lactones by P. aeruginosa isolates from chronic lung infections associated with cystic fibrosis. FEMS Microbiol Lett. 2000;184:273–8.
Holden MTG, Seth-Smith HMB, Crossman LC, Sebaihia M, Bentley SD, Cerdeño-Tárraga AM, et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J Bacteriol. 2009;191:261–77.
Darling P, Chan M, Cox AD, Sokol PA. Siderophore production by cystic fibrosis isolates of Burkholderia cepacia; 1998.
Ginther JL, Mayo M, Warrington SD, Kaestli M, Mullins T, Wagner DM, et al. Identification of Burkholderia pseudomallei near-neighbor species in the Northern Territory of Australia. PLoS Negl Trop Dis. 2015;9:e0003892.
Drevinek P, Vosahlikova S, Cinek O, Vavrova V, Bartosova J, Pohunek P, et al. Widespread clone of Burkholderia cenocepacia in cystic fibrosis patients in the Czech Republic. J Med Microbiol. 2005;54:655–9.
Zlosnik JEA, Zhou G, Brant R, Henry DA, Hird TJ, Mahenthiralingam E, et al. Burkholderia species infections in patients with cystic fibrosis in British Columbia, Canada. 30 years’ experience. Ann Am Thorac Soc. 2015;12:70–8.
LiPuma JJ, Spilker T, Coenye T, Gonzalez CF. An epidemic Burkholderia cepacia complex strain identified in soil. Lancet. 2002;359:2002–3.
Sahl JW, Vazquez AJ, Hall CM, Busch JD, Tuanyok A, Mayo M, et al. The effects of signal Erosion and Core genome reduction on the identification of diagnostic markers. MBio. 2016;7:e00846–16.
Ghosh S, Dureja C, Khatri I, Subramanian S, Raychaudhuri S, Ghosh S. Identification of novel small RNAs in Burkholderia cenocepacia KC-01 expressed under iron limitation and oxidative stress conditions. Microbiology. 2017;163:1924–36.
Martínez-Ocampo F, Fernández López MG, Lozano-Aguirre Beltrán LF, Popoca-Ursino EC, Ortiz-Hernández ML, Sánchez-Salinas E, et al. Draft genome sequence of Burkholderia cenocepacia strain CEIB S5-2, a methyl parathion- and p-Nitrophenol-degrading bacterium, isolated from agricultural soils in Morelos, Mexico. Genome Announc. 2016;4(2).
Daligault HE, Davenport KW, Minogue TD, Bishop-Lilly KA, Broomall SM, Bruce DC, et al. Whole-genome assemblies of 56 burkholderia species. Genome Announc. 2014;2(6).
Ho Y-N, Huang C-C. Draft genome sequence of Burkholderia cenocepacia strain 869T2, a plant-beneficial Endophytic bacterium. Genome Announc. 2015;3:e01327–15.
Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5.
Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods. 2016;8:12–24.
Agnoli K, Schwager S, Uehlinger S, Vergunst A, Viteri DF, Nguyen DT, et al. Exposing the third chromosome of Burkholderia cepacia complex strains as a virulence plasmid. Mol Microbiol. 2012;83:362–78.
Gomes MC, Tasrini Y, Subramoni S, Agnoli K, Feliciano JR, Eberl L, et al. The afc antifungal activity cluster, which is under tight regulatory control of ShvR, is essential for transition from intracellular persistence of Burkholderia cenocepacia to acute pro-inflammatory infection. PLoS Pathog. 2018;14:e1007473.
Zhou CE, Smith J, Lam M, Zemla A, Dyer MD, Slezak T. MvirDB--a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 2007;35(Database issue):D391–4.
Baldwin A, Sokol PA, Parkhill J, Mahenthiralingam E. The Burkholderia cepacia epidemic strain marker is part of a novel genomic island encoding both virulence and metabolism-associated genes in Burkholderia cenocepacia. Infect Immun. 2004;72:1537–47.
Urban TA, Goldberg JB, Forstner JF, Sajjan US. Cable Pili and the 22-Kilodalton Adhesin are required for Burkholderia cenocepacia binding to and transmigration across the squamous epithelium. Infect Immun. 2005;73:5426–37.
Sajjan US, Sylvester FA, Forstner JF. Cable-Piliated Burkholderia cepacia binds to cytokeratin 13 of epithelial cells; 2000.
Sajjan US, Forstner JF. Role of a 22-kilodalton pilin protein in binding of Pseudomonas cepacia to buccal epithelial cells. Infect Immun. 1993;61:3157–63.
LiPuma JJ, Spilker T, Gill LH, Campell PW, Liu L, Mahenthiralingam E. Disproportionate distribution of Burkholderia cepacia complex species and transmissibility markers in cystic fibrosis. Am J Respir Crit Care Med. 2001;164:92–6.
Pouyssegur J, Stoeber F. Genetic control of the 2-keto-3-deoxy-d-gluconate metabolism in Escherichia coli K-12: kdg regulon. J Bacteriol. 1974;117:641–51.
Søndberg E, Jelsbak L. Salmonella Typhimurium undergoes distinct genetic adaption during chronic infections of mice. BMC Microbiol. 2016;16:30.
Lu Y, Rashidul IM, Hirata H, Tsuyumu S. KdgR, an IClR family transcriptional regulator, inhibits virulence mainly by repression of hrp genes in Xanthomonas oryzae pv. Oryzae. J Bacteriol. 2011;193:6674–82.
Horinouchi M, Hayashi T, Kudo T. Steroid degradation in Comamonas testosteroni. J Steroid Biochem Mol Biol. 2012;129:4–14.
Holert J, Alam I, Larsen M, Antunes A, Bajic VB, Stingl U, et al. Genome sequence of Pseudomonas sp. strain Chol1, a model organism for the degradation of bile salts and other steroid compounds. Genome Announc. 2013;1:e00014–2.
Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A. 2007;104:1947–52.
Ruff J, Denger K, Cook AM. Sulphoacetaldehyde acetyltransferase yields acetyl phosphate : purification from Alcaligenes defragrans and gene clusters in taurine degradation. Biochem J. 2003;369(Pt 2):275–85.
Molinero N, Ruiz L, Sánchez B, Margolles A, Delgado S. Intestinal Bacteria interplay with bile and cholesterol metabolism: implications on host physiology. Front Physiol. 2019;10:185.
Ponnusamy D, Clinkenbeard KD. Role of Tellurite resistance operon in filamentous growth of Yersinia pestis in macrophages. PLoS One. 2015;10:e0141984.
Ponnusamy D, Hartson SD, Clinkenbeard KD. Intracellular Yersinia pestis expresses general stress response and tellurite resistance proteins in mouse macrophages. Vet Microbiol. 2011;150:146–51.
Mangalea MR, Plumley BA, Borlee BR. Nitrate sensing and metabolism inhibit biofilm formation in the opportunistic pathogen Burkholderia pseudomallei by reducing the intracellular concentration of c-di-GMP. Front Microbiol. 2017;8:1353.
Sass AM, Schmerk C, Agnoli K, Norville PJ, Eberl L, Valvano MA, et al. The unexpected discovery of a novel low-oxygen-activated locus for the anoxic persistence of Burkholderia cenocepacia. ISME J. 2013;7:1568–81.
Angus AA, Agapakis CM, Fong S, Yerrapragada S, Estrada-de los Santos P, Yang P, et al. Plant-associated symbiotic burkholderia species lack hallmark strategies required in mammalian pathogenesis. PLoS One. 2014;9:e83779.
Levy A, Gonzalez IS, Mittelviefhaus M, Clingenpeel S, Paredes SH, Miao J, et al. Genomic features of bacterial adaptation to plants. Nat Genet. 2017;50(1):138–50.
Xie S-X, Kato Y, Asano Y. High yield synthesis of nitriles by a new enzyme, Phenylacetaldoxime Dehydratase, from Bacillus sp. strain OxB-1. Biosci Biotechnol Biochem. 2001;65:2666–72.
Spaepen S, Vanderleyden J, Remans R. Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol Rev. 2007;31:425–48.
Irmisch S, Zeltner P, Handrick V, Gershenzon J, Köllner TG. The maize cytochrome P450 CYP79A61 produces phenylacetaldoxime and indole-3-acetaldoxime in heterologous systems and might contribute to plant defense and auxin formation. BMC Plant Biol. 2015;15:128.
Esteban-Torres M, Reverón I, Mancheño JM, de Las RB, Muñoz R. Characterization of a feruloyl esterase from lactobacillus plantarum. Appl Environ Microbiol. 2013;79:5130–6.
de Vries RP, VanKuyk PA, Kester HCM, Visser J. The Aspergillus Niger faeB gene encodes a second feruloyl esterase involved in pectin and xylan degradation and is specifically induced in the presence of aromatic compounds. Biochem J. 2002;363:377.
Schmidt S, Blom JF, Pernthaler J, Berg G, Baldwin A, Mahenthiralingam E, et al. Production of the antifungal compound pyrrolnitrin is quorum sensing-regulated in members of the Burkholderia cepacia complex. Environ Microbiol. 2009;11:1422–37.
Suvorova IA, Tutukina MN, Ravcheev DA, Rodionov DA, Ozoline ON, Gelfand MS. Comparative genomic analysis of the hexuronate metabolism genes and their regulation in gammaproteobacteria. J Bacteriol. 2011;193:3956–63.
Coenye T, Laevens S, Willems A, Ohle M, Hannant W, Govan JRW, et al. Burkholderia fungorum sp. nov. and Burkholderia caledonica sp. nov., two new species isolated from the environment, animals and human clinical samples. Int J Syst Evol Microbiol. 2001;51:1099–107.
Coenye T, Goris J, Spilker T, Vandamme P, LiPuma JJ. Characterization of unusual bacteria isolated from respiratory secretions of cystic fibrosis patients and description of Inquilinus limosus gen. Nov., sp. nov. J Clin Microbiol. 2002;40:2062–9.
Nally E, Groah SL, Pérez-Losada M, Caldovic L, Ljungberg I, Chandel NJ, et al. Identification of Burkholderia fungorum in the urine of an individual with spinal cord injury and augmentation cystoplasty using 16S sequencing: copathogen or innocent bystander? Spinal Cord Ser Cases. 2018;4:85.
Springman AC, Jacobs JL, Somvanshi VS, Sundin GW, Mulks MH, Whittam TS, et al. Genetic diversity and multihost pathogenicity of clinical and environmental strains of Burkholderia cenocepacia. Appl Environ Microbiol. 2009;75:5250–60.
Schwager S, Agnoli K, Köthe M, Feldmann F, Givskov M, Carlier A, et al. Identification of Burkholderia cenocepacia strain H111 virulence factors using nonmammalian infection hosts. Infect Immun. 2013;81:143–53.
Uehlinger S, Schwager S, Bernier SP, Riedel K, Nguyen DT, Sokol PA, et al. Identification of specific and universal virulence factors in Burkholderia cenocepacia strains by using multiple infection hosts. Infect Immun. 2009;77:4102–10.
Vergunst AC, Meijer AH, Renshaw SA, O’Callaghan D. Burkholderia cenocepacia creates an intramacrophage replication niche in zebrafish embryos, followed by bacterial dissemination and establishment of systemic infection. Infect Immun. 2010;78:1495–508.
Huber B, Feldmann F, Köthe M, Vandamme P, Wopperer J, Riedel K, et al. Identification of a novel virulence factor in Burkholderia cenocepacia H111 required for efficient slow killing of Caenorhabditis elegans. Infect Immun. 2004;72:7220–30.
Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–74.
Taylor DE. Bacterial tellurite resistance. Trends Microbiol. 1999;7:111–5.
Kolpen M, Hansen CR, Bjarnsholt T, Moser C, Christensen LD, van Gennip M, et al. Polymorphonuclear leucocytes consume oxygen in sputum from chronic Pseudomonas aeruginosa pneumonia in cystic fibrosis. Thorax. 2010;65:57–62.
Wongwanich S, Chotanachan P, Kondo E, Kanai K. Multifactorial pathogenic mechanisms of Burkholderia pseudomallei as suggested from comparison with Burkholderia cepacia. Southeast Asian J Trop Med Public Health. 1996;27:111–8.
Kolpen M, Kragh KN, Bjarnsholt T, Line L, Hansen CR, Dalbøge CS, et al. Denitrification by cystic fibrosis pathogens – Stenotrophomonas maltophilia is dormant in sputum. Int J Med Microbiol. 2015;305:1–10.
Moreno-Vivián C, Cabello P, Martínez-Luque M, Blasco R, Castillo F. Prokaryotic nitrate reduction: molecular properties and functional distinction among bacterial nitrate reductases. J Bacteriol. 1999;181:6573–84.
Hammer PE, Burd W, Hill DS, Ligon JM, van Pée K-H. Conservation of the pyrrolnitrin biosynthetic gene cluster among six pyrrolnitrin-producing strains. FEMS Microbiol Lett. 1999;180:39–44.
Legras JL, Chuzel G, Arnaud A, Galzy P. Natural nitriles and their metabolism. World J Microbiol Biotechnol. 1990;6:83.
Etesami H, Alikhani HA, Hosseini HM. Indole-3-acetic acid (IAA) production trait, a useful screening to select endophytic and rhizosphere competent bacteria for rice growth promoting agents. MethodsX. 2015;2:72–8.
Howden AJM, Rico A, Mentlak T, Miguet L, Preston GM. Pseudomonas syringae pv. Syringae B728a hydrolyses indole-3-acetonitrile to the plant hormone indole-3-acetic acid. Mol Plant Pathol. 2009;10:857–65.
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9.
Kim M, Oh H-S, Park S-C, Chun J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64:346–51.
Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:vey016.
Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, et al. antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017;45:W36–41.
Acknowledgments
The authors acknowledge the IRD itrop HPC (South Green Platform) at IRD Montpellier for providing HPC resources that have contributed to the research results reported within this paper. URL: https://bioinfo.ird.fr/- http://www.southgreen.fr. The LABGeM (CEA/Genoscope & CNRS UMR8030), the France Génomique and French Bioinformatics Institute national infrastructures are acknowledged for support within the MicroScope annotation platform. The authors are grateful to Marie Simonin for her helpful comments on earlier versions of the manuscript. We also gratefully acknowledge the helpful insights of Annette Vergunst concerning B. cenocepacia’s virulent behavior.
Funding
The authors gratefully acknowledge financial support from the CGIAR research program (CRP) RICE. The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
AW analyzed and interpreted the genomic data. LM and GB were major contributors in writing the manuscript. EK and EN made substantial contributions during study conception and data interpretation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
recA based phylogeny for 302 B. cenocepacia strains. The recA sequence of B. pseudomallei K96243 was used to root the tree. The evolutionary history was inferred using the Neighbor-Joining method. The associated taxa that clustered together in > 95% of replicate trees in the bootstrap test (1000 replicates) are displayed as black dots on the tree branches. The color ranges delineate the two recA lineages: IIIA (orange) and IIIB (blue). The colored strip indicates the isolation source (when known) of the respective strains: clinical (red), plant (green) and environmental (grey). The strains which were included in whole-genome analyses are marked by a black arrowhead. The outermost blue histogram is representative of the genomic completeness for the respective strains according to the NCBI annotation. In increasing bar size order: contig, scaffold, chromosome and complete. An interactive version in full quality is available online (https://itol.embl.de/tree/912033414356601565192630#).
Additional file 2: Figure S2.
BEAST-generated phylogenetic tree of 31 B. cenocepacia strains. A Bayesian analysis using the BEAST v1.10.2 software was used to generate this tree. The input data is the same as for Fig. 1. A General Time Reversible model (GTR; Gamma distributed rates with invariant sites and 5 discrete gamma categories) was used as substitution model. A strict clock model was applied and the tree prior was calculated using the Yule model. Finally, the MCMC length of chain was set to 1.107 and the burn-in value was set to 1.106 for analysis. The resulting consensus tree showing mean branch lengths was constructed using a maximum clade credibility prediction.
Additional file 3: Figure S3.
Phylogenetic tree of 31 B. cenocepacia strains. The input data is the same as for Fig. 1. The evolutionary history was inferred using the Neighbor-Joining method. The associated taxa that clustered together in > 95% of replicate trees in the bootstrap test (1000 replicates) are displayed as black dots on the tree branches. The tree is drawn to scale. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site. The analysis involved 31 nucleotide sequences. All positions with less than 95% site coverage were eliminated. That is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position. There were a total of 1,118,599 positions in the final dataset.
Additional file 4: Table S1.
Core-genome of the 31 B. cenocepacia strains. The 1065 genes used in the MLSA approach are detailed with their function, average gene length and nucleotide sequence.
Additional file 5: Table S2.
Detailed whole genome comparison data. Percentage identity and percentage coverage values for the ANIb and ANIm approaches as well as percentage identity for the dDDH approach. Taxonomic classification of strains 869 T2, DS 22E-1 and DWS 37E-2 based on their genomic alignment with B. seminalis, B. pseudomultivorans and B. latens respectively as estimated by ANIb.
Additional file 6: Table S3.
Additional genomic information on all available B. cenocepacia strains. Each strain is detailed with its specific affiliation, genome completeness, genomic size, GC content, genomic architecture, number of genomic scaffolds, gene count and protein count. The 31 strains used in the study are highlighted in yellow.
Additional file 7: Table S4.
Specific core-genomes of B. cenocepacia and Burkholderia sp. nov. Each gene is detailed with its function, abundance within the species and average length. Their presence within a genome is marked with an “X”. The genes further described in the study are highlighted in yellow.
Additional file 8: Table S5.
Distribution across taxa of virulence facilitating and environmental fitness improving genes. For each gene, their product and their function in virulence or environmental fitness are described. The nucleotide sequence of each gene was aligned against a database of 304 B. cenocepacia genomes using BLAST. The percentage of occurrence among strains of each major taxon (B. cenocepacia, Burkholderia sp. nov. and the third undefined taxon) is given. The table also shows if a gene was found in a least one outgroup strain.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Wallner, A., King, E., Ngonkeu, E.L.M. et al. Genomic analyses of Burkholderia cenocepacia reveal multiple species with differential host-adaptation to plants and humans. BMC Genomics 20, 803 (2019). https://doi.org/10.1186/s12864-019-6186-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-6186-z