
Abstract
Background
Phomafungin is a recently reported broad spectrum antifungal compound but its biosynthetic pathway is unknown. We combed publicly available Phoma genomes but failed to find any putative biosynthetic gene cluster that could account for its biosynthesis.
Results
Therefore, we sequenced the genome of one of our Phoma strains (F3723) previously identified as having antifungal activity in a high-throughput screen. We found a biosynthetic gene cluster that was predicted to synthesize a cyclic lipodepsipeptide that differs in the amino acid composition compared to Phomafungin. Antifungal activity guided isolation yielded a new compound, BII-Rafflesfungin, the structure of which was determined.
Conclusions
We describe the NRPS-t1PKS cluster ‘BIIRfg’ compatible with the synthesis of the cyclic lipodepsipeptide BII-Rafflesfungin [HMHDA-L-Ala-L-Glu-L-Asn-L-Ser-L-Ser-D-Ser-D-allo-Thr-Gly]. We report new Stachelhaus codes for Ala, Glu, Asn, Ser, Thr, and Gly. We propose a mechanism for BII-Rafflesfungin biosynthesis, which involves the formation of the lipid part by BIIRfg_PKS followed by activation and transfer of the lipid chain by a predicted AMP-ligase on to the first PCP domain of the BIIRfg_NRPS gene.
Similar content being viewed by others
Background
Despite the general reluctance and the slowly changing attitude of pharmaceutical and biotech industries to explore secondary metabolites of plants and microbes for pharmaceutical applications during the last two decades [1], more than a third of recently approved medicines are still natural products or have been derived from lead compounds found in living organisms [2,3,4,5,6]. In the field of antibacterial and antifungal compounds, inputs from natural product biology are particularly indispensable. Recent sequencing outputs from many microbes including bacteria and fungi support their potential role as a rich source pool for compounds with broad pharmacological relevance.
Nonribosomal peptides and polyketides represent a large class of natural products. Despite their immense structural and functional diversity, they are synthesized by strikingly similar multimodular enzymes called nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), respectively [7,8,9]. The sequences of both types of enzymes, NRPSs and PKSs, consist of modules where each module is thought to be responsible for catalyzing the attachment of a specific substrate on-to the growing chain in an assembly-line like manner [7, 8]. Typically, amino acids (NRPS) or simple carboxylic acids (PKS) are the substrates added by one module. A module consists of essential (core) domains but it is possible that it harbors additional auxiliary domain(s). The immense structural diversity of nonribosomal peptides and polyketides can be achieved by varying the number and/or order of modules with different combinations of both core domains and auxiliary domains [7, 8].
In the case of NRPSs, a typical module has at least three core domains, an adenylation domain (A domain), a peptidyl carrier protein (PCP; also known as thiolation domain, i.e. T domain) and a condensation domain (C domain). The A domain selects and activates the cognate amino acid by adenylation [10, 11]. The activated amino acid adenylate is then transferred to a PCP, which transports the activated intermediate to a C domain [12]. The PCP domain carries a phosphopantetheinyl at a conserved serine (Ser) residue, which supports the transportation of substrates between the active sites of the domains. The C domain finally catalyzes the formation of the peptide bond between the thioester group of the elongating peptide chain from the earlier module with the amino group of the current module. It has been inferred that the enzyme has an acceptor site for the nucleophile and a donor site for the electrophile [13,14,15,16].
For PKSs, a typical module contains an acyl carrier protein (ACP), a ketosynthase (KS), an acyltransferase (AT) domain and a thioesterase (Te) domain, which all together catalyse the linear extension of a polyketide intermediate by two carbon atoms [17,18,19]. The building block of a polyketide chain is usually an acyl-CoA or a carboxyacyl-CoA which is loaded on the AT domain. The AT domain transfers the activated building block chain to the neighbouring ACP domain [20]. The ACP domain, like the PCP domain of NRPSs, is 4′-phosphopantetheinylated and transfers the intermediate to the active site (cysteine) of the upstream KS domain. The KS domain catalyses a Claisen-like condensation reaction by forming a carbon-carbon bond between the thioester carbonyl of the ACP-bound polyketide chain and the alpha carbon atom of the extending unit. This decarboxylative condensation step yields a ketide chain with a β-keto group [8, 21]. The β-keto group is optionally modified by additional auxiliary domains like ketoreductase (KS), dehydratase (DH), or enoyl reductase (ER) domains before the growing polyketide chain is released as a linear or cyclic product via the C-terminal thioesterase (Te; termination) domain [8, 17]. Of note, most of the fungal PKS belong to modular iterative type I-PKS, which is a single, giant protein consisting of only one module using the same set of domains of this module in an iterative manner to build the final polyketide [7, 21, 22].
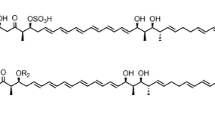
Recently, cyclic lipodepsipeptides, a sub-class of nonribosomal peptides, have attracted a great deal of attention for discovery of new antibiotics [23, 24]. These peptides contain one or more ester bonds in addition to amide bonds. Phomafungin (1) is a known cyclic lipodepsipeptide which is produced by a widespread tropical Phoma sp. It was initially identified, isolated and characterized based on a Candida albicans fitness test profile (CaFT) [25]. The compound was shown to have broad-spectrum antifungal activity against human fungal pathogens including Candida sp. Its structure resembles other cyclic lipodepsipeptides like Syringomycin E [26], Pseudomycin A [27], Cormycin A [28], and Phaeofungin [29]. It consists of a closed ring of eight amino acids (Homoserine-Threonine-Asparagine-Serine-Glycine-Glutamate-Homoserine-Alanine) linked to a fatty acid lipid hydrocarbon tail of β-hydroxy-γ-methyl-hexadecanoic acid (HMHDA) as shown in Fig. 1. Until recently, there has been no further research on this compound and there is no reported biosynthetic gene cluster for its synthesis.

Structures of Phomafungin (1) and BII-Rafflesfungin (2)
In this study, we report the discovery of a new cyclic lipodepsipeptide variant, BII-Rafflesfungin, from a marine Phoma species. It structurally resembles Phomafungin but differs in the amino acid composition of the closed ring structural subunit; it also retains the expected antifungal activity. Analysis of the genome sequence identified a combined NRPS-t1PKS biosynthetic gene cluster consistent with a role in the biosynthesis of this new compound.
Results
Sequencing and sequence annotation of the genome from Phoma strain F3723
The discovery of Phomafungin with distinct antifungal activity [25] without any identification of its biosynthetic genes or enzymes motivated us to analyse the publicly available Phoma genomes: Phoma herbarum (GCA_001599375.1) and, more recently, Phoma koolunga (GCA_004151575.1, GCA_004151145.1, GCA_004151165.1). Unfortunately, no NRPS/PKS was detected that could be speculated as the correct gene cluster for the synthesis of Phomafungin (for details see Additional File 1: Table S1). Because we did not have access to the original Phoma strain from which Phomafungin was isolated, we tested extracts derived from Phoma strains in a large strain collection [5] for antifungal activity and found that the extract of one strain, F3723, had growth inhibitory activity against Candida albicans (data not shown).
We used PacBio SMRT sequencing to determine the genome sequence of Phoma strain F3723. The 18S rRNA of F3723 is 99.8% identical with 18S rRNA of Phoma MJ76 (E-value = 0.0, Accession: HM590661) confirming the original species annotation in the library [5]. This finding is further supported by the phylogenetic analysis based on the standard protein coding marker gene, beta-tubulin, where F3723 clusters together with all other Phoma species (Additional File 2: Figure S1).
The resulting sequences including their protein-coding stretches, predicted by using AUGUSTUS (version 3.2.2) [30] (11,055 predicted protein sequences) and GeneMark-ES (version4.10) [31, 32] (11,687 predicted protein sequences), were analysed both with our in-house ANNOTATOR pipeline [33] as well as with antiSMASH (Version 3.0.3) [34] to detect any biosynthetic gene clusters consistent with the production of Phomafungin. There was no exact match but we found a single almost complete gene cluster having one NRPS gene with eight A domains which is consistent with the presence of eight amino acid residues in Phomafungin. Next to this cluster, as shown in Fig. 2a, we found genes encoding a type I-PKS module which could possibly synthesize the lipid part of Phomafungin. Of note, a type II thioesterase domain containing protein (orf-a) and an AMP-dependent ligase (orf-i) were also found in the vicinity of the NRPS/PKS genes.

The predicted biosynthetic gene cluster BIIRfg. a Gene organization of the predicted biosynthetic gene cluster. Red arrows show the biosynthetic genes while yellow ones are other genes present in the vicinity. The direction of arrows corresponds to that of the reading frame. The neighboring orfs of NRPS and PKS genes are labelled a, b, c, d, e, f, g, h, i, j and k. b Domain structure of the NRPS gene: The green ovals labelled An (n = 1..8) represent adenylation domains. The orange ovals labelled Cn (n = 1..8) represent condensation domains. The predicted condensation domain, which was not predicted by antiSMASH, is shown in yellow oval. The last condensation domain is labelled as CT and is shown in red oval. The pink ovals labelled En (n = 1,2) represent epimerization domains. Cyan ovals labelled PCPn (n = 1..9) represent peptidyl carrier protein (PCP) domains. The constituent modules in the cluster are marked from 1 to 10. c Domain structure of PKS gene: T1-PKS module consists of a beta-ketoacyl synthase (KS) domain shown in green, acyltransferase (AT) domain in orange, dehydratase (DH) domain in magenta, methyltransferase (cMT) domain in grey, enoyl reductase (ER) domain in cyan, ketoreductase (KR) in yellow and acyl carrier protein (ACP) domain in red
Detailed analysis of the NRPS gene architecture
According to the antiSMASH output, the NRPS gene in the cluster consists of 27 domains organized in 10 modules (M); M1 = PCP1, M2 = C1-A1-PCP2, M3 = C2-A2-PCP3, M4 = C3-A3-PCP4, M5 = C4-A4-PCP5, M6 = C5-A5-PCP6, M7 = C6-A6-PCP7-E1, M8 = C7-A7-PCP8-E2, M9 = A8-PCP9, M10 = CT. The initiation module, M1, consists of a single PCP domain which is sufficient to accept the fatty acid intermediate for further elongation of the peptide chain by subsequent downstream domains [35,36,37,38]. Each of the modules M2-M9 is thought to be responsible for catalyzing the attachment of one amino acid to the growing chain of the compound, in total eight amino acids. In accordance with their functionality, modules M2-M9 are expected to be repetitive blocks of C-A-PCP domains. We observed that modules M2-M8 were complete but module 9 (M9) lacked the C domain. Modules 7 and 8 (M7 and M8) harbor additional E domains M7 = C6-A6-PCP7-E1 and M8 = C7-A7-PCP8-E2 besides the typical C-A-PCP domains. The termination module M10 consists of the terminal condensation domain CT which is believed to help in cyclization of the peptide as reported previously in different studies [37, 38]. Unlike bacterial NRPS systems, where the terminating domain is usually a thioesterase domain, fungi typically have this type of specialized CT domain [39,40,41].
We were able to identify the missing C domain in module 9 based on a HMMER search [42] within our ANNOTATOR [33] environment with a profile created from the other eight C domains of the cluster against the translated protein sequence of the cluster region between E2 (end of module M8) and A8 (module M9). We got a single significant hit (E-value = 8.5e-82, see details in Additional File 3: Figure S2). Figure 3 shows an alignment of all nine C-domains from the cluster. Of note, all known functionally important residues were conserved. This suggests that the NRPS gene encodes a complete enzyme that could synthesize a depsipeptide consisting of eight amino acids. Its predicted complete structure (28 domains, 10 modules) is shown in Fig. 2b.

Alignment of all nine condensation domains of the NRPS gene cluster. The alignment also includes the C8 condensation domain which was not predicted by antiSMASH. The conserved motif HHxxxDG is shown in red box. For C1, C7, C8 and C9 (the last condensation domain which acts as termination domain) the first H (His) is replaced by S (Ser) but the functionally important second H (His) residue of the motif is found conserved in all the condensation domain sequences
Identification of specificity conferring codes for the adenylation domains reveals that the NRPS cluster is not compatible with the biosynthesis of Phomafungin
Deep sequence analysis of all A domains of the NRPS gene was done to identify the specificity determining codes of the A domains [43, 44]. For this purpose, all A-domains from the NRPS gene were aligned, using MAFFT [45], with the phenylalanine activating adenylation domain (PheA) of gramicidin S synthetase (GrsA; P0C061 as reference sequence [11, 46,47,48]. This alignment was used to predict amino acid residues responsible for substrate specificity. Table 1 summarizes these amino acids also known as Stachelhaus codes (according to the GrsA Phe numbering) [11] of the NRPS A domains, A1 to A8. To our great surprise, domains A4, A5 and A6 display the identical Stachelhaus code ‘DVQTVMAI’ and, therefore, are expected to activate the same amino acid. However, Phomafungin does not contain three identical amino acid residues. Thus, the NRPS cluster described here is not compatible with the biosynthesis of the peptide part of Phomafungin. Consequently, we hypothesized that F3723 may synthesize a novel variant of Phomafungin.
Towards the structure of the new compound BII-Rafflesfungin (2)
Since the extract derived from F3723 was previously shown to possess antifungal activity against Candida albicans, we used a C. albicans whole-cell assay (see Methods, “Antifungal assay”) to guide purification of the bioactive compound. This yielded a light yellowish oil, which we denote as BII-Rafflesfungin, with an ESIMS [M + H]+ peak at m/z 1002 and a molecular formula of C44H75N9O17 based on HRESIMS measurement. Notably, the mass of the new compound is 28 units smaller than that reported for Phomafungin, which would be consistent with the absence of two -CH2- groups [25].
The 13C NMR showed a total of 44 carbon resonances, comprising 19 methylenes (5 overlapped), 10 methines, 4 methyls, and 11 carbonyl carbon atoms. The observed 11 carbonyl signals resonating at δ 168.8–174.4 in the 13C NMR, together with the observed large methylene signals observed at δ 1.22–1.31 in the 1H NMR, suggested a lipopeptide moiety (Table 2). Further analysis of the HSQC, HMBC, and COSY spectra (see Methods section and Additional file 4: Figures. S3-S7) revealed the presence of 8 amino acid (an alanine (Ala), a glutamic acid (Glu), an asparagine (Asn), three serines (Ser), a threonine/allo-threonine (Thr/allo-Thr), and a glycine (Gly)) moieties, incorporating a fatty acyl (FA) group resembling the arrangement in Phomafungin [25]. The observed 28 mass units (corresponding to two -CH2- groups) lesser in BII-Rafflesfungin compared to Phomafungin are consistent with the replacement of two homoserine (Homoser) units in Phomafungin by two serine (Ser) units in BII-Rafflesfungin. The fatty acyl group in BII-Rafflesfungin was identified as β-hydroxy-γ-methyl hexadecanoic acid (HMHDA) based on the 1D, 2D NMR, and HRESIMS data being identical to those of Phomafungin [25].
We were able to establish a partial sequence of BII-Rafflesfungin as Gly-HMHDA-Ala-Glu-Asn through the HMBC couplings from α-protons and/or α-NH to carbonyl carbons of adjacent residues (Fig. 4). But, based on this method, the exact positions of the three serine and the threonine/allo-threonine residues in the peptide could not be determined. The planar structure of BII-Rafflesfungin was subsequently confirmed by HRESIMS/MS analysis of the hydrolysed product (linear peptide) which gave a pseudo molecular ion [M + H]+ at m/z 1020. The HR-MS/MS analysis of this linear peptide produced both b- and y-type fragments and is shown in Fig. 5. The b series fragment ions observed at m/z 945, 844, 757, 670, 583, 469, and 340 indicate sequential losses of Gly, Thr, Ser, Ser, Ser, Asn, Glu. Therefore, we establish the sequence of BII-Rafflesfungin as HMHDA-Ala-Glu-Asn-Ser-Ser-Ser-Thr-Gly with predicted structure as shown in (2) in Fig. 1. To note, the predicted structure of BII-Raflesfungin is in full agreement with the partial findings from the sequence analysis of the NRPS cluster.

Cosy and HMBC correlations of BII-Rafflesfungin. The sequential positions of the three serines and of threonine could not be determined based on these methods

MS2 fragments of the hydrolysed product (linear peptide)
To conclude, there is experimentally verified evidence that the Phoma species reported in the present study produces a new cyclic lipodepsipeptide that we named BII-Rafflesfungin. Its planar structure is as shown in (2) in Fig. 1. The related biosynthetic gene cluster was named as ‘BIIRfg’ and the NRPS gene responsible for the biosynthesis of the cyclic peptide core of BII-Rafflesfungin was named as ‘BIIRfg_NRPS’. The sequence of BIIRfg was submitted to Genbank (Accession number MK043052).
Revisiting the Stachelhaus codes of the BIIRfg NRPS adenylation domains: discovery of 6 new Stachelhaus codes
Even without knowing the exact sequence of BII-Rafflesfungin, we were able to predict some new Stachelhaus codes [9] (see Table 1) from the amino acid composition and the NRPS cluster sequence for BII-Rafflesfungin. The identical signatures for A4, A5, and A6 should code for serine as this is the only amino acid with three occurrences in the sequence. Further, the signature code for the A7 domain is ‘DAQTIMAI’ which is very similar to the code ‘DVQTVMAI’ for the serine-activating domain. Given that serine and threonine are structurally similar with polar uncharged side chains, we predicted that the A7 domain is a threonine-activating domain. In addition, we were able to predict A2 as a glutamic acid-activating domain based on similarity with the known codes [41]. Also, the NRPSPredictor2 [44] suggests the most probable Stachelhaus code to be Glu.
Based on the experimental structural elucidation of the new compound BII-Rafflesfungin, we were able to fill in all the gaps in Table 1 regarding the information about which adenylation domain selects/activates which amino acid. The predicted Stachelhaus codes of the adenylation domains A2 and A7 are in good agreement with the experimental findings. Most importantly, the three identical Stachelhaus codes for adenylation domains A4, A5 and A6 code for the same amino acid serine.
To our best knowledge, none of the new Stachelhaus codes found in our analysis for 6 amino acids (Ala, Glu, Asn, Ser, Thr, Gly; Table 1) was described before. Our result is fully supported by theoretical considerations and experimental findings.
Attempts towards the stereochemistry: sequence analysis of functional subtypes of condensation domains
In order to identify the subtypes of the eight condensation domains and to predict the stereochemistry of the amino acids, we carried out HMMER searches against the HMM models of DCL, LCL, starter and dual type of C domains. The HMM models were created from the alignments provided by Rausch et al. [49]. Unfortunately, the result of this analysis was inconclusive because all the C domains were highly scored by the DCL HMM model (Additional file 5: Table S2). This result suggests that all amino acids seem to be D amino acids. However, there is a contradictory line of evidence. For further investigation, we performed a phylogenetic tree analysis of the functional distribution of all the C domains using MEGA7 [50] (Maximum Likelihood (ML) tree). Surprisingly, all the C domains of the NRPS gene were found bundled with dual type C domains despite of being highly scored by the DCL type HMMER models (Additional file 6: Figure S8).
Of note, the NRPS gene harbors two epimerization domains (M7, Ser; M8, Thr) which typically epimerize the amino acid of the growing peptide chain into D configuration. Both domains seem to be functionally intact: We find the functionally important motif/active site HHxxxDxVSW [51] conserved in both sequences (Additional file 7: Figure S9). Interestingly, a HMMER search with the sequences of both epimerization domains E1(M7) and E2 (M8) against enzyme classification (EC) profiles [52, 53] resulted in significant hits to aspartate racemaces (EC 5.1.1.13) with E-values of 6.1 e-17 and 4.8 e-15, respectively. Therefore, we speculate that Ser7 and Thr 8 undergo stereoisomerization. But, to conclude, we cannot deduce the exact functional subtypes of the C domains of the NRPS gene and the stereochemistry of the amino acids from sequence-analytic considerations. This issue requires additional experimental clarification that is described below.
Experimental determination of the stereochemistry of BII-Rafflesfungin
In order to determine the absolute configuration of the amino acid residues, BII-Rafflesfungin (2) was hydrolyzed with 6 N HCl and the acid hydrolysate was derivatized with Nα-(2,4-dinitro-5-fluorophenyl)-L- alaninamide (L-FDAA, Marfey’s reagent). The LCMS analysis of L-FDAA of (2) in comparison with the corresponding derivatives of D- and L-amino acid standards (Marfey’s method [54], see also Methods “Acid hydrolysis and derivatization of BII-Rafflesfungin”) indicated the exclusive presence of L-Ala, D/L-Ser, D-allo-Thr, Gly, L-Asn and L-Glu (Additional file 8: Figure S10). In the case of Ser, both the D- and the L-forms were identified. The observed ratio of L-serine: D-serine was 2:1 in the extracted ion chromatogram (EIC) suggesting the presence of two L-serine and one D-serine amino acids (Additional file 8: Figure S10). Of note, the FDAA-Asn residue was determined by comparison with the retention time of FDAA-Asp instead of FDAA-Asn due to the conversion of asparagine to aspartic acid during acid hydrolysis. We did not determine the absolute configuration of HMHDA.
There is full agreement between the architecture of the BIIRfg_NRPS gene (Fig. 2b) and the experimental findings for the stereochemistry of BII-Rafflesfungin. The NRPS cluster contains three modules (M5, M6, M7) for catalyzing the attachment of the amino acid serine to the growing chain of the compound but only one of them (M8) harbors an epimerization (E) domain. This is a perfect match with the experimental finding that L-serine and D-serine were found in a ratio 2:1. The only other module that contains an epimerization domain is module M8 and this explains the finding of allo-Thr in the D configuration. All the other amino acid residues are found to be in the L configuration. To conclude, based on the combination of the experimental stereochemistry data and the analysis of the NRPS gene architecture, a cyclo-[HMHDA-L-Ala-L-Glu-L-Asn-L-Ser-L-Ser-D-Ser-D-allo-Thr-Gly] lipodepsipeptide structure (2) was established for BII-Rafflesfungin.
The BIIRfg gene cluster contains a single type I polyketide synthase (t1-PKS), an AMP-dependent ligase, and a type II thioesterase domain containing protein in addition to the BIIRfg NRPS gene
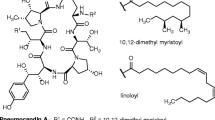
Blastp searches and in-depth sequence analysis using ANNOTATOR [33] with the deduced coding sequences of the BIIRfg cluster were performed to find the gene functions encoded by neighboring ORFs of the BIIRfg NRPS gene. The best hits for each of the ORFs are shown in Table 3. There are three important findings that are possibly related to the biosynthesis of BII-Rafflesfungin: (i) There is a single type I polyketide synthase (t1-PKS) module localized next to the BIIRfg NRPS cluster (Fig. 2c). This module shows the presence of a beta-ketoacyl synthase (KS), acyltransferase (AT), dehydratase (DH), methyltransferase (cMT), enoyl reductase (ER) and ketoreductase (KR) domains. The initial observation does not show the presence of an acyl carrier protein (ACP) domain which is required to complete the PKS module. However, a blastn search of the nucleotide sequence of the predicted PKS region against the NCBI NT database resulted in a full-length hit to a putative polyketide synthase (Fpo_905) from Fusarium poae strain NRRL 26941 (E-value = 0.0). The domain organization of Fpo_905 is KS-AT-DH-cMT-ER-KR-ACP. A blastp search using the protein sequence of the ACP domain from Fpo_905 alone against the BIIRfg_PKS sequence resulted in a significant hit to a 57 amino acids long sequence located at the C-terminus of the PKS (E-value = 3e-29, 84% identity). A HMMER3 search on the same region against Pfam-30 database finds the PP-binding domain (Phosphopantetheine attachment site, PF00550) with an E-value of 4.1e-11. Therefore, we conclude, that the PKS gene cluster (named as BIIRfg_PKS) is complete and has a gene organization as shown in Fig. 2c. Notably, the BIIRfg_PKS cluster harbors a SAM-dependent methyltransferase. A HMMER3 search with its sequence shows that it belongs to class I methyltransferases (Methyltransf_23, PF13489, E-value = 3.3e-16). We find all functionally important residues [55] conserved (data not shown). This type of enzyme is compatible with the production of the lipid part of BII-Rafflesfungin, where there is a methyl group attached to Cγ. (ii) Based on homology searches, orf-i is predicted to be an AMP-dependent ligase. A blastp search with orf-i against the NCBI non-redundant database finds VlmC, the reported AMP-dependent ligase from the verlamelin biosynthetic cluster (E-value = 0.0), and EcdI, the AMP-dependent ligase from the echinocandin biosynthesis cluster (E-value = 2e-122). The predicted AMP-ligase orf-i (573 AA) shows the presence of an AMP-binding domain (PF00501, E-value = 8.2e-45) and an AMP-binding enzyme C-terminal domain (PF13193, Evalue = 2.2e-15) similar to VlmC (580 AA) and EcdI (559 AA). An alignment of the predicted AMP-ligase orf-i along with EcdI and VlmC is provided in Additional file 9: Figure S11. (iii) orf-a (250AA) harbors a type II thioesterase domain (PF00975, E-value = 4.2e-7) which belongs to the alpha-beta hydrolase clan (CL0028). This finding is further supported by structural HHPred hits to 2K2Q_B (Surfactin synthetase thioesterase subunit: SrfAD) with an E-value of 4.8e-24 and to 3FLA_A (Rifamycin:RifR alpha-beta hydrolase thioesterase) with an E-value of 2.7e-17. The catalytic active site residues reported for SrfAD and RifR were also found to be conserved in orf-a. An alignment of the predicted TEII from BII-Rafflesfungin cluster (orf-a) along with SrfAD and RifR is shown in Additional file 10: Figure S12. The alignment highlights the conserved active site triad using red triangles in all the three sequences, S82-D192-H217 in orf-a; S94-D200-H228 in RifR [56], and S86-D189-H216 in SrfAD [57].
To conclude, with a single type I polyketide synthase (t1-PKS), an AMP-dependent ligase, a type II thioesterase domain containing protein, and the described NRPS gene in place, the composition of the BIIRfg gene cluster is consistent with the biosynthesis of the cyclic lipodepsipeptide BII-Rafflesfungin.
BII-Rafflesfungin has anti-fungal activity
We tested the effect of BII-Rafflesfungin on the growth of Candida albicans strain (SC5314) using the Clinical and Laboratory Standards Institute (CLSI) guidelines [58]. The widely used antifungal amphotericin B was used as a positive control. Exponentially-growing Candida albicans cells were treated with either BII-Rafflesfungin or amphotericin B at different concentrations and the growth as measured by OD600 nm was recorded after 48 h. Consistent with published data [58], amphotericin B completely inhibited the growth of C. albicans cells at 125 nM, BII-Rafflesfungin completely inhibited the growth of C. albicans cells at 32 μM (see Additional File 11: Figure S13).
Furthermore, we expanded the antifungal activity evaluation of BII-Rafflesfungin to other yeast strains and filamentous fungi; and the results are summarised in Table 4. The dose response curves are shown in supplementary (Additional file 12: Figure S14). BII-Rafflesfungin inhibited the growth of two additional strains of Candida (ATCC 10231 and ATCC 90028) and Saccharomyces cerevisiae (Additional file 12 Figure S14 (a) and Table 4). The IC50 for the C. albicans strains ATCC10231 and ATCC 90028 were 2.4 μM and 4.6 μM, respectively. For the S. cerevisiae strain (BY4741), the IC50 was 2.7 μM. BII-Rafflesfungin is a potent inhibitor of Aspergillus fumigatus growth, with an IC50 of 1.2 μM; however, it is 6-fold less active against A. brasiliensis (Additional file 12 Figure S14 (b).
To determine whether BII-Rafflesfungin has cytocidal or cytostatic activity, we tested the ability of Candida albicans (SC5314) cells to recover after pre-treatment with either BII-Rafflesfungin (100 μM) or DMSO for 6 h. We used Amphotericin B (10 μM; a cytocidal inhibitor) and Itraconazole (100 μM; a cytostatic inhibitor) as controls. Treatment with all three compounds for 6 h completely inhibited the growth of yeast cells (Additional file 13: Figure S15 A). We then washed off the compound and transferred the cells onto drug-free YPD agar plates. As expected Itraconazole-treated cells were able to grow after their transfer to drug-free YPD agar plates (Additional file 13: Figure S15 B). Similar to cells treated with Amphotericin B, BII-Rafflesfungin-treated cells failed to grow on YPD agar plates indicating that BII-Rafflesfungin has cytocidal activity (Additional file 13: Figure S15 B).
We also evaluated the mammalian cytotoxicity of BII-Rafflesfungin against 2 established cell lines, A549 lung carcinoma cells and HepG2 liver carcinoma cells (Table 4; Fig. S14 (c)). BII-Rafflesfungin was cytotoxic against both cell lines with an IC50 of 16.5 μM and 13.8 μM, respectively.
To conclude, BII-Rafflesfungin has potent broad spectrum anti-fungal activity but moderate level of mammalian cytotoxicity.
Discussion
With a continuous increase in the number of fungal genomes being sequenced, there is a substantial evidence about the abundance of various secondary metabolites which are synthesized by different NRPS/PKS gene clusters. Several of these fungal secondary metabolites are cyclic lipopeptides like destruxins [59], aureobasidine [60], beauvericine [61], apicidin [62] or echinocandins [35]. We have now identified another cyclic depsipeptide in a fungal genome, BII-Rafflesfungin, which is a novel analogue of Phomafungin [25]. We identified the NRPS-t1PKS gene cluster, which we name BIIRfg, responsible for the synthesis of BII-Rafflesfungin. The vicinity of BIIRfg_NRPS was explored to identify a polyketide synthase, BIIRfg_PKS gene, an AMP-dependent ligase, and a type II thioesterase (TEII) which completes the BIIRfg cluster for the biosynthesis of BII-Rafflesfungin. Of note, type II thioesterases are often found to be encoded within PKS and NRPS gene clusters; they fulfill diverse functions, including removal of aberrant residues blocking the megasynthase (repair function), and various roles in substrate selection or, release of intermediates and products [56, 57, 63].
Sequence analysis of BIIRfg identified ten modules consisting of 27 domains which were part of a single NRPS gene. The initial module M1 lacks the A domain as well as the C domain which is consistent with the other known NRPS clusters for lipopeptides like echinocandin (EcdA;AFT91378) [35, 37, 64], emericellamide (EasA; XP_660149) [36] and verlamelin (vlmS;AB862312) [38]. It has been reported that, in such cases, the initial module accepts the activated fatty acid intermediate at the PCP domain for further elongation of the actual peptide by downstream modules [37].
At the outset, we had no independent information about the structure of BII-Rafflesfungin. Since the specificities of many of the A domains have been biochemically characterized [11, 46, 65] and several prediction algorithms have been implemented to predict the specificity determining codes of A domains [34, 46, 66,67,68,69,70,71,72], it is possible to analyse the amino acid sequences of some A domains in order to predict which amino acid would be activated by the corresponding A domain. Therefore, we have investigated the sequences of all the A domains and tried to infer the specificity of binding amino acids from the signature sequence encoded in the A domains. The sequence segments representing A domains are actually Acyl-CoA synthetase NRPS A domains and belong to the Luciferase superfamily of enzymes [73]. Adenylation domains are organized two-partite: (i) a N-terminal core domain and (ii) a C-terminal sub-domain. Several characteristic conserved motifs of A domains (these are mA1:LTYxEL, mA2:LKAGxAYVPID, mA3:LAYxxYTSGTTGxPKG, mA4:FdxS, mA5:NxYGPTE, mA6:GELxIxGxGLARGYW, mA7:YKTGDQ, mA8:GrxDxQVKIRGxRVELEEVE, mA9:LpxYMIP and mA10:NGKIDR) have been well established in various studies [11, 74]. The positioning of the respective substrate α-amino acid is governed by the highly conserved aspartate residue in the A4 motif of the core domain and the lysine residue in the A10 motif of the sub-domain [75]. Originally, it was established that the approximately 100 amino acid residues in the region between core motifs A4 and A5 have ~ 10 critical residues (in the case of the gramicidin S synthetase (GrsA; P0C061), these 10 residues are: D235, A236, W239, T278, I299, A301, A322, I330, C331, also including K517), which represent the signature sequence of an A domain and determine its substrate specificity. But later, Challis et al. [46] explained that Cys331 and Lys 517 can be excluded from the signature sequence as Cys331 points away from the specificity pocket in GrsA [11] while Lys 517 is strictly conserved and does not participate in substrate discrimination. Hence, we took only eight residues in consideration as the signature sequence for the adenylation domains. This set of eight amino acids present at such specific locations is commonly referred to as Stachelhaus code [11]. The sequence positions of these codes are determined based on their proximity to the conserved sequence motifs mA4 and mA5.
The collinearity rule for NRPS genes implies that the order of different modules of an NRPS is coherent with the amino acid sequence of the corresponding peptide product [76]. However certain exceptions/variations to this rule have already been reported [77, 78]. The Stachelhaus codes from the A domains’ sequence for BIIRfg_NRPS combined with the collinearity rule helped to predict some A domain – amino acid assignments. The presence of identical signature codes in A4, A5, and A6 coincides with the observation of three serine residues in BII-Rafflesfungin. Two other codes (for A2 and A7) were predicted based on a sequence similarity approach and these predictions were later confirmed by HRESIMS data.
The original antiSMASH analysis left a C domain in the second last module missing. This domain was later identified using a HMMER search (see details in the Results section and in Additional file 3: Figure S2) which completed the BIIRfg cluster. The list of C domains belonging to the NRPS gene therefore appears to be complete. Various studies on the C domain phylogeny [49, 79, 80] illustrated that these condensation domains have different functional subtypes like:
-
(i)
the LCL (donor C acceptor) domain (it catalyses a peptide bond between two L-amino acids),
-
(ii)
the DCL domain (it catalyses a peptide bond to link a L-amino acid to the elongation peptide ending with a D-amino acids),
-
(iii)
the starter C Domain (it catalyses the acylation of the first amino acid with a β-hydroxyl-carboxylic acid) and,
-
(iv)
the dual E/C domain (it catalyses the peptide bond formation and cyclization of cysteine (Cys), serine (Ser) and threonine (Thr) residues.
There are no clear sequence distinctions between LCD or DCD type of condensation domains [79]. Despite being best scored by the DCL HMMs (Additional file 5: Table S2), the ML tree clusters the C domains from BIIRfg_NRPS with the dual type which however is very close to the DCL type. Similar pattern in clustering of DCL and dual type domains along with other types of C domain have been reported earlier [80] suggesting that these two types of domains are closely related. In fact, biochemical experiments had provided insights that dual C domains have catalytic roles for condensation and epimerization and act as DCL catalysts [81].
Although all the C domains in BIIRfg_NRPS cluster with the dual type, their amino acid sequences do not show the presence of the ‘HHI/LxxxxGD’ motif which is known to be present in the N terminal region of dual C domains along with the ‘HHxxxDG’ motif [49, 81]. The presence of these different core motifs in the C domains was first described by Crecy-Lagard et al. [82] and Marahiel et al. [51] and was subsequently updated by Rausch et al. [49]. As can be seen from the alignment (Fig. 3), all the C domains (with the exception of C1, C7, C8 and C9 (CT)) harbor the conserved ‘HHxxxDG’ motif; C1 and C9 have a ‘SHxxxDG’ motif while C7 and C8 show the presence of an alanine residue in place of the usual glycine residue in a ‘SHxxxDA’ motif. The second His-residue was found conserved in all these condensation domains which is known to be essential for the catalytic function of these domains [83, 84].
Based on all the genomic and experimental information, it is still not trivial to predict the stereochemistry of the amino acids in the final peptide. Nevertheless, it has been reported that traditionally E domains are associated with the modules which add D-amino acids in final peptides like surfactin [85], verlamelin [38] and other cyclic lipopeptides like bacillomycin, locillomycin and fengycin [86]. The epimerization domains in the BIIRfg_NRPS gene observed here also harbor the same ‘HHxxxDxVSW’ motif (where the second histidine is functionally important) [49, 79, 87, 88] (see alignment in Additional File 7: Figure S9). In the case of BII-Rafflesfungin, we were able to show that Ser and allo-Thr that are recognized by A6 (M7) and A7 (M8) are in D-configuration; i.e., there are D-Ser6 and D-allo-Thr7 in the peptide product.
Biosynthesis of BII-Rafflesfungin
Based on our combined computational and experimental results, we propose the following mechanism for the biosynthesis of BII-Rafflesfungin: The lipid part of the compound, β-hydroxy-γ-methyl hexadecanoic acid (HMHDA), is assembled by the BIIRfg_PKS cluster. The cluster itself contains all the necessary domains including a methyltransferase. A similar scenario was described for the biosynthesis of the fungal compound pneumocandin where the present PKS is thought to be responsible for the synthesis of the dimethylmyristate side chain [37, 89]. Based on the current knowledge from different pathways such as echinocandin B, emericellamide, pneumocandin and verlamelin biosynthesis [37, 38, 64, 89], we propose that the lipid moiety is released from the PKS module and activated to form an acyladenylate by orf-i, the predicted AMP-dependent ligase, and, subsequently, loaded on to the first PCP domain of the BIIRfg_NRPS gene to initiate the peptide synthesis as shown in Fig. 6. The CT domain, the last condensation domain of BIIRfg_NRPS, acts like a type I thioesterase. It terminates the peptide synthesis and releases the cyclic lipodepsipeptide BII-Rafflesfungin. We can only speculate on the function of the predicted thioesterase II, in many cases, it has a repair function by removing aberrant residues blocking the megasynthase [63].

Proposed biosynthetic pathway for the synthesis of BII-Rafflesfungin
Mechanism of action of BII-Rafflesfungin
BII-Rafflesfungin displayed antifungal activity against Candida albicans, Saccharomyces cerevisiae and Aspergillus fumigatus. BII-Rafflesfungin’s cytotoxic mode of inhibition suggests that it perturbs the membrane integrity of yeast cells. Similar mode of action was also proposed for Phomafungin, based on its C. albicans fitness test profile [25]. The long hydrophobic chain may facilitate its insertion into the plasma membrane and the polar cyclic peptide unit might affect the membrane structure. For Phomafungin, it was speculated that the primary effect on the plasma membrane is affected by changes in sphingolipid content and Ca2+ concentration [25]. Interestingly, cyclic depsipeptides Syringomycin and Syringopeptins from Pseudomonas inhibit the growth of yeast cells by binding to the plasma membrane [90,91,92]. Another cyclic depsipeptide, Aureobasidin, produced by the fungus Aureobasidium pullulan affects membrane structure by inhibiting inositol phosphorylceramide (IPC, sphingolipid) synthase, a key enzyme involved in sphingolipid biosynthesis [93]. Chemogenomic profiling, a fitness-based assay involving barcoded deletion strains in yeast, could give insights into BII-Rafflesfungin’s mode-of -action [94].
Conclusions
We have discovered a biosynthetic gene cluster predicted to synthesize BII-Rafflesfungin, which is a novel analogue of the known compound Phomafungin. The two compounds have the same amino acid composition with the exception of the presence of two homo-serine residues in Phomafungin instead of two common serines in BII-Rafflesfungin. This is the first study stating the presence of a NRPS-t1PKS gene cluster in Phoma sp for the synthesis of a novel analogue of Phomafungin. The compound BII-Rafflesfungin is biologically important as it shows antifungal activity.
The NRPS-t1PKS cluster ‘BIIRfg’ was found to be complete by the software tool antiSMASH except one C domain in the second last module of BIIRfg_NRPS gene and one ACP domain in the BIIRfg_PKS gene which were later identified by in-depth sequence analysis. The order of the amino acids in the final peptide product was experimentally verified using NMR studies and novel specificity conferring codes were predicted. Further studies using the available biochemical assays [95,96,97,98,99] to confirm the amino acid specificity for the A domains would be insightful. The mechanism of synthesis of BII-Rafflesfungin is proposed which involves the formation of the lipid part by BIIRfg_PKS followed by activation and transfer of the lipid chain by the predicted AMP-ligase on to the first PCP domain of BIIRfg_NRPS gene. This initiates the peptide synthesis by BIIRfg_NRPS gene which successively add different amino acids to the growing peptide and in the end releases the final cyclic lipodepsipeptide, BII-Rafflesfungin.
Methods
Fungal strain
The fungal strain was isolated from a black sea cucumber near the coastal area of Raffles Marina Clubhouse, Singapore. The strain was initially numbered as F3723. The collected black sea cucumber was blended and particles of size 105–210 μm were resuspended in sterile water. Serial dilutions of this suspension were then plated on marine agar supplemented with 50 mg/L tetracycline and allowed to grow at 24 °C for up to 30 days. Fungal colonies growing on the isolation plates were sub-cultured onto marine agar to obtain pure cultures. F3723 was maintained as frozen mycelia in 10% glycerol at -80 °C.
DNA isolation and sequencing
F3723 was grown for 5 days at 24 °C in YMG media (10 g/L malt extract, 4 g/L yeast extract and 4 g/L glucose), and the mycelia were harvested by centrifugation at 4000 g for 10 min. High-quality and molecular weight genomic DNA (> 20 kb) was obtained using the modified CTAB method [100]. Cells were re-suspended in 2X CTAB buffer containing 2% hexadecyltrimethylammonium bromide (CTAB; Sigma), 1.5 M NaCl, 25 mM EDTA, 100 mM Tris-HCl pH 8.8 and 0.1% polyvinylpyrrolidone (Sigma) and homogenised. Proteins were then removed using 1% SDS and proteinase K (25 mg/mL) before treated with RNaseA (10 mg/mL) for the removal of RNA from the total nucleic acids. The DNA was further purified with 25:24:1 phenol chloroform isoamyl alcohol (Sigma), followed by 24:1 chloroform isoamyl alcohol (Sigma). Extracted DNA was re-suspended in 50 μL 10 mM TE buffer pH 8.0 and additional purification step was carried out using Mag-Bind® RxnPure Plus (OMEGA bio-tek) as per manufacturer’s protocol. The quality of DNA was verified using a NanoDropTm 2000 spectrophotometer (Thermo Scientific), Qubit™fluorometric quantitation with Qubit™ 3.0 Fluorometer (Thermo Scientific) and agarose gel electrophoresis. Purified genomic DNA was sheared to approximately 20 kb using a g-Tube (Covaris). A SMRTbell library was prepared according to manufacturer’s instructions, loaded with a MagBead bound library protocol onto two SMRTCells at 0.125 and 0.3 nM, and sequenced using the P5-C3 chemistry on the PacBio RSII instrument (Pacific Biosciences) with a 180 min movie time. Two additional SMRTCells were run at 0.2 and 0.4 nM and sequenced using the P6-C4 chemistry on the PacBio RSII instrument with a 240 min movie time. De novo assembly was performed with the Hierarchical Genome Assembly Process 3 (HGAP3) [101] in the SMRT Analysis suite (version 2.3) using all default parameters. In total, 288,789 reads were collected, with a mean read length of 13,441 bp, giving a total of 3,881,628,628 bp of raw sequence. De novo assembly of the raw sequence resulted in 122 polished contigs. The total length of these 122 assembled contigs was 35,411,911 bp; the largest contig size was 2,158,956 bp, and the N50 contig length was 978,069 bp. Given the final genome assembly size, therefore, the overall sequencing coverage was ~ 110 × .
BII-Rafflesfungin: extraction and isolation
The culture broths (160 × 50 mL, total 8 L) of Phoma (F3723) were combined and centrifuged to separate the supernatant and the mycelia. The combined mycelia were freeze-dried, extracted two times with MeOH (4 L), filtered and, then, were evaporated to dryness using rotary evaporation. One litre of water was added to the dried methanolic extract (94 g) and charged to a 10 cm × 8 cm Sepra C18-E (50 μm, 65A, phenomenex) column. The column was eluted by isocratic gradient of 20, 50, 80, and 100% aqueous methanol. Each fraction was submitted to antifungal activity testing. The antifungal activity was found to be concentrated in the 80% aqueous methanol fraction, which was then concentrated under reduced pressure and yielded 0.25 g of a partially enriched fraction. The 80% aqueous methanol dried extract was dissolved in 2.5 mL of DMSO and separated by C18 reversed-phase preparative HPLC (solvent A: H2O + 0.1% HCOOH, solvent B: ACN + 0.1% HCOOH; flow rate: 30 mL/min, gradient conditions: 65:35 isocratic for 5 min; 35 to 70% of solvent B over 85 min, followed by 70 to 100% of solvent B over 10 min, and finally isocratic at 100% solvent B for 10 min to give BII-Rafflesfungin (2, 10 mg).
Chemical structural data
The 1H, 13C, HSQC, COSY, and HMBC spectra of the compound are provided in Additional file 4: Figures. S3 to S7.
BII-Rafflesfungin (2) yellowish oil; [α]D + 65.3 (c 0.2, MeOH); UV (MeOH) λmax (log ε) end absorption nm; HRESIMS m/z 1002.5371 (calcd for C44H75N9O17 + H, 1002.5354); 1H and 13C NMR data, see Table 2.
General experimental procedures
JASCO P-2000 digital polarimeter was used to record the optical rotations while GE Healthcare Ultrospec 9000 spectrophotometer was used to obtain the UV spectra.
Bruker DRX-400 NMR spectrometer with Cryoprobe was used to collect the NMR spectra. 5-mm BBI (1H, G-COSY, multiplicity-edited G-HSQC, and G-HMBC spectra) or BBO (13C spectra) probe heads equipped with z-gradients were used.
Agilent 1260 Infinity Preparative-Scale LC/MS Purification System and Agilent 6130B single quadrupole mass spectrometer for LC and LC/MS Systems were used to perform the preparative HPLC analysis.
Agilent UHPLC 1290 Infinity coupled to Agilent 6540 accurate-mass quadrupole time-of-flight (QTOF) mass spectrometer which was equipped with a splitter and an ESI source were used to acquire the HRESIMS and MS/MS spectra. For over 15 min, under standard gradient condition of 100% water with 0.1% formic acid to 100% acetonitrile with 0.1% formic acid, the analysis was performed with a C18 4.6 × 75 mm, 2.7 μm column at flowrate of 2 mL/min. The operating parameters for QTOF were the same as in [6].
Nα-(2,4-Dinitro-5-fluorophenyl)-L-alaninamide (L-FDAA) and amino acid standards were purchased from Sigma Aldrich except D-allo-threonine which was purchased from Chem Cruz. Aspartic acid and glutamic acid were converted from asparagine and glutamine (Sigma Aldrich), respectively.
Hydrolysis of BII-Rafflesfungin
To a stirred solution of BII-Rafflesfungin (2, 1 mg) and 1 mL of distilled water, 1 mL of 0.01 M NaOH was added. The mixture was stirred for 19 h at room temperature. The reaction mixture was charged on a small column of C-18 silica gel and washed with water, and the linear peptide was then eluted with MeOH. The MeOH fraction was concentrated and dried using rotary evaporator to yield 0.8 mg of the opened form of the linear peptide, as light yellowish oil. HRESIMS m/z 1020.5470 (calcd for C44H77N9O18 + H, 1020.5459). HRESIMS/MS: m/z 945.5151 (calcd for C42H73N8O16, 945.5139), 844.4669 (calcd for C38H66N7O14, 844.4662), 757.4343 (calcd for C35H61N6O12, 757.4342), 681.2686 (calcd for C24H41N8O15, 681.2686), 670.4028 (calcd for C32H56N5O10, 670.4022), 583.3701 (calcd for C29H51N4O8, 583.3701), 552.2255 (calcd for C19H34N7O12, 552.2260), 469.3270 (calcd for C25H45N2O6, 469.3272), 438.1828 (calcd for C15H28N5O10, 438.1831), 340.2844 (calcd for C20H38NO3, 340.2846).
Acid hydrolysis and derivatization of BII-Rafflesfungin
BII-Rafflesfungin (0.4 mg) was dissolved in 0.5 mL of 6 N HCl and heated at 100 °C for 8 h. The reaction mixture was cooled to room temperature and evaporated to dryness. The dried acid hydrolysate was added in 100 μL of H2O, 100 μL of 1% Nα-(2,4-Dinitro-5-fluorophenyl)-L-alaninamide (FDAA) in acetone, and 40 μL of 1 M sodium bicarbonate. The mixture was heated at 40 °C, with stirring for 1 h. The reaction mixture was then cooled to room temperature, neutralized with 20 μL of 2 M HCl, and dried in vacuo. The residue was dissolved in H2O (200 μL) and centrifuged to remove insoluble material before LCMS analysis [Zorbax Eclipse Plus 2.1 × 50 mm 1.8 μm, elution with 20 min linear gradient of 10–45% of solvent B (ACN 0.1% FA)]. The retention time (tR, min) for the standard amino acids DAA-amino acid derivatives were as follows: L-alanine (8.661), D-alanine (10.377), L-serine (6.379), D-serine (6.660), glycine (7.922), L-glutamic acid (7.831), D-glutamic acid (8.807), L-aspartic acid (7.100), D-aspartic acid (7.900), L-threonine (6.775), D-threonine (8.816), L-allo-threonine (6.941), D-allo-threonine (7.845) (Additional file 8: Figure S10).
Species annotation and biosynthetic cluster detection
Blast [102] based taxonomic analysis of the 18S rRNA was done which supports the species annotation of F3723. In addition, the beta-tubulin gene from F3723 was scanned against NT database using a blastn search. The most similar sequences were selected and incorporated into a multiple sequence alignment using ClustalO [103]. A maximum likelihood tree was generated from the alignment using Mega7 with JTT matrix under discrete gamma distribution with four categories [50].
The assembled Phoma genome was analysed by a locally installed antiSMASH (version 3.0.3) [34, 104]. Protein coding genes were predicted by using AUGUSTUS (version 3.2.2, default parameters with Fusarium graminearum as training set) [30] and GeneMark-ES (version4.10, default parameters) [31, 32]. The predicted protein sets were further analysed by ANNOTATOR tools [33]. Manual annotation efforts were helpful to detect missing function predictions.
Phylogenetic analysis of the C domains
All the sequences of the different subtypes of C domains were obtained from the NaPDoS server [80]. The amino acid sequences of all C domains from BIIRfg cluster were aligned with the NaPDos dataset using MUSCLE [105]. The ML tree was constructed using Mega7 with JTT matrix under discrete gamma distribution with four categories [50].
Antifungal assays
The Minimal Inhibitory Concentration (MIC) of C. albicans (SC5314) cells was determined using the CLSI guidelines [58]. Cells were treated with various concentrations (0 μM – 64 μM, see Additional file 11: Figure S13) of BII-Rafflesfungin and Amphotericin B (0 nM – 1000 nM, Fig. S13) in RPMI 1640 medium in duplicates in a 96-well microplate. The microplate was photographed after 48 h of incubation at 35 °C. The least concentration of the compound that caused an optically clear well was computed as its MIC.
Antifungal activity was also determined against five strains: three yeasts, Candida albicans ATCC 10231, Candida albicans ATCC 90028 and Saccharomyces cerevisiae BY4741; and two moulds, Aspergillus brasiliensis ATCC 16404 and Aspergillus fumigatus ATCC 46645. The activity was evaluated by determining the IC50 value using a rapid microbroth dilution method in 96- or 384-well microplate format [106]. Logarithmically growing yeast (Candida albicans/ Saccharomyces cerevisiae) cells in Sabouraud Dextrose Broth (SDB) medium were treated with various concentrations (0, 0.47, 0.94, 1.88, 3.75, 7.5, 15 and 30 μM) of BII-Rafflesfungin in triplicates. Cultures of Candida albicans strains ATCC 10231 and ATCC 90028 were incubated at 25 °C and 35 °C, respectively. Saccharomyces cerevisiae cultures were incubated at 30 °C. The growth (OD600nm) was recorded after 24 h incubation.
Frozen stocks of Aspergillus conidial suspensions were diluted in SDB medium to final concentrations of approximately 2.5 × 103 spores/mL (ATCC 16404) or 2.5 × 104 spores/mL (ATCC 46645). These conidial suspensions were then added to various concentrations (3 nM to 100 μM) of BII-Rafflesfungin in 384-well microplates in triplicates. The microplates were subsequently incubated for 72 h at 24 °C prior to OD600nm measurement.
For the cytocidal/cytostatic activity test, C. albicans (SC5314) cells at a starting OD600 nm of 0.6 were incubated with either DMSO or BII-Rafflesfungin (100 μM) or Amphotericin B (10 μM) or Itraconazole (100 μM) in duplicate in YPD medium at 30 °C with shaking (220 rpm). OD600nm values of the cultures after 6 h were obtained. Subsequently, the cells were washed twice, normalized and diluted in YPD medium. Equal volumes (3 μl) were spotted on YPD agar plates. Growth was recorded after incubation of the plates at 30 °C for 2 days.
Cytotoxicity of BII-Rafflesfungin on the A549 human lung carcinoma cell line and the liver carcinoma cell line HepG2 were determined as described before [6]. A549 cells were seeded at 1500 cells per well, and HepG2 cells were seeded at 2500 cells per well in a 384-well microplate. Cells were treated with a series of BII-Rafflesfungin concentrations ranging from 0.8 μM to 100 μM and incubated for 72 h at 37 °C in the presence of 5% CO2. Cytotoxic effect of the compound was measured using the PrestoBlue™ cell viability reagent (Life Technologies). Following incubation of the microplates with the dye for 2 h, the fluorescence reading (Excitation / Emission: 560 nm /590 nm) was recorded using the Tecan Infinite M1000 Pro reader.
References
Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. 2015;14:111–29.
Cragg GM, Grothaus PG, Newman DJ. New horizons for old drugs and drug leads. J Nat Prod. 2014;77:703–23.
Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–35.
Newman DJ, Cragg GM. Drugs and drug candidates from marine sources: an assessment of the current "state of play". Planta Med. 2016;82:775–89.
Ng SB, Kanagasundaram Y, Fan H, Arumugam P, Eisenhaber B, Eisenhaber F. The 160K natural organism library, a unique resource for natural products research. Nat Biotechnol. 2018;36:570–3.
Sirota FL, Goh F, Low KN, Yang LK, Crasta SC, Eisenhaber B, Eisenhaber F, Kanagasundaram Y, Ng SB. Isolation and identification of an Anthracimycin analogue from Nocardiopsis kunsanensis, a halophile from a Saltern, by genomic mining strategy. J Genomics. 2018;6:63–73.
Salomon CE, Magarvey NA, Sherman DH. Merging the potential of microbial genetics with biological and chemical diversity: an even brighter future for marine natural product drug discovery. Nat Prod Rep. 2004;21:105–21.
Schwarzer D, Marahiel MA. Multimodular biocatalysts for natural product assembly. Naturwissenschaften. 2001;88:93–101.
Hutchinson CR. Polyketide and non-ribosomal peptide synthases: falling together by coming apart. Proc Natl Acad Sci U S A. 2003;100:3010–2.
Conti E, Stachelhaus T, Marahiel MA, Brick P. Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 1997;16:4174–83.
Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol. 1999;6:493–505.
Weber T, Baumgartner R, Renner C, Marahiel MA, Holak TA. Solution structure of PCP, a prototype for the peptidyl carrier domains of modular peptide synthetases. Structure. 2000;8:407–18.
Keating TA, Marshall CG, Walsh CT, Keating AE. The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains. Nat Struct Biol. 2002;9:522–6.
Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure. 2007;15:781–92.
Stachelhaus T, Mootz HD, Bergendahl V, Marahiel MA. Peptide bond formation in nonribosomal peptide biosynthesis. Catalytic role of the condensation domain. J Biol Chem. 1998;273:22773–81.
Weber T, Marahiel MA. Exploring the domain structure of modular nonribosomal peptide synthetases. Structure. 2001;9:R3–9.
Dutta S, Whicher JR, Hansen DA, Hale WA, Chemler JA, Congdon GR, Narayan AR, Hakansson K, Sherman DH, Smith JL, et al. Structure of a modular polyketide synthase. Nature. 2014;510:512–7.
Khosla C. Harnessing the biosynthetic potential of modular polyketide synthases. Chem Rev. 1997;97:2577–90.
Smith JL, Skiniotis G, Sherman DH. Architecture of the polyketide synthase module: surprises from electron cryo-microscopy. Curr Opin Struct Biol. 2015;31:9–19.
Yuzawa S, Backman TWH, Keasling JD, Katz L. Synthetic biology of polyketide synthases. J Ind Microbiol Biotechnol. 2018.
Chen H, Du L. Iterative polyketide biosynthesis by modular polyketide synthases in bacteria. Appl Microbiol Biotechnol. 2016;100:541–57.
Fujii I. Functional analysis of fungal polyketide biosynthesis genes. J Antibiot (Tokyo). 2010;63:207–18.
Bionda N, Pitteloud JP, Cudic P. Cyclic lipodepsipeptides: a new class of antibacterial agents in the battle against resistant bacteria. Future Med Chem. 2013;5:1311–30.
Bionda N, Fleeman RM, de lF-N, Rodriguez MC, Reffuveille F, Shaw LN, Pastar I, Davis SC, Hancock REW, Cudic P. Identification of novel cyclic lipopeptides from a positional scanning combinatorial library with enhanced antibacterial and antibiofilm activities. Eur J Med Chem. 2016;108:354–63.
Herath K, Harris G, Jayasuriya H, Zink D, Smith S, Vicente F, Bills G, Collado J, Gonzalez A, Jiang B, et al. Isolation, structure and biological activity of phomafungin, a cyclic lipodepsipeptide from a widespread tropical Phoma sp. Bioorg Med Chem. 2009;17:1361–9.
Grilley MM, Stock SD, Dickson RC, Lester RL, Takemoto JY. Syringomycin action gene SYR2 is essential for sphingolipid 4-hydroxylation in Saccharomyces cerevisiae. J Biol Chem. 1998;273:11062–8.
Di GD, Camoni L, Marchiafava C, Ballio A. Biological activities of pseudomycin a, a lipodepsinonapeptide from Pseudomonas syringae MSU 16H. Phytochemistry. 1997;45:1385–91.
Scaloni A, Dalla SM, Amodeo P, Mannina L, Vitale RM, Segre AL, Cruciani O, Lodovichetti F, Greco ML, Fiore A, et al. Structure, conformation and biological activity of a novel lipodepsipeptide from Pseudomonas corrugata: cormycin a. Biochem J. 2004;384:25–36.
Singh SB, Ondeyka J, Harris G, Herath K, Zink D, Vicente F, Bills G, Collado J, Platas G, Gonzalez d, V et al.: Isolation, structure, and biological activity of Phaeofungin, a cyclic lipodepsipeptide from a Phaeosphaeria sp. using the genome-wide Candida albicans fitness test. J Nat Prod 2013, 76:334–345.
Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–44.
Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33:6494–506.
Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–90.
Eisenhaber B, Kuchibhatla D, Sherman W, Sirota FL, Berezovsky IN, Wong WC, Eisenhaber F. The recipe for protein sequence-based function prediction and its implementation in the ANNOTATOR software environment. Methods Mol Biol. 2016;1415:477–506.
Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Muller R, Wohlleben W, et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–43.
Cacho RA, Jiang W, Chooi YH, Walsh CT, Tang Y. Identification and characterization of the echinocandin B biosynthetic gene cluster from Emericella rugulosa NRRL 11440. J Am Chem Soc. 2012;134:16781–90.
Chiang YM, Szewczyk E, Nayak T, Davidson AD, Sanchez JF, Lo HC, Ho WY, Simityan H, Kuo E, Praseuth A, et al. Molecular genetic mining of the Aspergillus secondary metabolome: discovery of the emericellamide biosynthetic pathway. Chem Biol. 2008;15:527–32.
Huttel W. Structural diversity in echinocandin biosynthesis: the impact of oxidation steps and approaches toward an evolutionary explanation. Z Naturforsch C. 2017;72:1–20.
Ishidoh K, Kinoshita H, Nihira T. Identification of a gene cluster responsible for the biosynthesis of cyclic lipopeptide verlamelin. Appl Microbiol Biotechnol. 2014;98:7501–10.
Du L, Lou L. PKS and NRPS release mechanisms. Nat Prod Rep. 2010;27:255–78.
Kopp F, Marahiel MA. Macrocyclization strategies in polyketide and nonribosomal peptide biosynthesis. Nat Prod Rep. 2007;24:735–49.
von DH: A survey of nonribosomal peptide synthetase (NRPS) genes in Aspergillus nidulans. Fungal Genet Biol 2009, 46 Suppl 1:S45-S52.
Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195.
Li W, Rokni-Zadeh H, De VM, Ghequire MG, Sinnaeve D, Xie GL, Rozenski J, Madder A, Martins JC, De MR. The antimicrobial compound xantholysin defines a new group of Pseudomonas cyclic lipopeptides. PLoS One. 2013;8:e62946.
Mizuno CM, Kimes NE, Lopez-Perez M, Auso E, Rodriguez-Valera F, Ghai R. A hybrid NRPS-PKS gene cluster related to the bleomycin family of antitumor antibiotics in Alteromonas macleodii strains. PLoS One. 2013;8:e76021.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Challis GL, Ravel J, Townsend CA. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem Biol. 2000;7:211–24.
De B, I, de Kock MJ, Yang M, de WP, van Beek TA, Raaijmakers JM: Genome-based discovery, structure prediction and functional analysis of cyclic lipopeptide antibiotics in Pseudomonas species. Mol Microbiol 2007;63:417–28.
Zhang W, Li Y, Qian G, Wang Y, Chen H, Li YZ, Liu F, Shen Y, Du L. Identification and characterization of the anti-methicillin-resistant Staphylococcus aureus WAP-8294A2 biosynthetic gene cluster from Lysobacter enzymogenes OH11. Antimicrob Agents Chemother. 2011;55:5581–9.
Rausch C, Hoof I, Weber T, Wohlleben W, Huson DH. Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol Biol. 2007;7:78.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Marahiel MA, Stachelhaus T, Mootz HD. Modular peptide Synthetases involved in nonribosomal peptide synthesis. Chem Rev. 1997;97:2651–74.
Desai DK, Nandi S, Srivastava PK, Lynn AM. ModEnzA: accurate identification of metabolic enzymes using function specific profile HMMs with optimised discrimination threshold and modified emission probabilities. Adv Bioinforma. 2011;2011:743782.
Srivastava PK, Desai DK, Nandi S, Lynn AM. HMM-ModE--improved classification using profile hidden Markov models by optimising the discrimination threshold and modifying emission probabilities with negative training sequences. BMC Bioinformatics. 2007;8:104.
Salib MN, Molinski TF. Cyclic Hexapeptide dimers, Antatollamides a and B, from the ascidian Didemnum molle. A tryptophan-derived auxiliary for l- and d-amino acid assignments. J Org Chem. 2017;82:10181–7.
Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28:329–35.
Claxton HB, Akey DL, Silver MK, Admiraal SJ, Smith JL. Structure and functional analysis of RifR, the type II thioesterase from the rifamycin biosynthetic pathway. J Biol Chem. 2009;284:5021–9.
Koglin A, Lohr F, Bernhard F, Rogov VV, Frueh DP, Strieter ER, Mofid MR, Guntert P, Wagner G, Walsh CT, et al. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature. 2008;454:907–11.
Rex JH: Reference method for broth dilution antifungal susceptibility testing of yeasts : approved standard. Wayne PA: Clinical and Laboratory Standards Institute.; 2008.
Wang B, Kang Q, Lu Y, Bai L, Wang C. Unveiling the biosynthetic puzzle of destruxins in Metarhizium species. Proc Natl Acad Sci U S A. 2012;109:1287–92.
Slightom JL, Metzger BP, Luu HT, Elhammer AP. Cloning and molecular characterization of the gene encoding the Aureobasidin a biosynthesis complex in Aureobasidium pullulans BP-1938. Gene. 2009;431:67–79.
Xu Y, Orozco R, Wijeratne EM, Gunatilaka AA, Stock SP, Molnar I. Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem Biol. 2008;15:898–907.
Jin JM, Lee S, Lee J, Baek SR, Kim JC, Yun SH, Park SY, Kang S, Lee YW. Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. Mol Microbiol. 2010;76:456–66.
Kotowska M, Pawlik K. Roles of type II thioesterases and their application for secondary metabolite yield improvement. Appl Microbiol Biotechnol. 2014;98:7735–46.
Huttel W, Youssar L, Gruning BA, Gunther S, Hugentobler KG. Echinocandin B biosynthesis: a biosynthetic cluster from Aspergillus nidulans NRRL 8112 and reassembly of the subclusters Ecd and Hty from Aspergillus pachycristatus NRRL 11440 reveals a single coherent gene cluster. BMC Genomics. 2016;17:570.
Stachelhaus T, Marahiel MA. Modular structure of peptide synthetases revealed by dissection of the multifunctional enzyme GrsA. J Biol Chem. 1995;270:6163–9.
Ansari MZ, Yadav G, Gokhale RS, Mohanty D. NRPS-PKS: a knowledge-based resource for analysis of NRPS/PKS megasynthases. Nucleic Acids Res. 2004;32:W405–13.
Bachmann BO, Ravel J. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217.
Blin K, Medema MH, Kottmann R, Lee SY, Weber T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2017;45:D555–9.
guero-Chapin G, Molina-Ruiz R, Maldonado E, de la RG, Sanchez-Rodriguez A, Vasconcelos V, Antunes A. Exploring the adenylation domain repertoire of nonribosomal peptide synthetases using an ensemble of sequence-search methods. PLoS One. 2013;8:e65926.
Prieto C, Garcia-Estrada C, Lorenzana D, Martin JF. NRPSsp: non-ribosomal peptide synthase substrate predictor. Bioinformatics. 2012;28:426–7.
Prieto C. Characterization of nonribosomal peptide Synthetases with NRPSsp. Methods Mol Biol. 2016;1401:273–8.
Rottig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. NRPSpredictor2--a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–7.
Gulick AM. Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–27.
Lee TV, Johnson LJ, Johnson RD, Koulman A, Lane GA, Lott JS, Arcus VL. Structure of a eukaryotic nonribosomal peptide synthetase adenylation domain that activates a large hydroxamate amino acid in siderophore biosynthesis. J Biol Chem. 2010;285:2415–27.
Sussmuth RD, Mainz A. Nonribosomal peptide synthesis-principles and prospects. Angew Chem Int Ed Engl. 2017;56:3770–821.
Schwarzer D, Finking R, Marahiel MA. Nonribosomal peptides: from genes to products. Nat Prod Rep. 2003;20:275–87.
Luo C, Liu X, Zhou X, Guo J, Truong J, Wang X, Zhou H, Li X, Chen Z. Unusual biosynthesis and structure of Locillomycins from Bacillus subtilis 916. Appl Environ Microbiol. 2015;81:6601–9.
Rouhiainen L, Jokela J, Fewer DP, Urmann M, Sivonen K. Two alternative starter modules for the non-ribosomal biosynthesis of specific anabaenopeptin variants in Anabaena (cyanobacteria). Chem Biol. 2010;17:265–73.
Roongsawang N, Lim SP, Washio K, Takano K, Kanaya S, Morikawa M. Phylogenetic analysis of condensation domains in the nonribosomal peptide synthetases. FEMS Microbiol Lett. 2005;252:143–51.
Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR. The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS One. 2012;7:e34064.
Balibar CJ, Vaillancourt FH, Walsh CT. Generation of D amino acid residues in assembly of arthrofactin by dual condensation/epimerization domains. Chem Biol. 2005;12:1189–200.
De Crecy-Lagard V, Marliere P, Saurin W. Multienzymatic non ribosomal peptide biosynthesis: identification of the functional domains catalysing peptide elongation and epimerisation. C R Acad Sci III. 1995;318:927–36.
Bergendahl V, Linne U, Marahiel MA. Mutational analysis of the C-domain in nonribosomal peptide synthesis. Eur J Biochem. 2002;269:620–9.
Roche ED, Walsh CT. Dissection of the EntF condensation domain boundary and active site residues in nonribosomal peptide synthesis. Biochemistry. 2003;42:1334–44.
Kraas FI, Helmetag V, Wittmann M, Strieker M, Marahiel MA. Functional dissection of surfactin synthetase initiation module reveals insights into the mechanism of lipoinitiation. Chem Biol. 2010;17:872–80.
Luo C, Liu X, Zhou H, Wang X, Chen Z. Nonribosomal peptide synthase gene clusters for lipopeptide biosynthesis in Bacillus subtilis 916 and their phenotypic functions. Appl Environ Microbiol. 2015;81:422–31.
Bloudoff K, Schmeing TM. Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: discovery, dissection and diversity. Biochim Biophys Acta. 2017;1865:1587–604.
Konz D, Marahiel MA. How do peptide synthetases generate structural diversity? Chem Biol. 1999;6:R39–48.
Chen L, Yue Q, Zhang X, Xiang M, Wang C, Li S, Che Y, Ortiz-Lopez FJ, Bills GF, Liu X, et al. Genomics-driven discovery of the pneumocandin biosynthetic gene cluster in the fungus Glarea lozoyensis. BMC Genomics. 2013;14:339.
Bensaci MF, Gurnev PA, Bezrukov SM, Takemoto JY. Fungicidal activities and mechanisms of action of Pseudomonas syringae pv. Syringae Lipodepsipeptide Syringopeptins 22A and 25A. Front Microbiol. 2011;2:216.
Takemoto JY, Zhang L, Taguchi N, Tachikawa T, Miyakawa T. Mechanism of action of the phytotoxin syringomycin: a resistant mutant of Saccharomyces cerevisiae reveals an involvement of Ca2+ transport. Microbiology. 1991;137:653–9.
Zhang L, Takemoto JY. Syringomycin stimulation of potassium efflux by yeast cells. Biochimica et Biophysica Acta-Biomembranes. 1989;987:171–5.
Aeed PA, Young CL, Nagiec MM, Elhammer AP. Inhibition of inositol phosphorylceramide synthase by the cyclic peptide aureobasidin a. Antimicrob Agents Chemother. 2009;53:496–504.
Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci U S A. 2004;101:793–8.
Duckworth BP, Wilson DJ, Aldrich CC. Measurement of nonribosomal peptide Synthetase Adenylation domain activity using a continuous hydroxylamine release assay. Methods Mol Biol. 2016;1401:53–61.
Ishikawa F, Miyamoto K, Konno S, Kasai S, Kakeya H. Accurate detection of Adenylation domain functions in nonribosomal peptide Synthetases by an enzyme-linked immunosorbent assay system using active site-directed probes for Adenylation domains. ACS Chem Biol. 2015;10:2816–26.
Kittila T, Schoppet M, Cryle MJ. Online pyrophosphate assay for analyzing Adenylation domains of nonribosomal peptide Synthetases. Chembiochem. 2016;17:576–84.
Maruyama C, Niikura H, Takakuwa M, Katano H, Hamano Y. Colorimetric detection of the Adenylation activity in nonribosomal peptide Synthetases. Methods Mol Biol. 2016;1401:77–84.
McQuade TJ, Shallop AD, Sheoran A, Delproposto JE, Tsodikov OV, Garneau-Tsodikova S. A nonradioactive high-throughput assay for screening and characterization of adenylation domains for nonribosomal peptide combinatorial biosynthesis. Anal Biochem. 2009;386:244–50.
Allen GC, Flores-Vergara MA, Krasynanski S, Kumar S, Thompson WF. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc. 2006;1:2320–5.
Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–9.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, de Los Santos ELC, Kim HU, Nave M, et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Balouiri M, Sadiki M, Ibnsouda SK. Methods for in vitro evaluating antimicrobial activity: a review. J Pharm Anal. 2016;6:71–9.
Funding
The authors acknowledge financial support from Agency for Science, Technology and Research (A*STAR) Singapore but the funding body did not participate in the design of the study and collection, analysis and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets generated and analysed during the current study are available in GenBank (Accession Number MK043052). Regarding the availability of the strain, please contact Ng Siew Bee (ngsb@bii.a-star.edu.sg).
Author information
Authors and Affiliations
Contributions
FG, VN, CYL and SBN cultivated the Phoma sp., isolated the DNA for sequencing and prepared the extract from Phoma sp. fermentation. HZ, AL, and SLC performed sequencing and assembly. CEN, KNL and YK isolated the BII-Rafflesfungin compound from the extract and defined its chemical structure. MA, SC, SBN and PA performed the antifungal assay. SC and SBN tested the cytotoxicity. SS, FE and BE performed the bioinformatics analysis. SBN, YK, PA, FE and BE were involved in the project design. SS, CEN, SLC, PA, YK, SC, SBN, FE and BE took part in writing the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. antiSMASH results (links) for publicly available full genome assemblies of Phoma sp. in NCBI (Feb 2019). (PDF 22 kb)
Additional file 2:
Figure S1. Circular representation of the phylogenetic tree of protein coding marker beta-tubulin for inferring the taxonomic classification of strain F3723. (PDF 147 kb)
Additional file 3:
Figure S2. Prediction of the missing C domain in Module 9. (PDF 551 kb)
Additional file 4:
Figure S3. 1H NMR spectrum (DMSO-d6, 400 MHz) of BII-Rafflesfungin. Figure S4. 13C NMR spectrum (DMSO-d6, 100 MHz) of BII-Rafflesfungin. Figure S5. HSQC spectrum (DMSO-d6, 400 MHz) of BII-Rafflesfungin. Figure S6. COSY spectrum (DMSO-d6, 400 MHz) of BII-Rafflesfungin. Figure S7. HMBC spectrum (DMSO-d6, 400 MHz) of BII-Rafflesfungin. (PDF 196 kb)
Additional file 5:
Table S2. HMMER search results against the HMM models of DCL, LCL, starter and dual type of C domains with all 9 C domains as query sequences. (PDF 78 kb)
Additional file 6:
Figure S8. Phylogenetic tree analysis of the functional distribution of C domains using MEGA7 (Maximum Likelihood (ML) tree). (PDF 23 kb)
Additional file 7:
Figure S9. Multiple sequence alignment of the two epimerization domains E1 and E2 from the BIIRfg_NRPS gene. (PDF 124 kb)
Additional file 8:
Figure S10. Results of the LCMS analysis of the absolute configuration of BII-Rafflesfungin by Marfey’s method. (PDF 111 kb)
Additional file 9:
Figure S11. Multiple sequence alignment of the predicted AMP-dependent ligase for BII-Rafflesfungin biosynthesis along with VlmC (AMP-dependent ligase from Verlamelin biosynthesis) and EcdI (AMP-dependent ligase from Echinocandin B biosynthesis). (PDF 353 kb)
Additional file 10:
Figure S12. Multiple sequence alignment of the predicted Type II thioesterase (orf-a) along with the known TEII of Surfactin-Synthetase (SrfAD) and Rifamycin (RifR). (PDF 215 kb)
Additional file 11:
Figure S13. BII-Rafflesfungin inhibits the growth of yeast cells/standard CLSI test with Amphotericin B as positive control. (PDF 224 kb)
Additional file 12:
Figure S14. Growth inhibitory effects of BII Rafflesfungin against yeasts (a), Aspergillus species (b) and mammalian cell lines (c). (PDF 100 kb)
Additional file 13:
Figure S15. BII-Rafflesfungin has cytocidal activity. (PDF 40 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sinha, S., Nge, CE., Leong, C.Y. et al. Genomics-driven discovery of a biosynthetic gene cluster required for the synthesis of BII-Rafflesfungin from the fungus Phoma sp. F3723. BMC Genomics 20, 374 (2019). https://doi.org/10.1186/s12864-019-5762-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5762-6