
Abstract
Background
The new genomic technologies have provided novel insights into the genetics of interactions between vectors, viruses and hosts, which are leading to advances in the control of arboviruses of medical importance. However, the development of tools and resources available for vectors of non-zoonotic arboviruses remains neglected. Biting midges of the genus Culicoides transmit some of the most important arboviruses of wildlife and livestock worldwide, with a global impact on economic productivity, health and welfare. The absence of a suitable reference genome has hindered genomic analyses to date in this important genus of vectors. In the present study, the genome of Culicoides sonorensis, a vector of bluetongue virus (BTV) in the USA, has been sequenced to provide the first reference genome for these vectors. In this study, we also report the use of the reference genome to perform initial transcriptomic analyses of vector competence for BTV.
Results
Our analyses reveal that the genome is 189 Mb, assembled in 7974 scaffolds. Its annotation using the transcriptomic data generated in this study and in a previous study has identified 15,612 genes. Gene expression analyses of C. sonorensis females infected with BTV performed in this study revealed 165 genes that were differentially expressed between vector competent and refractory females. Two candidate genes, glutathione S-transferase (gst) and the antiviral helicase ski2, previously recognized as involved in vector competence for BTV in C. sonorensis (gst) and repressing dsRNA virus propagation (ski2), were confirmed in this study.
Conclusions
The reference genome of C. sonorensis has enabled preliminary analyses of the gene expression profiles of vector competent and refractory individuals. The genome and transcriptomes generated in this study provide suitable tools for future research on arbovirus transmission. These provide a valuable resource for these vector lineage, which diverged from other major Dipteran vector families over 200 million years ago. The genome will be a valuable source of comparative data for other important Dipteran vector families including mosquitoes (Culicidae) and sandflies (Psychodidae), and together with the transcriptomic data can yield potential targets for transgenic modification in vector control and functional studies.
Similar content being viewed by others


Background
Arboviruses (arthropod-borne viruses) are a taxonomically diverse group that include some of the most important emerging and re-emerging pathogens of wildlife, livestock and human beings worldwide [1,2,3]. Among arbovirus vectors, the majority of recent genomic studies have been carried out on the mosquitoes Aedes (Stegomyia) aegypti (L.) and Aedes (Stegomyia) albopictus (Skuse) due to their involvement in human to human transmission of a wide-range of arboviruses, their relative ease of colonization and recent technical advances in genome sequencing and annotation technologies [4]. While these studies have led to major advances in control of the arboviruses these species transmit and our understanding of what drives susceptibility to infection [5,6,7], tools and resources for use with many other vector groups remain neglected.
To date, no genomic analyses have been carried out for vectors of non-zoonotic arboviruses that are pathogenic to livestock and wildlife. Among the most important of these vector groups are Culicoides biting midges (Diptera: Ceratopogonidae), species of which transmit internationally important arboviruses of ruminants, equines and deer [8,9,10]. In recent years, unprecedented outbreaks of Culicoides-borne arboviruses such as bluetongue virus (BTV) have inflicted huge economic losses on the livestock sector of Europe (e.g. estimate of US$ 1.4 billion in France in 2007) through clinical disease and accompanying restrictions against the movement of livestock imposed to limit virus spread [11, 12]. Globally, BTV outbreaks initiate non-tariff trade barriers restricting the movement of livestock and livestock germplasm, and cause a decrease in ruminant productivity in regions as diverse as India and the USA [13]. Globally, the economic impact of bluetongue has been estimated to US$ 3 billion [12].
Culicoides are notoriously difficult to culture under laboratory conditions and their small size of approximately 1.5 mm body length renders them far from ideal subjects for transcriptomic and genome manipulation studies [14]. Only one of 14 confirmed vector species of Culicoides is currently colonized, Culicoides sonorensis Wirth and Jones [8, 15]. Culicoides sonorensis colonies have already provided valuable insights into the genetic basis of vector competence for BTV [16], a de novo transcriptome [17], and have been used to construct a physical map of the C. sonorensis genome which consists of four chromosomes [18, 19].
Susceptibility to infection and transmission of arboviruses by Culicoides is determined in part by the heritability of barriers to virus dissemination following ingestion of the bloodmeal [13, 20, 21]. Experimental evidence of a midgut infection barrier, a midgut escape barrier and a haemocoel dissemination barrier in C. sonorensis (formerly C. variipennis or C. v. sonorensis [22]) have been defined [23]. There is no evidence to date of salivary gland barriers preventing transmission in any species of Culicoides [24], as has been inferred in several species of mosquitoes [20]. Previous studies of the genetic basis for vector competence of C. sonorensis for BTV, using maternal inheritance properties, identified a 90 kd protein and used antibodies to this protein to isolate and characterize a cDNA clone encoding a glutathione S-transferase class delta enzyme [25]. Differential gene regulation in response to BTV infection, however, has not been investigated to date.
In this study, we sequence, de novo assemble, annotate and explore the first full genome of C. sonorensis. We then use transcriptomic analyses both to improve gene prediction within the genome build, and to elucidate differential gene expression associated with Culicoides competence for a BTV serotype 1 strain. The full genome sequence and transcriptome analyses are important resources for further studies of this phylogenetic group which is separated from the other major Dipteran vector families by at least 220 million years [26]. In addition to being a valuable resource for comparative study of vector phylogenomics, the provision of Culicoides genomes is of interest as a group that demonstrates several unique features, including a great capacity for long-distance dispersal by semi-passive flight and the ability to reach huge population density under suitable conditions. In addition, their close association with livestock raises questions concerning both host preference and vector competence for arboviruses. Many of these questions can be readily addressed by understanding genetic diversity within populations which will be enhanced by the provision of comparative genomic data, but also in the long term by yielding targets for transgenic modification. The present genome provides a resource to facilitate all these studies.
Methods
Samples
All C. sonorensis used in this study originated from the ‘AA’ colony which was originally established in 1955 at the Kerrville, Texas laboratory of the US Department of Agriculture [27]. Since 1969, this colony has been maintained at The Pirbright Institute without any additional outbreeding [28]. Within this period, two selection bottlenecks of ≤10 individuals were performed to increase susceptibility to BTV serotype 4 and African horse sickness virus (AHSV) serotype 9 infection (Mellor, Pers Comm). Following the second selection in the 1990’s [29], the colony strain was renamed as PIR-s-3. No attempt was made to further reduce heterozygosity prior to using individuals from the PIR-s-3 line in this study. All C. sonorensis used were 3–4 days old and had not been fed sucrose following emergence from pupae.
DNA extraction
The genomes of C. sonorensis males and females were sequenced separately. Adults were separated by sex under a stereomicroscope and stored in 95% ethanol at room temperature prior to use. In order to obtain sufficient amount of DNA for genomic sequencing, DNA was extracted from 375 males in three pools of 100 and one pool of 75 individuals, and from 150 females in one pool of 100 and one pool of 50 individuals. Pooled samples were homogenised twice for 30 s in 75 μl of phosphate buffered saline using a Tissuelyser™ (Qiagen, UK) at a frequency of 25 Hz/s and a 3 mm stainless-steel ball bearing (DeJay Distribution Ltd., UK) within 2 ml screw-topped tubes. Genomic DNA extraction was conducted using gravity-flow anion-exchange tips (Qiagen’s Genomics Tip 20/G) with extraction of DNA up to 150 kb in size and a maximum of 20 μg of product. A user-developed protocol for mosquitoes and other insects was followed (see Additional file 1: Supplementary Methods). The DNA obtained from each extraction was pooled by sex and the concentration of the resulting samples was evaluated using a Qubit® 2.0 Fluorometer (ThermoFisher Scientific, UK) with the Qubit® DNA BR Assay Kit (ThermoFisher Scientific, UK). The integrity of the genomic DNA was visualized using a 5 μl sample on a 1% (weight/volume w/v) agarose gel.
Genome sequencing and de novo assembly
8–9 μg of high molecular weight (HMW) genomic DNA were used to sequence the genome of male and female C. sonorensis separately. One paired-end (PE) libraries of insert sizes 200 bp a mate-pair (MP) library with an average fragment size of 4.4 kb were sequenced with 150 bp paired-end from both female and male pools were run on Illumina HiSeq 2000 and 2500 sequencing systems. Library construction and sequencing was performed at The Earlham Institute (Norwich, UK). Read pair quality was assessed with FastQC v0.10.1. Results suggested that 6 bp should be removed from the start of each read, and in the case of MP data the last 40–50 bp had unexpectedly high levels of k-mer duplication. Both PE and MP reads were filtered to limit their size to 100 bp after removal of the first 6 bp. MP data was processed with NextClip v1.0 [30] to select reads which contained both ends of the original fragment. This created a dataset of reads with variable length, which were subsequently limited to 100 bp.
PE and MP libraries were initially assembled using Velvet v1.2.08 [31] with a k-mer length of 91. The N50 of the assembly was maximized using an expected coverage of 27, a coverage cut-off of six, and with scaffolding option set to ‘no’. This initial assembly was filtered to remove short (< 4 kb) and repeated contigs (> 90% sequence identity to a larger contig) using BLAT v34 and a custom script to count identical bases, considering any overlaps between matches. The assembly was then scaffolded using SSPACE v2.0 [32] and the 400 bp PE library, using three passes of decreasing stringency. Any remaining contigs smaller than 500 bp were removed (9913 total). The resulting assembly was then scaffolded a second time using the 4.4 kb MP library with SSPACE v2.0 (Fig. 1). This assembly was assessed using FRCbam v1.0 [33] and Reapr v1.0.15 [34], and the output from Reapr was processed with GapFiller v1.11 [35]. The final assembly was assessed using FRCbam v1.0 to confirm that an improvement in the reported error rate had occurred. Genome size was estimated using the number of total trimmed nucleotides which were used in the assembly divided by the maximum of the per-position coverage frequency distribution, after reads were mapped back to the assembly using BWA-MEM [36]. Kmer spectra were assessed using KAT (https://kat.readthedocs.io/en/latest). Redundant contigs were identified using Redundans [37].

Differential gene expression analyses between females susceptible to full BTV infection and refractory females. a Scatterplot of the pairwise comparison of the gene expression levels between the two phenotypes. The average gene expression across all genes is displayed by the blue line. b Volcano plot displaying fold changes in expression of the 165 differentially expressed genes between Culicoides sonorensis that are susceptible to infection and those that are refractory to full infection with BTV
Data (reads, assembly and annotation) have been deposited in the ENA database under the accession number PRJEB19938. The genome used in the subsequent analyses is the latest assembly without redundancy removal.
Gene model prediction and annotation
Prediction and annotation of the genome assembly was conducted using MAKER v2.31.6 [38]. The genome annotation was carried out using the transcriptome data reported in this study and transcriptome data from previous studies on C. sonorensis [17, 39]. In addition, we used the genome annotations of ten species of Anopheles, Ae. aegypti, Belgica antarctica Jacobs, Culex quinquefasciatus Say, Drosophila melanogaster Mg., Lutzomyia longipalpis (Lutz & Neiva, 1912) and Phlebotomus papatasi (Scopoli) (see Additional file 1: Supplementary Methods for the species and genome assembly versions used). SNAP v2006-7-28 [40] and AUGUSTUS v2.5.5 [41] were used to predict ab initio gene models. SNAP v2006-7-28 was trained using 500 models and AUGUSTUS v2.5.5 using the best 1000 models extracted from the initial MAKER v2.31.6 output, as recommended in the software documentation. Coverage of conserved proteins was assessed using the CEGMA v2.4 [42] and BUSCO v3.0.2 [43] pipelines. Analysis of Gene Ontology annotation was performed with Blast2GO v3 [44]. The orthology of the annotated genes was established using the Ensembl Compara Gene Trees pipeline [45], using the genomes of C. sonorensis and 17 other arthropod species as input (see Additional file 1: Supplementary Methods). Genes were functionally annotated using InterProScan, which computationally identifies the presence of domain, motif or family signatures within a protein sequence and infers functional descriptors (taken from the Gene Ontology (GO)).
Infection experiments with bluetongue virus
The virus used to infect C. sonorensis was a western topotype strain of BTV serotype 1 (GIB2007/01) [46]. Virus stock was mixed 1:1 with defibrinated horse blood (TCS Biosciences, Buckingham, UK) with 6.2 Log10 (TCID50)/ml being the final infectious dose. All blood-feeding of C. sonorensis was conducted using a Hemotek membrane-based system (Discovery Workshops, Accrington, UK). A total of 150 and 145 C. sonorensis in two replicates were fed on the blood:BTV-1 suspension and survived the extrinsic incubation period of 8 days at 25 °C. During this period of incubation, 10% (w/v) sucrose solution was offered via a cotton wool pad. Each individual was then decapitated using disposable needles and the head and remainder of the body of each were stored separately in RNAlater™ (Thermofisher Scientific, UK) at − 20 °C.
RNA extraction
RNA extraction from C. sonorensis was performed following the TRIzol® (ThermoFisher Scientific, UK) protocol (see Additional file 1: Supplementary Methods). In brief, samples were homogenised in 100 μl of Schneider’s Drosophila media (Gibco™, Thermofisher Scientific, UK) and RNA was extracted using TRIzol® and chloroform, followed by a precipitation with isopropanol. The integrity of the RNA was analysed using a Bioanalyser 2100 and the concentration was estimated on a Qubit® 2.0 Fluorometer (ThermoFisher Scientific, UK) with the Qubit® DNA BR Assay Kit (ThermoFisher Scientific, UK).
RNA was extracted from individual heads of C. sonorensis fed with blood:BTV-1 as described for the bodies (Additional file 1: Supplementary Methods). The corresponding bodies (abdomen and thorax) of the BTV-1-positive and BTV-1-negative heads were pooled separately for RNA extraction. In addition, the RNA from 100 C. sonorensis fed on horse blood 3 days post-emergence and left without access to sucrose for 8 days, and from 100 C. sonorensis fed on a 10% (w/v) sucrose solution from 3 to 8 days following emergence, were also extracted and their transcriptomes sequenced as controls.
Quantification of infection with BTV-1
We used RT-qPCR to detect and quantify the presence of disseminated BTV-1 viral RNA in the heads of blood:BTV-1 fed C. sonorensis. For this, the RNA was first reverse-transcribed into DNA using the ProtoScript II First Strand cDNA Synthesis kit (New England Biolabs) (see Additional file 1: Supplementary Methods for a detailed protocol). A reaction with no enzyme was included as a no-RT negative control. The resulting cDNA was used as template in RT-qPCRs for the detection of BTV using SYBR Green assays (Additional file 1: Supplementary Methods). The reactions were performed using the BTV specific primers BTVuni 291-311F 5’ GCTTTTGAGGTGTACGTGAAC 3′ and BTVuni 381-357R 5’ TCTCCCTTGAAACTCTATAATTACG 3′ [47]. No-template and no-RT reactions were included as negative controls. To verify that negative reactions were not due to a lack of cDNA in the sample, we also performed RT-qPCR reactions for each sample using C. sonorensis specific primers for the Vacuolar ATPase gene (Vac-ATPase forward 5’ GCTGCTGCTGCCATCATTTT 3′ and Vac-ATPase reverse 5’ CCGGTCGCATCACTGACATA 3′). All reactions were performed in duplicate. Samples with and without BTV-1 RNA were identified using the quantification cycle (Cq) values of the reactions.
For the purposes of this study, we considered vector competent individuals to be those with disseminated infections that include replication in the head capsule, as this has been demonstrated to allow the isolation of BTV-1 [46]. We refer as refractory individuals to those without detectable BTV after a suitable incubation period (8 days), those with BTV midgut infections that had not disseminated within the insect, those where the process of dissemination had not been completed (i.e. those with haemocel dissemination barriers, midgut escape and infection barriers) and those still retaining inactivated BTV following the infective bloodmeal [24]. In the case of this study, we used a Cq value of 27 to differentiate between vector competent and vector refractory (see Results).
Transcriptome sequencing and analyses
In total, eight transcriptomes were sequenced: two biological replicates each for i) BTV-competent; ii) BTV-refractory; iii) blood-fed; and, iv) sucrose-fed. Library construction and sequencing was performed at Edinburgh Genomics (The University of Edinburgh, Scotland). A total of 2.5 μg of RNA from each sample was used to construct TrueSeq libraries, which were sequenced using 50 base paired-end (PE) reads on an Illumina HiSeq 2500. Each library was run in two lanes.
The quality of the raw sequence data was analyzed in FastQC v0.11.2. Reads were aligned to the genome assembly (the version of the genome that has not been cleaned of the redundant scaffolds) with TopHat v2.0.6 [48] using the gene model annotations generated from the genome as reference. The resulting bam files were assembled with Cufflinks v2.2.1 [49] and merged into a single transcriptome with Cuffmerge v2.2.1. Quantification of gene and transcript expression and comparison of the expression levels was performed with Cuffquant v2.2.1 and Cuffdiff v2.2.1, respectively. The Cuffquant results for each transcriptome (two biological + two sequencing lane runs) were grouped per experimental conditions (blood-fed, sucrose-fed, vector competent and vector refractory) to compare the expression profiles between these different experimental conditions with Cuffdiff. Differential expression analyses were then explored with the R package CummeRbund v2.9.3 [50]. Differentially expressed (DE) genes with a significant change in expression level were identified using an α value of 0.05, which establishes the filtering value of the multiple-testing corrected q-values. Identification and functional classification of the DE genes was carried out using BLAST and InterProScan searches in Blast2Go version 4.0.2. Enrichment and gene set enrichment analyses were performed on the sets of DE genes between the different conditions using the interface provided in Blast2Go to the Fisher’s Exact Test implemented in FatiGO [51] and the Gene Set Enrichment Analysis (GSEA) package [52]. To perform the GSEA, all genes were ranked per the logarithmic fold-change, removing the infinite values. Transcriptome data have been deposited in the ENA database under the accession numbers ERR2171964-ERR2171979.
Validation of differentially expressed genes
Quantitative reverse transcription PCR (RT-qPCR) was used to validate the change in expression of four differentially expressed (DE) genes between the vector competent and refractory pools of C. sonorensis. These DE genes were selected based on previous identification in vector competence studies of C. sonorensis or because they have a direct functional link with antiviral response. The genes were: a gene sharing 73% identity with the antiviral helicase ski2 of Cx. quinquefasciatus (ski2; XP_001845019); a gene sharing 63% identity with glutathione S-transferase (gst; XP_001654620), a gene sharing 96% identity with glutathione S-transferase-1 from C. sonorensis (gst-1; AAB94639) and a gene sharing 49% identity with a gene encoding a Toll protein in Acyrthosiphon pisum (Hempitera: Aphididae) (XP_001948700). The RNA used to validate the change in expression were the two biological replicas of vector competent and refractory C. sonorensis transcriptomes sequenced.
Expression levels were normalized against three reference genes whose stability was ranked using three approaches, BestKeeper [53], geNorm [54], and NormFinder [55] (Additional file 2: Table S3). Gene-specific primers and probes were designed using Primer3Plus [56] (Additional file 2: Table S4; Additional file 1: Supplementary Methods). Primers were compared against the C. sonorensis genome using BLAST to ensure specificity to the target region. Amplification efficiency was tested by RT-qPCR with hydrolysis probes by generating a standard curve using triplicate technical replicates of five-fold serial dilutions of RNA template from teneral female C. sonorensis specimens. All RNA templates were treated with Turbo DNA-Free™ Kit (ThermoFisher Scientific, UK) prior to RT-qPCR to remove any DNA from the sample following the manufacture recommended protocol. Starting template total RNA concentration was evaluated using a Qubit® 3.0 Fluorometer (ThermoFisher Scientific, UK) with the Qubit® RNA HS Assay Kit (ThermoFisher Scientific, UK).
cDNA synthesis and quantitative amplification was performed in one reaction using the Superscript® III One-Step RT-qPCR System with Platinum® Taq DNA polymerase (ThermoFisher Scientific, UK) (see Additional file 1: Supplementary Methods for a detailed protocol). Reactions were performed following the fast cycling programme as described in the manual (Additional file 1: Supplementary Methods). PCR amplification efficiency of each primer-probe-target combinations was calculated via the linear regression of Cq as the log2 of the relative RNA template concentration using the ggplot2 package [57] in R. Reactions which exhibited amplification efficiency between 90 and 110% with an R2 value of ≥0.98 were considered valid. Relative expression levels for the gene of interest between vector competent and refractory C. sonorensis were calculated using the ΔΔCq method [58].
Molecular evolution analyses
Homologues of the ski2 and gst-1 genes in other insect species were identified using BLAST in Blast2Go. The top BLAST hits from Ae. aegypti, Cx. quinquefasciatus or An. gambiae were then used to identify the orthologues in other vector species in VectorBase [59]. We verified that the sequences used started with Methionine and ended with a stop codon, whenever possible. The protein sequences identified were aligned using the mode expresso of T-Coffee [60]. Poorly aligned regions (positions with score 0–6) were trimmed from the alignment and the resulting alignments were used in the evolutionary analyses (see Additional file 1: Supplementary Methods for more details and commands used to align the sequences; Additional file 3: Supplementary Alignment which include the sequences before and after alignment in fasta format). Genetic distances and the diversity were estimated using the software MEGA v7.0.26 [61].Phylogenetic analyses were performed in MrBayes v3.2 [62] using the protein alignment as input and the models of evolution that best fitted our alignments were identified using ProtTest 3.4.2 [63]. Two independent analyses of 2 million generations with four chains (one cold and three heated) were conducted. Trees and parameters were sampled every 50th generation and the first 10,000 were discarded. The remaining trees were used to estimate the consensus and Bayesian posterior probabilities of each branch. The ski2 from Saccharomyces cerevisiae Meyen ex E.C. Hansen and the gst-1 from Pediculus humanus L. were used as outgroups.
Results
Genome sequencing, construction and annotation
Assembly of the Illumina reads resulted in a genome of 189,075,353 bp (189 Mb) assembled in 7974 scaffolds. In contrast, the total genome size estimated using the method of Schell et al. [36] (from the maximum depth of read coverage) was 304.74 Mb (Additional file 2: Table S11). The N50 of the genome assembly was 89,502 and the proportion of Ns was of 2.63% (Table 1). Analysis with RepeatModeler [64] identified a total of 793 repeat regions encompassing ~ 14% of the genome. When low-complexity regions are included, the total repeat content is 29.7% including 2 Mb of Type II transposons and 5 Mb of Type I transposons (mostly LINE elements). The GC content of the C. sonorensis genome is 28% in the complete assembly and 34% in the exons (Table 1). This is low when compared to the closest fully sequenced relative to C. sonorensis (Belgica antarctica (Diptera: Chironomidae): total GC content = 39%) and a range of other Diptera genomes (Table 1).
Analyses on the completeness of the assembly identified 98.79% (100% including partial matches) of 248 Core Eukaryotic Genes Mapping Approach (CEGMA) genes and over 97% of the Benchmarking Universal Single-Copy Orthologs (BUSCO) gene set for Insecta (97.1%), Arthropoda (97.7%) and Metazoa (97.3%), with additional genes from these sets (0.7–0.9%) found in fragmentary form. Of these genes, 65.5% (Metazoa)-66.3% (Insecta, Arthropoda) were found in single copy, compared with 89.3% of Insecta BUSCO genes that were present in single copy in the Aedes aegypti assembly, and a higher number for some other assemblies (Additional file 2: Table S12). There is a duplication level of 30.8% within the Culicoides genome assembly, which is high in comparison to other genomes. This could be the result of genetic variation among/within the sequenced genomes from the pool of individuals and the representation within the assembly of alternative alleles.
Annotation of the Illumina assembled genome resulted in 15,612 protein-coding genes and 21,336 transcripts, which is comparable to the number of protein-coding genes in other species of Diptera (Table 1). The mean gene, intron and exon lengths in C. sonorensis are 5040 bp, 828 bp, and 480 bp, respectively. The average number of introns per gene in C. sonorensis is 6.7. A total of 2991 genes have been estimated to be alternatively spliced and the maximum number of splice variants per gene (19) is comparable to Ae. aegypti and An. gambiae.
To assess redundancy, we generated the K-mer spectra of the assembly and removed potentially redundant contigs using Redundans. This generated an alternative assembly of 156 Mb, with 3839 contigs, and an N50 scaffold length of 109,184 bp. Coverage of the BUSCO data sets was not changed, with values for Insecta, Arthropoda, Metazoa and Eukaryota within +/− 0.1% of the original assembly. The proportion of duplicate BUSCOs, however, was reduced from 30.8% (unreduced Illumina assembly) to 12.1–13.2% (reduced assembly) (Table 2), confirming the presence of BUSCO gene family members within the redundant contigs. Part of the redundancy occurs in repetitive elements of the genome, including satellite, simple and tandem repeats, transposons, LTRs and low complexity regions, which are reduced by approximately 20% after analyses. In addition, 20% of the genes are affected by redundancy, and the number of genes after the redundancy is collapsed becomes 12,453. However, of the 3159 gene models that are removed, 80% are still present in the Redundans-reduced assembly and thus are likely to represent miss-assemblies due to heterozygosity within the sequencing material. Of those genes reported in this study, 62 were affected by redundancy but only 2 did not have a high confidence BLAST hit in the reduced genome assembly.
Homology to genes annotated in other species was detected for 93% of the genes annotated in C. sonorensis, with an average of 19 homologous genes and nine orthologues identified per gene; these numbers are comparable to the other species used in the analysis. Over 90% of the C. sonorensis genes were linked to their homologues via the ancestral nodes of Arthropoda (6609 genes), Neoptera (6361 genes) or Diptera (1225 genes).
InterProScan was used to detect protein domains and assign GO terms to genes. Overall, a similar number of domains and high-level GO terms were associated with at least one gene as in most other sequenced insects (Additional file 2: Table S13), although a larger number of specific GO terms tend to have been applied in genomes that have been manually annotated for gene function. In total, 46,329 GO term assignments were made. This compares to 45,600 assignments for An. gambiae, and 103,897 assignments for D. melanogaster (in which many terms have been manually assigned based on the literature, in addition to automatic assignments). Of 149 high-level categories in the GO (“GO Slim”), 137 were present in C. sonorensis as opposed to 140 categories represented in the genome of An. gambiae and 144 in D. melanogaster. GO terms assigned to more genes in C. sonorensis than either An. gambiae and D. melanogaster include: ion binding (2869 genes, as opposed to 2543 in An. gambiae and 2349 in D. melanogaster), oxidoreductase activity (703 genes, as opposed to 622 in An. gambiae and 632 in D. melanogaster), kinase activity (367 genes as opposed to 315 in An. gambiae and 363 in D. melanogaster), hydrolase activity acting on glycosyl bonds (112 genes as opposed to 105 in An. gambiae and 107 in D. melanogaster) and tRNA metabolic process (101 genes as opposed to 86 in An. gambiae and D. melanogaster). These results suggest an increase in the number of genes encoding these functions in C. sonorensis compared with other species, although the possibility that such genes are selectively over-represented in the current C. sonorensis assembly cannot be ruled out.
To identify tandemly arrayed genes, we looked for genes that contained common domain architectures (as defined by the set of InterPro-defined domains found within each gene) and were located within 100,000 nucleotides of each other. One thousand, two hundred and eighty one genes were found in such arrays. The longest such array contained 10 copies of a gene containing a chitin-binding domain. The longest array of a protein with this architecture comprises just two copies in D. melanogaster, and there are no such arrays in Ae. aegypti or An. gambiae, although there are various longer arrays of proteins containing this domain alongside others in these species (Additional file 2: Table S14). The second longest such array contains 9 copies of a gene encoding a protein with arrestin and immunoglobulin domains; the homologous proteins are found in 10 tandem copies in Ae. aegypti and D. melanogaster, and four tandem copies in An. gambiae (Additional file 2: Table S14).
Transcriptome sequencing and analysis
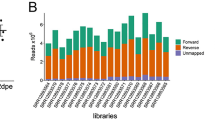
Paired-end sequencing of transcriptomes resulted in an average of 50 million reads with a length of 50 bp for each sample sequenced. Phred scores exceeded 34 in all cases. The transcriptomes from the different conditions were merged with Cuffmerge prior to a comparative analysis of their gene expression profiles. The number of genes on the merged transcriptome is 17,263 and the number of transcripts is 35,813. Of the genes present in the transcriptome, 15,630 had already been annotated in the genome. One thousand, eight hundred and sixteen transcripts, mapping to 1635 loci, were novel. The proportion of gene loci with a single transcript is 62.7% (10,826), but 93.3% among the genes that had not been annotated on the genome. BLAST searches against NCBI’s Arthropod non-redundant (nr) protein database resulted in 92.4% of the isoforms (33,095) and 90% of the genes matching to a database entry (15,268 of the genes had one or more hits).
Differential expression of genes between competent and refractory Culicoides sonorensis
The RT-qPCR for the detection of BTV RNA in individual heads identified 85 (56%) C. sonorensis in replicate one and 53 (37%) in replicate two that produced Cq values ≤27 (Additional file 4: Figure S1). We used the Cq value 27 as a cut off for differentiating between vector competent and refractory individuals as the distribution of values was bimodal and 27 was the local minimum (Additional file 4: Figure S1). Sixty-five (44%) C. sonorensis in replicate one and 92 (63%) in replicate two produced Cq values of > 27 or no Cq value. Replicate one had a significantly greater proportion of disseminated infections than replicate two (Fishers exact test; df = 1, p = 0.0007). Three and nine individuals from the first and second experiment replicates, respectively, were false negatives, that is, samples that were negative for BTV-1 but that also failed to amplify the C. sonorensis genes. This indicated that the RNA extraction failed and were discarded from further analyses. Transcriptome studies were therefore conducted using 147 and 136 female C. sonorensis, from the two biological replicates of the infection study, respectively.
Gene expression profiles showed that more genes had their expression upregulated in refractory C. sonorensis than in competent individuals (Fig. 1a). A total of 165 genes demonstrated significant differential expression (DE) (Fig. 1b; Additional file 2: Table S1) of which 123 (75%) had been annotated and 42 (25%) represented novel genes (Additional file 2: Table S2). A total of 94 (57%) were upregulated in refractory individuals, while 71 were downregulated (43%) (Additional file 2: Table S1). One hundred and twenty of the 165 genes showing DE had one or more BLAST hits in the non-redundant (nr) protein database from NCBI (Additional file 2: Table S2) and 88% of these corresponded closely to genes from other members of the suborder Nematocera. Of the 165 genes, 63 were associated with gene ontology (GO) terms (Additional file 4: Figure S2). However, enrichment analysis showed no significant results.
Ski2 (XLOC_006435) and glutathione S-transferase-1 (CSON011559) showed an increase in expression levels in the females that were resistant to full dissemination, the log2 fold change being − 1.10 (P = 0.00035; q-value = 0.00381) and − 1.07 (P = 0.00055, q-value = 0.00555), respectively (Fig. 2; Additional file 2: Table S1). In contrast, the Toll-like (XLOC_000522) gene and the glutathione s transferase (CSON010973) were expressed at higher levels in the samples that were susceptible to a fully disseminated BTV infection (log2 fold change 1.21, P = 0.00005 and q-value = 0.00072) and (log2 fold change 2.07, P = 0.00005 and q-value = 0.00072) (Fig. 2; Additional file 2: Table S1).

Expression bar plots for candidate genes identified as differentially expressed at significant level between vector competent (orange bars) and refractory (blue bars) samples to full BTV infection. Toll corresponds to the Toll-like gene, ski2 is the antiviral helicase ski2, gst is glutathione s transferase, and gst-1 is the glutathione s transferase-1
The direction and degree of change in expression levels of the four genes identified as potentially influencing C. sonorensis competence for BTV (Ski2, gst-1, gst and Toll-like) was confirmed using RT-qPCR and the same samples. The reference genes used were CytB5, RpL13 and RpS8, which showed the highest level of stability of those tested (Additional file 2: Tables S3 and S4). The direction of up and down regulation of the genes and the log-2 fold change was similar in all cases as that detected in the RNAseq data (Additional file 2: Table S5).
Molecular evolution of ski2 antiviral helicase and glutathione s transferase-1
The top BLAST hit for the C. sonorensis ski2 was the gene from Cx. quinquefasciatus (XP_001845019.1 = CPIJ003293). There are 37 genes identified as orthologous to Cx. quinquefasciatus ski2 in 36 species in VectorBase. Thirty five are 1-to-1 orthologues and there are two orthologous genes in An. maculatus (Additional file 2: Table S6a), although these two orthologues align each to the 5′- and 3′-ends of the Ski2 gene, suggesting that the Ski2 gene model is split in two scaffolds in this species. The mean evolutionary diversity estimated using JTT model with gamma distribution value α = 0.5 (as estimated with ProtTest) and pairwise deletion across the sequences from Diptera species (no outgroups included) was 0.513, and pairwise distances between protein sequences ranged between 0.000 (An. coluzzi v. An. gambiae) and 1.198 (G. fuscipes fuscipes v. An. maculatus). The distance of the ski2 protein between C. sonorensis and mosquitoes was 0.752, with sandflies was 0.834, and with Brachycera had a mean distance of 0.928. The mean evolutionary distance within the Nematocera was 0.272, and 0.171 within the Brachycera. The evolutionary relationship of this protein is consistent with species-level phylogeny (Fig. 3a).

Bayesian phylogenies of ski2 (a) and gst-1 (b) genes. Tip labels correspond to the Ensembl Identifier followed by the species name (in the case of C. sonorensis, the number that precedes correspond to the RNA transcript); colouring corresponds to the Nematocera in red and Brachycera in green. Labels on branches show the posterior probability values
In the gst-1 gene from C. sonorensis, the most similar BLAST hit is that of C. variipennis (synonym of C. sonorensis; AAB94639.1) [25]. The second closest was the gst-1 copy of An. gambiae (AGAP004164), for which 61 orthologues have been identified in 31 other species in VectorBase (Additional file 2: Table S6b). The mean evolutionary distance between the gst-1 protein of the Diptera species, estimated with the same model parameters as for ski2 protein, was 0.647. The mean distance between the gst-1 protein of C. sonorensis and that of mosquitoes was 0.199, while its evolutionary mean distance to Brachycera was 0.866. The pairwise distances ranged between 0.000 (observed between several sequences) and 2.914 (between the copy of M. domestica MDOA012670 and An. coluzzi ACOM0333224). The mean evolutionary distance within the Nematocera was 0.141, and 0.722 within the Brachycera. The phylogeny of the gst-1 protein recovers the two main clades in Diptera, Brachycera and Nematocera, but there is little resolution within these clades (Fig. 3b).
Immune response related genes in C. sonorensis
All D. melanogaster genes from FlyBase that had InterPro hits containing Toll, Imd and JAK/STAT associated to their names and all GO terms matching to the Toll, Imd and JAK/STAT pathway terms were used as reference to identify the homologues in C. sonorensis (Additional file 2: Tables S7, and S8; Additional file 4: Figure S3). Of the 42 genes identified in the Toll pathway of D. melanogaster 36 were also identified in C. sonorensis; eight of the 11 genes of the Imd signalling pathway in D. melanogaster were identified in the C. sonorensis genome and 10 of the 13 D. melanogaster JAK/STAT pathway genes were identified. There are four instances in which the same C. sonorensis genes were identified as different immune-related genes. Two C. sonorensis genes, CSON012766 and CSON015181, were predicted by the Compara workflow to be homologous to both Dorsal and Dif. These two genes had 99.6% similarity at protein sequence level and 96% similarity at DNA sequence level, and are located in different scaffolds. The similarity to D. melanogaster Dorsal was 65% and to Dif was 61%. Four C. sonorensis genes, CSON001282, CSON001790, CSON007335, CSON011712, were homologous to both Toll (FBgn0262473) and Tehao (FBgn0026760). Blastp searches identified all four genes as having leucine-rich repeats and Toll/Interleukin-1 receptor homology (TIR) domains, and the similarity with Toll is about 38–40% at the protein sequence level. Two C. sonorensis genes, CSON003218, CSON008584, are homologous to Peptidoglycan recognition protein LC (Fbgn0035976) and Peptidoglycan recognition protein LF (Fbgn0035977). Using blastp searches, the two genes showed partial similarity higher than 50% to several PGRPs at the protein level, not being able to differentiate between them using BLAST. Finally, one single C. sonorensis gene (CSON004570) is homologous to Unpaired 1, 2 and 3 (FBgn0004956, FBgn0030904, FBgn0053542). Using blastp, the C. sonorensis protein had partial identities of 25–30% to these three Unpaired proteins of D. melanogaster. Similar to the cases above, it is not possible to identify the exact identity of the gene without further analyses.
Additional searches for immune related genes in C. sonorensis were carried out using blastp with Ae. aegypti and Cx. quinquefasciatus gene sequences from the ImmunoDB database (http://cegg.unige.ch/Insecta/immunodb) as queries (Additional file 2: Tables S9 and S10). Four annotated genes in C. sonorensis were identified as potential anti-microbial peptides (AMPs) belonging to the gene subfamilies Attacin, Cecropin, Defensin and Diptericin (Additional file 2: Table S9a); however, the analyses did not identify more than a single gene within each of the AMP subfamilies. Eight potential Toll-like receptors were identified using blastp although there were three instances in which the highest identities were different depending on the species used to provide input data (Additional file 2: Table S10), reflecting the large difference in sequence between the homologous genes in Ae. aegypti and Cx. quinquefasciatus. All five genes belonging to the Toll pathway (Additional file 2: Table S10) and all genes of the JAK/STAT signal transduction pathway were identified in C. sonorensis (Additional file 2: Table S10); while of the Imd pathway, one gene was identified in five of the eight subfamilies (Additional file 2: Table S10).
The Compara approach reconciles the gene tree (derived from sequence similarity metrics) with the species tree to identify likely orthologous relationships; but requires an initial clustering step (which can lead to the inclusion of sequences into the wrong trees) [45]. The only instance in which the two approaches identify a different C. sonorensis gene and the orthologue of a gene in the above pathways is that of Pelle (which activates the Toll Receptor), for which BLAST identifies CSON012655 as its homologue, and Compara identifies CSON013584 and CSON013585. There were other cases in which BLAST identifies a potential homologue while Compara does not (e.g. TNF-receptor-associated factor 6, Traf6, or tube) and genes Imd and Fadd, neither of the two approaches identify a homologue in C. sonorensis.
It should be noted that the results presented in this study come from automated pipelines. Although some cases have been manually verified, there are still genes which have not been identified in this study and other that need improvement in their annotation. It is expected that the annotation and curation of the genome will advance as it is used by the research community.
Discussion
This study has produced the first de novo genome assembly of the Dipteran family Ceratopogonidae, which includes important vectors of emerging and re-emerging veterinary arboviruses [8, 65]. The closest relative to this group for which a genome of comparable quality is available is the non-biting midge, Polypedilum vanderplanki Hinton (Family: Chironomidae) [66], which diverged from the Ceratopogonidae some 220 million years ago. This study provides a primary resource for comparative studies with other arbovirus vector species (e.g. mosquitoes [67]). It also will facilitate efforts to produce transgenic Culicoides to control Culicoides-borne pathogens, although the limitations on colony production for the other epidemiologically relevant species of Culicoides remains a major constraining factor [14, 68].
The genome assembly of C. sonorensis has a similar size to that of other Diptera species for which their genome is available, except for Ae. aegypti which has an unusually large genome. There is however some discrepancy between the estimated size using the method of Schell et al. [36], the size of the original assembly and the size once redundancy is removed. The difference in estimated and assembled sizes could be due to the high level of redundancy as this will spread the coverage across different regions of the genome assembly that would be otherwise collapsed after redundancy removal. Thus, the overestimate of the size can be ultimately linked to the heterogeneity of the starting DNA material used for sequencing the genome. Various measures (the high proportion of BUSCO genes present in multiple copy, and estimates of genome size and redundancy using read mapping and alignment approaches) indicate the presence of a high level of redundancy in the assembly (Table 2), which could result from genetic variation within and amongst individuals used to sequence the genome. However, the individuals come from a colony that has not had any outbreeding for over 40 years, which should have reduced the genetic variation among the individuals of the colony. When the redundancy is collapsed the number of scaffolds is reduced by half, the genome size is reduced by 40 Mb and the N50 increases, resulting in a less fragmented genome. However, the coverage of BUSCO genes was not affected but the number of duplicate gene copies was reduced by more than 50%. In addition, 20% of the genes were removed after the redundancy analyses, although 80% still had a high confidence hit gene in the collapsed genome, indicating that they had been duplicated during the assembly. Of those reported in the present study, 62 were affected by redundancy but 60 still had a homologous gene in the collapsed assembly. In our view, the redundancy analysis can be considered as an analysis of confidence in the gene models of the original genome assembly, and there is high confidence in 80% of the genes annotated while 20% of the gene models should be considered as low confidence as they could be the result of assembly artefacts, miss-predicted genes, or gene families with several copies difficult to distinguish. This suggests that a proportion of the redundancy was due to heterozygosity. In addition, another part of the redundancy corresponded to repetitive elements of the genome, and these were reduced by 20% in the collapsed genome. The low GC content of the genome, which is the lowest of any Diptera species sequenced so far, is likely to have had an impact in the assembly of particularly AT-rich regions. Thus, the discrepancy between the genome sizes before and after the redundancy is removed can be partly explained by the heterogeneity of the starting material for sequencing affecting the gene prediction, but also partially due to naturally occurring repeat regions. Whether these genes and repeat elements are real or an artefact of the assembly process can only be verified with further experimental work. Long read-sequencing approaches would likely help reduce this redundancy.
The original genome assembly is not wholly contiguous, however, other general features are consistent with similarly sized genomes from other species. Thus, features such as the repeat content, gene count and level of alternative splicing are within the ranges observed in An. gambiae and D. melanogaster. Coverage of well-conserved gene families is complete and comparable with other published assemblies (Additional file 2: Table S12). The gene set has been annotated with a comparable number of InterPro domains and high-level GO terms as An. gambiae and other Diptera, while the number of specific GO terms is comparable to other genomes that have not been extensively manually annotated with functional descriptions. The molecular functions of ion binding, oxidoreductase activity, kinase activity, hydrolase activity acting on glycosyl bonds, methyltransferase activity all highly represented, as is the tRNA metabolic process. In addition, the genes of the immune-related pathways have been identified and annotated, revealing complete or nearly-complete orthologous pathways. Overall, all these observations indicate that the present assembly and annotation of the C. sonorensis genome represents a reliable resource to use in genetic studies of biting midges. Nevertheless, it is still highly fragmented and the use of long-read sequencing technologies like PacBio would help improving the scaffolding. In addition, the use of this genomic resource by the community will help improve the annotation of the genes. This is the case for many of the genomes of vector species that have been neglected over the years.
A striking feature of the C. sonorensis genome is its GC content, which is among the lowest reported for Dipteran species (although close to identical to the non-biting midge P. vanderplanki [66] and other non-Dipteran arthropods like the pea aphid, Acyrthosiphon pisum Harris [69]). This is not just a feature of the repeated content, but extends into the protein-coding genes (which have a higher GC content than the non-coding regions, but less than in comparable species: Table 1). Recent comparative analyses of codon usage in Diptera and Hymenoptera showed an association between codon bias and high GC content in Diptera, but low GC content in Hymenoptera [70]. Thus, the low GC content observed in C. sonorensis could reflect a codon bias in comparison to other Diptera species. Functional investigations of GC content have also revealed that it has an impact on genome functioning and species ecology in microbes [71], vertebrates [72] and plants [73]. It is also known that the GC content has an impact in the efficiency of sequencing technologies [74]. Regions with low GC content have less coverage than more GC balanced ones. This characteristic of the C. sonorensis genome should be accounted for in future genomic studies, and appropriate experimental designs that take into account the low GC content should be used (e.g. [75]).
Studies investigating gene expression during infection with BTV identified four candidate genes whose regulation was correlated with vector competence, despite only using a single time-point at 8 days post-infection and pools of C. sonorensis exhibiting different degrees of virus dissemination. Interestingly, the elevated expression of gst-1 in refractory Culicoides observed in this study is consistent with a previously proposed genetic mechanism for C. sonorensis vector competence for BTV [13, 15, 24]. These studies identified a locus controlling vector competence in the AA colony, the origin colony of PIR-s-3 colony used in the present study. The former study used a maternally inherited 90kd protein consistent with genetic studies that suggested maternal inheritance of the competence controlling factor identified in unfertilized eggs of refractory females to isolate a clone that upon sequencing was identified as gst-1 [25]. Though the current study evaluated gene expression after exposure to BTV and the previous study compared genetically selected resistant and susceptible families using individuals from these families who were not exposed to BTV, both independently identified involvement of gst-1. Thus, the differential expression of the gene in the current study is confirmation of its involvement in BTV infection in AA colony, although its role requires functional validation.
From the evolutionary perspective, the phylogenies of ski2 and gst-1 reflect the accepted evolutionary history of the included Diptera species, although the phylogenetic tree of ski2 shows more resolution within Nematocera and Brachycera than the gst-1. It is also interesting to note the two contrasting modes of evolution of gst-1 between the two main clades of Diptera. The gst-1 protein in the Brachycera clade shows multiple duplications in comparison with the Nematocera and the estimated diversity in the Brachycera is higher (0.722) than that in the Nematocera (0.141). Within the Nematocera the level of diversity of the gst-1 protein is lower than in ski2.
Future studies of vector competence in C. sonorensis must characterize genes that facilitate BTV competence and differentiate these from differentially expressed genes that are the consequence of BTV infection. To determine variation in the mechanism and factors controlling BTV-vector competence in the AA and PIR-s-3 colonies observed in the present study, it will be necessary to carry out studies of different populations of C. sonorensis and other Culicoides vectors and non-vector species. Once controlling genes are identified it will be essential to assess the effect of polymorphisms, the various functions of the identified genes in the absence of the pathogen to evaluate factors that influence their frequencies, and compare these processes between different species of Culicoides [23].
The present paper presents the genome of C. sonorensis and its use in transcriptomic analyses to provide information that will help elucidate the transmission of viruses by vectors and provide new avenues of research to understand vector competence for BTV. We present a list of candidate genes that will be further explored to show their involvement in the transmission of BTV by C. sonorensis. Furthermore, the results also provide evidence for the involvement of genes belonging similar functional families in response to virus infection in different species belonging to different families of Diptera. These results show the importance of comparative analyses to interrogate the evolution of host-pathogen interactions, and the C. sonorensis genome will facilitate such studies.
Conclusions
Here we present the first annotated genome of the biting midge, C. sonorensis, a vector of economically important viruses of livestock, such as bluetongue virus (BTV) and Schmallenberg virus. This genome has been fundamental in gaining information about the genetics of BTV transmission. Thus, gene expression comparison between females susceptible to a full infection by BTV and those females refractory to full BTV infection, identified 165 genes that are candidates to be involved in vector competence in this species. Of these genes, the gst-1 gene was identified previously to be involved in BTV transmission, while the antiviral helicase ski2 involved in suppressing the replication of dsRNA viruses. Overall, the publication of the genome and transcriptomes will lead to advances in the control of economically relevant arboviruses of livestock. In addition, this genome fills a phylogenetic gap of more than 200 million years, and will be a useful source of comparative data for studies of vector competence across Diptera.
Abbreviations
- AMPs:
-
Antimicrobial peptides
- BLAST:
-
Basic local alignment search tool
- bp:
-
Base pair
- BTV:
-
Bluetongue virus
- BUSCO:
-
Benchmarking Universal Single-Copy Orthologs
- CEGMA:
-
Core Eukaryotic Genes Mapping Approach
- Cq :
-
Quantification cycle
- DE:
-
Differentially expressed
- GO:
-
Gene ontology
- GSEA:
-
Gene set enrichment analysis
- gst:
-
Glutathione S-transferase
- HMW:
-
High molecular weight
- Imd:
-
Immunodeficiency
- JAK/STAT:
-
Janus kinanses/signal transducer and activator of transcription
- kb:
-
Kilobase pairs
- MP:
-
Mate-pair
- PE:
-
Paired-end
- RT-qPCR:
-
Quantitative reverse transcription Polymerase Chain Reaction
- TCID50 :
-
50% Tissue culture infective dose
- w/v :
-
Weight/volume
- μl:
-
Microlitre
References
Weaver SC, Reisen WK. Present and future arboviral threats. Antivir Res. 2010;85:328–45.
Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Rus KR, Vipotnik TV, Vodusek VF, et al. Zika virus associated with microcephaly. N Engl J Med. 2016;374:951–8.
Pialoux G, Gauzere BA, Jaureguiberry S, Strobel M. Chikungunya, an epidemic arbovirosis. Lancet Infect Dis. 2007;7:319–27.
Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, Shamim MS, Machol I, Lander ES, Aiden AP, et al. De novo assembly of the Aedes aegypti genome using hi-C yields chromosome-length scaffolds. Science. 2017;356:92–5.
Alphey L. Genetic control of mosquitoes. Annu Rev Entomol. 2014;59:205–24.
Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, Loftus B, Xi ZY, Megy K, Grabherr M, et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007;316:1718–23.
Olson KE, Blair CD. Arbovirus-mosquito interactions: RNAi pathway. Curr Opin Virol. 2015;15:119–26.
Purse BV, Carpenter S, Venter GJ, Bellis G, Mullens BA. Bionomics of temperate and tropical Culicoides midges: knowledge gaps and consequences for transmission of Culicoides-borne viruses. Annu Rev Entomol. 2015;60:373–92.
Carpenter S, Mellor PS, Fall AG, Garros C, Venter GJ. African horse sickness virus: history, transmission, and current status. Annu Rev Entomol. 2017;62:343–58.
Savini G, Afonso A, Mellor P, Aradaib I, Yadin H, Sanaa M, Wilson W, Monaco F, Domingo M. Epizootic haemorragic disease. Res Vet Sci. 2011;91:1–17.
Carpenter S, Wilson A, Mellor PS. Culicoides and the emergence of bluetongue virus in northern Europe. Trends Microbiol. 2009;17:172–8.
Rushton J, Lyons N. Economic impact of bluetongue: a review of the effects on production. Vet Ital. 2015;51:401–6.
Tabachnick WJ. Culicoides variipennis and bluetongue-virus epidemiology in the United States. Annu Rev Entomol. 1996;41:23–43.
Nayduch D, Cohnstaedt LW, Saski C, Lawson D, Kersey P, Fife M, Carpenter S. Studying Culicoides vectors of BTV in the post-genomic era: resources, bottlenecks to progress and future directions. Virus Res. 2014;182:43–9.
Jones RH, Foster NM. Oral infection of Culicoides variipennis with bluetongue virus - development of susceptible and resistant lines from a colony population. J Med Entomol. 1974;11:316–23.
Tabachnick WJ. Genetic control of oral susceptibility to infection of Culicoides variipennis with bluetongue virus. Am J Trop Med Hyg. 1991;45:666–71.
Nayduch D, Lee MB, Saski CA. The reference transcriptome of the adult female biting midge (Culicoides sonorensis) and differential gene expression profiling during Teneral, blood, and sucrose feeding conditions. PLoS One. 2014;9:e98123.
Nunamaker RA, Brown SE, Murphy KE, Tabachnick WJ, Knudson DL. Physical mapping of the genome of the biting midge, Culicoides variipennis. Mol Biol Cell. 1998;9:452A.
Nunamaker RA, Brown SE, McHolland LE, Tabachnick WJ, Knudson DL. First-generation physical map of the Culicoides variipennis (Diptera : Ceratopogonidae) genome. J Med Entomol. 1999;36:771–5.
Mellor PS, Boorman J, Baylis M. Culicoides biting midges: their role as arbovirus vectors. Annu Rev Entomol. 2000;45:307–40.
Tabachnick WJ. Nature, nurture and evolution of intra-species variation in mosquito arbovirus transmission competence. Int J Environ Res Public Health. 2013;10:249–77.
Holbrook FR, Tabachnick WJ, Schmidtmann ET, McKinnon CN, Bobian RJ, Grogan WL. Sympatry in the Culicoides variipennis complex (Diptera : Ceratopogonidae): a taxonomic reassessment. J Med Entomol. 2000;37:65–76.
Carpenter S, Veronesi E, Mullens B, Venter G. Vector competence of Culicoides for arboviruses: three major periods of research, their influence on current studies and future directions. Rev Sci Techn Off Int Epiz. 2015;34:97–112.
Fu H, Leake CJ, Mertens PPC, Mellor PS. The barriers to bluetongue virus infection, dissemination and transmission in the vector, Culicoides variipennis (Diptera : Ceratopogonidae). Arch Virol. 1999;144:747–61.
Abdallah MA, Pollenz RS, Droog FN, Nunamaker RA, Tabachnick WJ, Murphy KE. Isolation and characterization of a cDNA clone coding for a glutathione S-transferase class delta enzyme from the biting midge Culicoides variipennis sonorensis Wirth and Jones. Biochem Genet. 2000;38:377–90.
Wiegmann BM, Trautwein MD, Winkler IS, Barr NB, Kim JW, Lambkin C, Bertone MA, Cassel BK, Bayless KM, Heimberg AM, et al. Episodic radiations in the fly tree of life. Proc Natl Acad Sci U S A. 2011;108:5690–5.
Jones RH. The laboratory colonization of Culicoides variipennis (coq). J Econ Entomol. 1957;50:107–8.
Boorman J. Maintenance of laboratory colonies of Culicoides variipennis (coq), Culicoides nubeculosus (mg) and Culicoides riethi Kieffer (Diptera, Ceratopogonidae). Bull Entomol Res. 1974;64:371–7.
Mellor PS, Rawlings P, Baylis M, Wellby MP. Effect of temperature on African horse sickness virus infection in Culicoides. In: Mellor PS, Baylis M, Hamblin C, Calisher CH, PPC M, editors. Archives of Virology. Wien: Springer-Verlag; 1998. p. 155–63.
Leggett RM, Clavijo BJ, Clissold L, Clark MD, Caccamo M. NextClip: an analysis and read preparation tool for Nextera long mate pair libraries. Bioinformatics. 2014;30:566–8.
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9.
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–9.
Vezzi F, Narzisi G, Mishra B. Reevaluating assembly evaluations with feature response curves: GAGE and Assemblathons. PLoS One. 2012;7:e52210.
Hunt M, Kikuchi T, Sanders M, Newbold C, Berriman M, Otto TD. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 2013;14:R47.
Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13:R56.
Schell T, Feldmeyer B, Schmidt H, Greshake B, Tills O, Truebano M, Rundle SD, Paule J, Ebersberger I, Pfenninger M. An annotated draft genome for Radix auricularia (Gastropoda, Mollusca). Genome Biol Evol. 2017;9:585–92.
Pryszcz LP, Gabaldon T. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 2016;44:e113.
Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinform. 2011;12:491.
Nayduch D, Lee MB, Saski CA. Gene discovery and differential expression analysis of humoral immune response elements in female Culicoides sonorensis (Diptera: Ceratopogonidae). Parasit Vectors. 2014;7:388.
Stanke M, Schoffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. Bmc Bioinform. 2006;7:62.
Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32:W309–12.
Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genornes. Bioinformatics. 2007;23:1061–7.
Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2.
Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–35.
Herrero J, Muffato M, Beal K, Fitzgerald S, Gordon L, Pignatelli M, Vilella AJ, Searle SMJ, Amode R, Brent S, et al. Ensembl comparative genomics resources. Database. 2016;2016:bav096.
Veronesi E, Antony F, Gubbins S, Golding N, Blackwell A, Mertens PPC, Brownlie J, Darpel KE, Mellor PS, Carpenter S. Measurement of the infection and dissemination of bluetongue virus in Culicoides biting midges using a semi-quantitative RT-PCR assay and isolation of infectious virus. PLoS One. 2013;8:e70800.
Shaw AE, Monaghan P, Alpar HO, Anthony S, Darpel KE, Batten CA, Guercio A, Alimena G, Vitale M, Bankowska K, et al. Development and initial evaluation of a real-time RT-PCR assay to detect bluetongue virus genome segment 1. J Virol Methods. 2007;145:115–26.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7:562–78.
Goff L, Trapnell C, Kelley D. cummeRbund: Analysis, exploration, manipulation, and visualization of Cufflinks high-throughput sequencing data. In: R package version 2180; 2013.
Al-Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics. 2004;20:578–80.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50.
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper - excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–15.
Hoerndli FJ, Toigo M, Schild A, Gotz J, Day PJ. Reference genes identified in SH-SY5Y cells using custom-made gene arrays with validation by quantitative polymerase chain reaction. Anal Biochem. 2004;335:30–41.
Ohl F, Jung M, Xu CL, Stephan C, Rabien A, Burkhardt M, Nitsche A, Kristiansen G, Loening SA, Radonic A, et al. Gene expression studies in prostate cancer tissue: which reference gene should be selected for normalization? J Mol Med. 2005;83:1014–24.
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JAM. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71–4.
Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer; 2009.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45.
Lawson D, Arensburger P, Atkinson P, Besansky NJ, Bruggner RV, Butler R, Campbell KS, Christophides GK, Christley S, Dialynas E, et al. VectorBase: a data resource for invertebrate vector genomics. Nucleic Acids Res. 2009;37:D583–7.
Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27:1164–5.
Smit AFA, Hubley R: RepeatModeler Open-1.0. 2008–2015 http://www.repeatmasker.org. 2015.
Carpenter S, Groschup MH, Garros C, Felippe-Bauer ML, Purse BV. Culicoides biting midges, arboviruses and public health in Europe. Antivir Res. 2013;100:102–13.
Gusev O, Suetsugu Y, Cornette R, Kawashima T, Logacheva MD, Kondrashov AS, Penin AA, Hatanaka R, Kikuta S, Shimura S, et al. Comparative genome sequencing reveals genomic signature of extreme desiccation tolerance in the anhydrobiotic midge. Nat Commun. 2014;5:4784.
Severson DW, Behura SK. Mosquito genomics: progress and challenges. Annu Rev Entomol. 2012;57:143–66.
Mills MK, Nayduch D, Michel K. Inducing RNA interference in the arbovirus vector, Culicoides sonorensis. Insect Mol Biol. 2015;24:105–14.
Richards S, Gibbs RA, Gerardo NM, Moran N, Nakabachi A, Stern D, Tagu D, Wilson ACC, Muzny D, Kovar C, et al. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010;8:e1000313.
Behura SK, Severson DW. Comparative analysis of codon usage Bias and codon context patterns between dipteran and hymenopteran sequenced genomes. PLoS One. 2012;7:e43111.
Foerstner KU, von Mering C, Hooper SD, Bork P. Environments shape the nucleotide composition of genomes. EMBO Rep. 2005;6:1208–13.
Galtier N, Piganeau G, Mouchiroud D, Duret L. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics. 2001;159:907–11.
Smarda P, Bures P, Horova L, Leitch IJ, Mucina L, Pacini E, Tichy L, Grulich V, Rotreklova O. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc Natl Acad Sci U S A. 2014;111:E4096–102.
Chen YC, Liu T, Yu CH, Chiang TY, Hwang CC. Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS One. 2013;8:e62856.
Oyola SO, Otto TD, Gu Y, Maslen G, Manske M, Campino S, Turner DJ, Macinnis B, Kwiatkowski DP, Swerdlow HP, et al. Optimizing Illumina next-generation sequencing library preparation for extremely AT-biased genomes. BMC Genomics. 2012;13:1.
Acknowledgements
We thank Eric Denison and James Barber in the insectary for their supply of reagents and insects for this study. We would like to thank the three reviewers and the editor for their positive comments and suggestions, which have helped improve this work.
Funding
This work was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) grant BB/J016721/1 and the core Strategic Programme Grant to The Pirbright Institute. These funding bodies had no role in the design of the study, collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
All sequencing data, genomic and transcriptomic, generated in this study has been deposited at the European Nucleotide Archive (ENA) database under the accession number PRJEB19938. Transcriptome data have been deposited in the ENA database under the accession numbers ERR2171964- ERR2171979.
Author information
Authors and Affiliations
Contributions
MF, SC and PK conceived the research project. RMH, MH, IMA, RS, LEH, AGU, EV and LC have performed the laboratory experiments and bioinformatic analyses. DN and CS have helped with the annotation of the immune-related genes. RMH, MF and SC have written the manuscript. RMH, AGU, MK, PK and SC have done the revisions to the manuscript. PK, WT, ND and CS have provided insightful revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Supplementary Methods. DNA extraction. Protocol followed to extract DNA from C. sonorensis samples. Genome annotation and comparative analysis. Details about the genomes of species used in the gene annotation and in the comparative analyses. RNA extraction. Protocol used to extract RNA from C. sonorensis. RT-qPCR for the detection of BTV-1 infection. Description of the method used to detect the presence of BTV-1 in C. sonorensis females after the experimental infections. RT-qPCR protocol for the validation of differentially expressed genes. Description of the protocol used to verify the change in expression of Toll-like protein, glutathione-s-transferase, glutathione-s-transferase 1 and ski2 antiviral helicase genes observed in the transcriptomes of vector-competent and refractory samples. Alignment of gst-1 and ski2. The commands used to align the protein sequences of gst-1 and ski2. These alignments were then used to run the phylogenetic analyses. (ZIP 117 kb)
Additional file 2:
Table S1. Differentially expressed genes between vector competent and vector refractory C. sonorensis females. Table S2. BLAST results for the differentially expressed genes between vector competent and refractory transcriptomes. Table S3. Ranking of the expression stability of the reference genes used in RT-qPCR. Table S4. Primers and probe used for RT-qPCR of ski2, gst, gst-1 and Toll-like. Table S5. Difference of expression levels in four genes (Toll-like, gst, gst-1 and ski2) between vector-competent and vector-refractory females assessed by RNAseq and RT-qPCR. Table S6. Orthology analysis of two of genes Ski2 (a) and gst-1 (b). Table S7. Immune pathways genes identified in C. sonorensis using the Ensembl Compara pipeline. Table S8. Homologue immune genes associated with Toll (a), Imd (b) and Jak/Stat (c). Table S9. Immune related genes identified in C. sonorensis using blastp. Table S10. Details of the blastp results. Table S11. Read mapping and estimate of assembly size according to the method of Schell et al... [36]. Table S12. BUSCO analysis results. Table S13. Number of distinct InterPro and GO terms annotated C. sonorensis and six other Diptera species. Table S14. Tandemly repeated gene arrays in C. sonorensis versus other insect species.
Additional file 3:
Supplementary Alignments. Zip file containing the gst-1 and ski2 CDS and protein sequences, the protein alignment obtained using T-Coffee as described in the Additional file 1: Supplementary Methods, and the protein alignment after removing variable positions (with a T-Coffee value < 6 as described in the Additional file 1: Supplementary Methods). (DOCX 267 kb)
Additional file 4:
Figure S1. Distribution of Cq values from RT-qPCR used to detect BTV-1 virus infection in the two virus-feeding experiments (in blue and orange) of C. sonorensis. The vertical, dashed line corresponds to a Cq value of 27 used to differentiate vector competent from refractory females. Figure S2. Classification and functional distribution of the genes differentially expressed between vector competent and refractory C. sonorensis per the Gene Ontology level 6. Blue: Molecular Function; Green: Cellular Component; Pink: Biological Process. Figure S3. Number of genes in the Toll (a), Imd (b) and Jak/Stat (c) pathways that have been duplicated or lost in different species of Diptera as identified using the Ensembl Compara pipeline. (DOCX 158 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Morales-Hojas, R., Hinsley, M., Armean, I.M. et al. The genome of the biting midge Culicoides sonorensis and gene expression analyses of vector competence for bluetongue virus. BMC Genomics 19, 624 (2018). https://doi.org/10.1186/s12864-018-5014-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5014-1