
Abstract
Background
The mud crab (Scylla paramamosain) is a euryhaline and commercially important species. MiRNAs participate in the regulation of many physiological activities.
Results
The miRNA transcriptome of the gills of S. paramamosain was used to investigate the expression profiles of miRNAs in response to a sudden drop in salinity. In total, seven known miRNAs and 43 novel miRNAs were identified, with 18 differentially expressed small RNAs. Fourteen thousand nine hundred fifty-one differentially expressed miRNAs target genes were screened by prediction. GO analysis of differentially expressed miRNAs target genes indicated that 578 genes associated with cellular processes, 523 associated with metabolic processes, and 422 associated with single-organism processes were the most strongly affected by a sudden drop in salinity from 23‰ to 3‰. KEGG pathway analysis showed 14 pathways were related to amino acid metabolism, which plays an important role in osmoregulation. Besides, several pathways were associated with starch and sucrose metabolism (ko00500), glycosaminoglycan degradation (ko00531), and galactose metabolism (ko00052).
Conclusions
S. paramamosain regulated osmotic pressure and energy balance by regulating target genes to adapt to a sudden changes in salinity. These results provided a basis for further investigations of miRNA-modulating networks underlying the osmoregulation of S. paramamosain.
Similar content being viewed by others

Background
The commercially important mud crab Scylla paramamosain (Crustacea: Decapoda: Portunidae: Syclla De Hann) is widely distributed along the coasts of southern China and other Indo-Pacific countries [23, 43, 47]. Due to its rapid growth, large size, popularity amongst consumers, and high market value, S. paramamosain is becoming an important farmed fish along the southeast coast of China [52, 28]. Mud crab production was 240,293 t in 2016 [10]. Salinity is a key abiotic parameter that influences the distribution, abundance, physiology, and well-being of crustaceans [4, 24, 33]. Although a euryhaline species, a sudden decrease in salinity, especially exceeding 10%, is often lethal to S. paramamosain. Previous studies have revealed that crustaceans regulate hemolymph osmotic pressure mainly by adapting to changes in salinity through ion channels [33] and free amino acids [35].
Small noncoding ribonucleic acid (snRNA), which include microRNA (miRNA), small interfering RNA (siRNA), and Piwi-associated RNA (piRNA), has been isolated from a wide variety of plant and mammalian cells. snRNA is involved in various biological and cellular processes by targeting specific miRNAs for degradation or translational repression [3, 8, 21, 48]. So far, most studies have focused on miRNAs, which are 19–23 nucleotides (nt) in length and formed as a single strand of RNA and highly phylogenetically conserved across almost all species [7, 32, 37, 49]. The molecular mechanisms of miRNAs underlying the silencing of target genes have been described [5, 7].
MiRNAs participate in the regulation of many physiological activities [7], including development [12, 15], immunity [18, 31], cell proliferation [6, 54], cell apoptosis [11, 41], and lipid metabolism [40], among others. Many studies have investigated the roles of miRNAs in aquatic animals, including Oreochromis niloticus [39], Oncorhynchus mykiss [29], Cyprinus carpio [27], Crassostrea gigas [56], and Chlamys farreri [9]. A large number of miRNAs of several aquatic crustaceans, including Daphnia magna [42], Marsupenaeus japonicus [34], Litopenaeus vannamei [50], Eriocheir sinensis [14], S. paramamosain [22], and Portunus trituberculatus [26], have been sequenced and mined.
In our previous study, transcriptomic analysis was conducted on the gills (an important organ in osmoregulation of marine crustaceans) of S. paramamosain to reveal the adaptive mechanisms in response to sudden salinity drop under normal conditions (23‰ salinity) and stress conditions caused by a sudden drop in salinity (3‰) [44]. The functional genomic studies of DEGs obtained from Wang et al., [44] allow for a better understanding of various physiological responses in marine crustaceans induced by a sudden drop in salinity. As we all know, microRNAs play a very important role in regulating the expression of functional genes. Therefore, on the basis of previous research, the miRNA transcriptome of the gills of S. paramamosain was examined under normal conditions (23‰ salinity) and stress conditions caused by a sudden drop in salinity (3‰) in this study. These results provide a basis for further investigations of miRNA-modulation of networks underlying osmoregulation of S. paramamosain.
Methods
Animals and salinity challenge experiment
A total of 300 randomly selected crabs with an average body weight of ~ 30 g were selected and maintained in a natural water environment with a salinity of 23‰ and a temperature of ~ 20 °C. The crabs were randomly assigned to one of six groups of 50 animals each and housed in six cement pools under identical physical and chemical conditions. The salinity of the seawater for three of the groups was adjusted to 3‰ from 23‰, and then decreased to 20‰. These three groups were defined as the LS (low salinity) group. The other three groups were defined as the CK groups, where the salinity of seawater was maintained at 23‰. All other conditions were the same as the LS group.
According to [44], there were four deaths in the CK group within 7 days and 24 deaths in the LS group. The LS death time was concentrated within 24, 48, and 72 h. In addition, the LS group showed hyperactivity within 48 h, and as time went by, the motility was diminished and normalized. The LS group did not have food over 72 h, and gradually started to eat over time. Conditions returned to normal after 120 h. In our study, the mud crabs had adjusted to a salinity of 3‰ in 120 h after a sudden drop in salinity. In order to study the molecular mechanism underlying this adaptation, we performed transcriptional profiling at 120 h [44]. The crabs in CK and LS group at 120 h were killed under the condition of alcohol anesthesia, and gills were quickly removed and used for subsequent sequencing and studies.
The construction and deep sequencing of small RNA libraries
Total RNA was extracted from S. paramamosain gills tissues with TRIzol reagent (Invitrogen Corporation, Carlsbad, CA, USA), as recommended by the manufacturer.
RNA fragments, 18–30 bases in length, were isolated from total RNA extracted from gills of three individual mud crabs as one sample after being separated through 15% denaturing gels to net three samples from each group. Then, sRNAs were excised from the gel and sequentially ligated to 3′- and 5′-adapters. After each ligation step, sRNAs were separated in 15% denaturing gels. The final purified ligation products were reversely transcribed into complementary DNA (cDNA) using reverse transcriptase (Finnzymes Oy, Espoo, Finland). The first strand cDNA was PCR amplified using Phusion* DNA polymerase (Finnzymes Oy). The purified DNA fragments were used for clustering and sequencing using the Illumina Hiseq 4000 sequencing system (Illumina, Inc., San Diego, CA, USA), which was performed by BGI Diagnosis Technology Co., Ltd. (Shenzhen, China).
miRNA identification
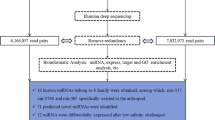
Raw data (raw tags) in the FASTQ format were first processed using self-written Perl and Python scripts (BGI, Shenzhen, China). In this step, clean data (clean tags) were processed by removing low-quality tags containing ploy-N, with 5′ primer contaminants, without 3′ primers or the insert tag, and with ploy A or T or G or C from the raw data. At the same time, the Q20, Q30, and GC contents of the raw data were calculated. Then, the length of sRNA tags within a certain range from clean tags was determined for downstream analysis. All clean tags were searched against the Rfam database (http://rfam.xfam.org//) for annotation. The tags annotated as tRNA, rRNA, small nucleolar RNA (snoRNA), and snRNA were discarded. Considering that there was no available information of the S. paramamosain genome, the remaining small RNA tags were compared to known miRNAs from all metazoan species in the miRBase 21.0 database (http://www.mirbase.org/) to identify conserved miRNAs. Only perfect matches were considered as conserved miRNAs. Tags that were not aligned to the miRBase database were used to predict novel miRNAs. The miREvo evolutionary analysis platform for next-generation sequencing and mirdeep2 software package were used to predict novel miRNAs by exploring the secondary structure, the Dicer cleavage site, and the minimum free energy of the former unannotated small RNA tags that could be mapped to reference sequences. Q_value < 0.001 and |log2 (foldchange) | > 1 were set as thresholds to identify significantly differential expression of miRNAs.
Target gene prediction and analysis
As there was no published information on the S. paramamosain genome, the assembled Unigenes from the S. paramamosain (https://www.ncbi.nlm.nih.gov/sra/SRP129841, SRA accession: SRP129841; Temporary Submission ID: SUB3501735) [44] were considered as candidate genes for target gene prediction. The RNAhybrid [20], miRanda [17], and TargetScan [2] web servers were used to predict the target genes of identified miRNAs. The predicted target genes were aligned using the Basic Local Alignment Search Tool (https://blast.ncbi.nlm.nih.gov/). Afterward, gene ontology (GO) analysis of the target genes was performed using the Gene Ontology Enrichment Analysis Software Toolkit (http://omicslab.genetics.ac.cn/GOEAST/; [55]).
Quantitative real-time PCR assay
MiRNAs expression levels were assayed by quantitative realtime PCR (qPCR) using a SYBR primescript™ miRNA RT-PCR kit (TaKaRa). Total RNA was isolated from the gills of mud crabs in the LS groups at 120 h after a sudden drop in salinity from 23‰ to 3‰ using RNAiso Plus total RNA extraction reagent (TaKaRa), while total RNA was isolated at 120 h from crabs in the CK groups maintained at a salinity of 23‰. The cDNA was synthesized using the Perfect Real Time version of the PrimerScript™ RT reagent kit with gDNA Eraser (Perfect Real Time) (TaKaRa) according to the manufacturer’s instructions. The 18S rRNA gene was selected as an internal control. Primers used in this study are listed in Table 1. The qPCR was conducted in 15 μl reaction volumes containing 300 nM of each primer and cDNA derived from 0.1 μg of total RNA. Cycling parameters were 95 °C for 2 min, and followed by 50 cycles of 95 °C for 10 s, 60 °C for 10 s and 72 for 40 s. All reactions were run in triplicate. The 2 -ΔΔCT method was employed in the analysis of relative quantification. Significant differences were examined by paired t-test in which p value< 0.01 and p value< 0.01 was considered to be statistically significant.
Results
Bgiseq-500 sequencing of small RNAs
To identify miRNAs of the mud crab in response to a sudden drop in salinity, small RNA libraries derived from the CK and LS groups were constructed and sequenced using the BGIseq-500 Next Generation Sequencing Platform. Raw tags were obtained from the three CK groups and three LS groups, respectively (Table 2). After removal of low quality sequences, contaminated with 5′ linkers, no 3′ linker sequence, no inserts, poly A included, shorter than 18 nt in length, clean tags (Table 2) of 18–30 nt in length were obtained. The clean tags of the CK and LS libraries were similar in size distribution and frequency, and most of the sequences were 21–23 nt in length (Fig. 1), of which those 22 nt in length were the most abundant (Fig. 1). After data filtering, the clean tags were compared with known small RNA databases, including miRBase [19], Rfam [30], siRNA, piRNA, snoRNA, etc. Other non-coding RNAs (rRNA, snoRNA, tRNA, and snRNA) and repeat sequences (Table 3) were annotated. The remaining tags of the CK and LS libraries were used for miRNA analysis, respectively.

The distribution of small RNAs relative to length in the CK and LS groups
The discovery of known, novel and differential expression of miRNAs
In the CK and LS libraries, there were a total of seven known miRNAs (miR-100-5p_1, miR-100-5p, miR-100, miR-7a, miR-7a_1, miR-7_2, and miR-7) of variable abundances. MiR-100-5p_1 was the most accumulated miRNA, with a total of 11,848,942 tags from the CK and LS libraries (5,794,803 tags in the CK group and 6,054,139 in the LS group). While the least abundant miRNA in the two libraries was miR-7 (1157 tags in the CK group and 6374 in the LS group) (Table 4).
A total of 43 novel miRNAs were identified in the CK and LS libraries, and displayed unequal expression levels (Table 4). The top five most abundant miRNAs were novel_mir1, miR-100-5p_1, miR-100-5p, miR-100, and novel_mir2, with 20,827,222, 11,848,942, 5,703,827, 670,379 and 433,445 tags detected between the two libraries. Besides, novel_mir18, novel_mir19, novel_mir22, novel_mir35, novel_mir40 and novel_mir47 were found only in the CK groups, while novel_mir45, novel_mir24, novel_mir48, and novel_mir31 were only found in the LS groups, with read counts all below 40 tags. The miRNAs of S. paramamosain were matched against the miRNAs of Portunus trituberculatus. The salinity regulation -related miRNAs in gills included mir_7 (known), novel_mir1, novel_mir4, and novel_mir8.
A comparison of miRNA expressed in response to a sudden drop in salinity (Q_value< 0.001 and |log2 (fold change)| > 1), indicated that a total of 18 miRNAs were differentially expressed small RNAs (DESs), with 10 miRNAs up-regulated and eight down-regulated (Table 4 and Additional file 1: Table S1). In the CK and LS libraries, the most up-regulated miRNA was novel_mir47 (log2.Fold_change = 6.542014656) and the most down-regulated miRNA was novel_mir45 (log2 Fold_change = − 6.093703464). Among the 18 miRNAs, the most abundant miRNAs were novel_mir7, novel_mir5, and miR-7 with 87,809, 80,241, and 7531 tags detected between the CK and LS libraries, respectively (Table 4).
Validation and target gene prediction of differentially expressed miRNAs
QPCR was adopted to validate the 18 differentially expressed miRNAs acquired from sequencing. The results showed that miR-7, novel_miR16, novel_miR18, novel_miR19, novel_miR22, novel_miR26, novel_miR35, novel_miR40, novel_miR45, novel_miR47 and novel_miR53 were differentially expressed up-regulated miRNAs (Fig. 2a & Additional file 1: Table S2), with ratio (LS / CK) ranging from 1.34 to 4.73; meanwhile, novel_miR5, novel_miR7, novel_miR24, novel_miR31, novel_miR34, novel_miR37 and novel_miR48 were differentially expressed down-regulated miRNAs, with ratio (CK / LS) ranging from 1.40 to 3.86 (Fig. 2b, Additional file 1: Table S2). The differences of the 18 differentially expressed miRNAs all reached the extremely significant level (P<0.01) (Fig. 2). The genes changes most according to qPCR results were novel_mir35 and novel_mir24 (Additional file 1: Table S2), among the up-regulated and down-regulated miRNAs, respectively. Though the trend (up or down) was nearly same as sequencing results (Additional file 1: Tables S1 & S2), the specific figure were different from each other. Perhaps because the sensitivity of the two different detection methods brought about the differences between sequencing and qPCR results. Moreover, novel_mir45 was down-regulated in sequencing results (Additional file 1: Table S1), but up-regulated in qPCR results (Fig. 2a & Additional file 1: Table S2). The reason was unknown.

Gene expression levels of all of 18 differentially expressed miRNAs for the comparison of CK and LS groups according qPCR analysis. a Up-regulated differentially expressed miRNAs; b Down-regulated differentially expressed miRNAs. The significant difference of miRNA expression between CK and LS was indicated with asterisks (**: P < 0.01)
Using RNAhybrid [20], the microRNA Target Prediction Tool (miRanda, [17]) was used to predict the target genes of miRNAs, combined with corresponding filtering conditions, such as free energy, score values, etc., to obtain an aggregation of the target genes of the identified miRNAs. A total of 65,354 unigenes of the S. paramamosain transcriptome were identified as miRNA targets (Fig. 3a), and 18 differentially expressed miRNAs had 14,951 target genes (Fig. 3b).

Target prediction by RNAhybrid and miRanda. a Statistics of total miRNAs target prediction. b Statistics of differentially expressed miRNAs target prediction
GO classification, and KEGG pathway analysis of differentially expressed miRNAs target genes
GO functional classification of 18 differentially expressed miRNAs target genes was performed using WEGO software [53]. According to the second-tier GO terms, there were three main GO categories: biological process (21), cellular component (17), and molecular function (12) (Fig. 4). In total, 21 processes were identified in the biological process category, with 578 DESs of target genes involved in cellular processes, 523 involved in metabolic processes, and 422 involved in single-organism processes (Fig. 4). These three biological processes were the most strongly affected in the gill of S. paramamosain by a sudden drop in salinity from 23‰ to 3‰ (Fig. 4). In the cellular component category, cell (421), cell parts (417), membrane (334), and membrane part (262) were the most common (Fig. 4). In the molecular function category, catalytic activity (559) and binding (471) were the most common (Fig. 4).

GO functional classification of differentially expressed miRNAs target genes from the gill of S. paramamosain in response to a sudden drop in salinity from 23‰ to 3%. The number of DEGs (presented as a square root value) is shown on the X axis, while GO terms are shown on the Y axis. All GO terms are grouped in to three ontologies: biological processes are shown in blue, cellular components in green, and molecular functions in red
KEGG pathway classification of differentially expressed miRNAs target genes was performed. Of all DESs of the target genes, 5019 were mapped in 319 KEGG pathways (Additional file 1: Table S2), which were classified into six categories (level 1) according to their biological functions, which included organismal systems, human diseases, environmental information processing, genetic information processing, metabolism, and cellular processes (Fig. 5a). Of the 319 pathways, 91 (28.53%), 79 (24.76%), 76 (23.82%), 34 (10.66%), 22 (6.90%), and 17 (5.33%) were related to metabolism, organismal systems, human diseases, environmental information processing, genetic information processing, and cellular processes, respectively (Fig. 5a and Additional file 1: Table S3). /Pathway enrichment analysis of the DESs of the target genes based on the top 20 of KEGG enrichment results (Fig. 5b and Additional file 1: Table S3) showed the largest number of pathways were related to metabolism, such as starch and sucrose metabolism (ko00500), glycosaminoglycan degradation (ko00531), and galactose metabolism (ko00052), which are closely related to energy metabolism. These results suggest that cell metabolism in the gill of S. paramamosain was altered tremendously in response to a sudden drop in salinity from 23‰ to 3%. Especially, glycometabolism associated closely with energy metabolism was enhanced. So changes in cell metabolism in the gills of mud crab were very important for adaptation to a sudden drop in salinity to maintain physiological activities.

KEGG pathway classification (a) and functional enrichment (b) of differentially expressed miRNAs target genes. a The numbers of DEGs are shown on the X axis, while the KEGG pathway terms are shown on the Y axis. All second pathway terms are grouped in the following top pathway terms, as indicated by different colors: OS, organismal systems; M, metabolism; HD, human diseases; GIP, genetic information processing; EIP, environmental information processing; and CP, cellular processing. b RichFactor is the ratio of DESs of target genes annotated in this pathway term to all gene numbers annotated in this pathway term. Greater richFator means greater intensiveness. The Q-value is the corrected p-value ranging from 0~ 1. A lower Q-value indicates greater intensiveness. The top 20 enriched pathway terms are displayed (Q-value < 0.05)
Discussion
Most crustaceans live in aquatic environments and are very sensitive to chemical changes in water. Changes in water salinity directly affect the growth, survival rate, and reproduction of crustaceans, which are mainly related to changes in osmotic pressure and ion concentrations in body fluids. Artificial breeding environment restrictions (i.e., mass water exchange and heavy rainfall) and desalinization will lead to environmental salinity-induced stress. A rapid change in salinity due to heavy rainfall and mass water exchange will cause acute stress, while long-term decrease in salinity of an estuary can cause chronic stress, although crustaceans can adapt to such low-salt environments. Most euryhaline crustaceans can maintain internal homeostasis. The internal balance mechanisms (compensatory reactions) of euryhaline crustaceans can maintain homeostasis in response to a sudden change in salinity. This process can be simplified as a change from “stress” to “adaptation” without “failure.” However, high-intensity salinity stress may result in serious adverse reactions of crustaceans, including reduced production, morbidity, or even death. S. paramamosain is an economical euryhaline large-sized marine crustacean. The salinity of water for culturing of S. paramamosain is generally < 25‰ and the lowest salinity is close to freshwater (such as around Shanghai, China). Mud crabs are mainly distributed throughout estuary areas with large changes in salinity. During the rainy season, the salinity of farm ponds greatly vary, which can adversely affect the culture of S. paramamosain, resulting in sever economic losses. Because S. paramamosain is a euryhaline species, its sensitivity to sudden changes of salinity is often overlooked. This study preliminarily explored the adaptive mechanism of S. paramamosain to a sudden drop in salinity via changes in miRNA profiles of the gills. It is worth noting that the gills of S. paramamosain in the LS group had adapted to the change in salinity to 3‰ for 120 h.
Previous studies have shown that miRNAs are widely involved in various biological processes in animals, including cell differentiation, proliferation, development, apoptosis, metabolism, signal transduction, immunity, and evolution. MicroRNAs can guide silencing complexes to cleave target genes or inhibit the translation of target genes by pairing with 3′UTR region [13, 36]. GO functional classification of the DESs of target genes in response to a sudden drop in salinity from 23‰ to 3% revealed that 91 (28.53%) were related to metabolism. Many pathways were associated with amino acid metabolism, such as degradation of valine, leucine, and isoleucine (ko00280), isoleucine biosynthesis (ko00290), histidine metabolism (ko00340), arginine and proline metabolism (ko00330), tryptophan metabolism (ko00380), arginine biosynthesis (ko00220), alanine, aspartate, and glutamate metabolism (ko00250), glycine, serine, and threonine metabolism (ko00260), lysine degradation (ko00310), cysteine and methionine metabolism (ko00270), tyrosine metabolism (ko00350), lysine biosynthesis (ko00300), phenylalanine metabolism (ko00360), and phenylalanine, tyrosine, and tryptophan biosynthesis (ko00400) (Fig. 5 and Additional file 1: Table S3). Free amino acids play important roles in osmoregulation [1, 25, 46]. The results of the current study implied that amino acid metabolism in the gills of S. paramamosain was altered to equilibrate changes in exoteric osmotic pressure. Besides, we also identified pathways closely associated with energy metabolism, such as starch and sucrose metabolism (ko00500), glycosaminoglycan degradation (ko00531), and galactose metabolism (ko00052), among others (Additional file 1: Table S3). Changes in salinity lead to increased rates of oxygen consumption in shrimp and crabs, as well as an increased demand for energy, accelerated metabolism, and physiological dysfunction, which can results in reduced immunity [38]. Because salinity affects respiratory metabolism and energy balance of crustaceans, a sudden change in salinity may lead to a change in the osmotic pressure of body fluids of crustaceans, thereby resulting in energy dysregulation, which requires consumption of additional energy in response to osmotic pressure adjustments.
Conclusion
Although S. paramamosain is an important economical cultured marine animal, few studies have investigated osmotic adjustments in response to a change in salinity. Studies on the capabilities of crustaceans to adapt to different salinities are limited, especially research at the transcriptome and protein levels. However, one such study by the author of this article is currently under review. In this study, high-throughput sequencing technology was used to obtain the miRNA profiles of the gills of a normal control group (23‰) and in response to a sudden drop in salinity (3‰). The results of this study identified seven known miRNAs and 43 novel miRNAs, with 18 DESs (differentially expressed small RNAs). A total of 14,951 DESs of target genes were obtained by miRNA target gene prediction. GO functional classification of differentially expressed miRNAs target genes that were the most strongly affected in the gills of S. paramamosain in response to a sudden drop in salinity from 23‰ to 3‰ indicated that 578 genes were associated with cellular processes, 523 with metabolic processes, and 422 with single-organism processes. KEGG pathway analysis of differentially expressed miRNAs target genes showed that most of the identified pathways were associated with metabolism, including 14 pathways related to amino acid metabolism, which plays an important role in osmoregulation. Besides, several pathways were associated with energy metabolism, such as starch and sucrose metabolism (ko00500), glycosaminoglycan degradation (ko00531), and galactose metabolism (ko00052), among others. The above pathways reveal that S. paramamosain regulates osmotic pressure and energy balance by regulating target genes to adapt to a sudden change in salinity from 23‰ to 3‰. This article for the first time preliminarily studied the adaptation mechanisms of S. paramamosain to a sudden drop in salinity. Furthermore, some novel miRNAs were discovered, which paves the way for further in-depth exploration on salinity adaptation of S. paramamosain.
Abbreviations
- DESs:
-
Differentially expressed small RNAs
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- RT-qPCR:
-
Quantitative reverse transcription PCR
- S. paramamosain :
-
Scylla paramamosain
- sRNAs:
-
Small RNAs
References
Abe H, Okuma E, Amano H, et al. Role of free d- and l-alanine in the Japanese mitten crab Eriocheir japonicus to intracellular osmoregulation during downstream spawning migration. Comp Biochem Phys A. 1999;123(1):55–9.
Agarwal V, Bell GW, Nam J, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:e05005.
Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5.
Anger K. Salinity as a key parameter in the larval biology of decapod crustaceans. Invertebr Reprod Dev. 2003;43(1):29–45.
Bartel DP. MicroRNA target recognition and regulatory functions. Cell. 2009;136(2):215–33.
Brennecke J, Hipfner DR, Stark A, et al. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113(1):25–36.
Bushati N, Cohen SM. MicroRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205.
Carrington JC, Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301:336–8.
Chen G, Zhang C, Jiang F, et al. Bioinformatics analysis of hemocyte miRNAs of scallop Chlamys farreri against acute viral necrobiotic virus (AVNV). Fish Shellfish Immunol. 2014;37(1):75–86.
China Fishery Statistical Yearbook, 2017, China's Ministry of Agriculture.
Deng D, Wang L, Chen Y, et al. MicroRNA-124-3p regulates cell proliferation, invasion, apoptosis, and bioenergetics by targeting PIM1 in astrocytoma. Cancer Sci. 2016;107(7):899–907.e73392.
Feng Y, Cao JH, Li XY, et al. Inhibition of miR-214 expression represses proliferation and differentiation of C2C12 myoblasts. Cell Biochem Funct. 2011;29(5):378–83.
Flynt AS, Lai EC. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet. 2008;9(11):831–42.
He L, Wang YL, Li Q, et al. Profiling microRNAs in the testis during sexual maturation stages in Eriocheir sinensis. Anim Reprod Sci. 2015;162:52–61.
Jalali S, Ramanathan GK, Parthasarathy PT, et al. Mir-206 regulates pulmonary artery smooth muscle cell proliferation and differentiation. PLoS One. 2012;7(10):e46808.
Jiang H, Wong WH. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25(8):1026–32.
John B, Enright AJ, Aravin A, et al. Correction: Human MicroRNA targets. PLoS Biol. 2005;3(7):e264.
Kim JK, Kim TS, Basu J, et al. MicroRNA in innate immunity and autophagy during mycobacterial infection. Cell Microbiol. 2017; https://doi.org/10.1111/cmi.12687.
Kozomara A, Griffithsjones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42(Database issue):D68–73.
Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34(Web Server issue):W451–4.
Lau NC, Seto AG, Kim J, et al. Characterization of the piRNA complex from rat testes. Science. 2006;313:363–7.
Li S, Zhu S, Li C, et al. Characterization of micro RNAs in mud crab Scylla paramamosain under Vibrio parahaemolyticus infection. PLoS One. 2013;8(8):e73392.
Lin Z, Hao M, Zhu D, Li S, Wen X. Molecular cloning, mRNA expression and nutritional regulation of a Δ6 fatty acyl desaturase-like gene of mud crab, Scylla paramamosain. Comp Biochem Physiol B Biochem Mol Biol. 2017;208-209:29–37.
Long X, Wu X, Zhao L, et al. Effects of salinity on gonadal development, osmoregulation and metabolism of adult male Chinese mitten crab, Eriocheir sinensis. PLoS One. 2017;12(6):e0179036.
Lu JY, Shu MA, Xu BP, et al. Mud crab Scylla paramamosain glutamate dehydrogenase: molecular cloning, tissue expression and response to hyposmotic stress. Fish Sci. 2015;81:175–86.
Lv J, Liu P, Gao B, et al. The identification and characteristics of salinity-related micro RNAs in gills of Portunus trituberculatus. Cell Stress Chaperones. 2016;21(1):63–74.
Ma J, Li Y, Wu M, et al. Serum immune responses in common carp (Cyprinus carpio L.) to paraquat exposure: the traditional parameters and circulating microRNAs. Fish Shellfish Immunol. 2018;76:133–42.
Ma LB, Zhang FY, Ma CY, et al. Scylla paramamosain (Estampador) the most common mud crab (genus Scylla) in China: evidence from mtDNA. Aquac. Res. 2006;37:1694–1698.
Mennigen JA, Zhang D. MicroTrout: a comprehensive, genome-wide miRNA target prediction framework for rainbow trout, Oncorhynchus mykiss. Comp Biochem Physiol Part D Genomics Proteomics. 2016;20:19–26.
Nawrocki EP, Burge SW, Bateman A, et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43(Database issue):D130–7.
O'Connell RM, Rao DS, Baltimore D. MicroRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312.
Qu K, Wang Z, Lin XL, et al. MicroRNAs: key regulators of endothelial progenitor cell functions. Clin Chim Acta. 2015;448:65–73.
Romano N, Zeng C. Importance of balanced Na+/K+ ratios for blue swimmer crabs, Portunus pelagicus, to cope with elevated ammonia-N and differences between in vitro and in vivo gill Na+/K+-ATPase responses. Aquaculture. 2011;318(1–2):154–61.
Ruan L, Bian X, Ji Y, et al. Isolation and identification of novel microRNAs from Marsupenaeus japonicus. Fish Shellfish Immunol. 2011;31(2):334–40.
Shinji J, Wilder MN. Dynamics of free amino acids in the hemolymph of Pacific whiteleg shrimp Litopenaeus vannamei, exposed to different types of stress. Fish Sci. 2012;78(6):1187–94.
Siomi H, Siomi MC. On the road to reading the RNA-interference code. Nature. 2009;457(7228):396–404.
Takacs CM, Giraldez AJ. MicroRNAs as genetic sculptors: fishing for clues. Semin Cell Dev Biol. 2010;21(7):760–7.
Tantulo U, Fotedar R. Comparison of growth, osmoregulatory capacity, ionic regulation and organosomatic indices of black tiger prawn (Penaeus monodon Fabricius, 1798) juveniles reared in potassium fortified inland saline water and ocean water at different salinities. Aquaculture. 2006;258(1–4):594–605.
Tao YF, Qiang J, Bao JW, et al. miR-205-5p negatively regulates hepatic acetyl-CoA carboxylase β mRNA in lipid metabolism of Oreochromis niloticus. Gene. 2018;660:1–7.
Tate R, Rotondo D, Davidson J. Regulation of lipid metabolism by microRNAs. Curr Opin Lipidol. 2015;26(3):243–4.
Thompson BJ, Cohen SM. The hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126(4):767–74.
Ünlü ES, Gordon DM, Telli M. Small RNA sequencing based identification of MiRNAs in Daphnia magna. PLoS One. 2015;10(9):e0137617.
Walton ME, Le VL, Lebata JH, Binas J, Primavera JH. Seasonal abundance, distribution and recruitment of mud crabs (Scylla spp.) in replanted mangroves. Estuar Coast Shelf Sci. 2006;66:493–500.
Wang H, Tang L, Wei HL, et al. Transcriptomic analysis of adaptive mechanisms in response to sudden salinity drop in the mud crab, Scylla paramamosain. BMC Genomics. 2018;19:421.
Wang L, Feng Z, Wang X, et al. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8.
Wang WN, Wang AL, Bao L, et al. Changes of protein-bound and free amino acids in the muscle of the freshwater prawn Macrobrachium nipponense in different salinities. Aquaculture. 2004;233(1):561–71.
Wang Z, Sun B, Zhu F. Epigallocatechin-3-gallate inhibit replication of white spot syndrome virus in Scylla paramamosain. Fish Shellfish Immunol. 2017;67:612–9.
Watanabe T, Takeda A, Tsukiyama T, et al. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev. 2006;20:1732–43.
Wheeler BM, Heimberg AM, Moy VN, et al. The deep evolution of metazoan microRNAs. Evol Dev. 2009;11(1):50–68.
Xi QY, Xiong YY, Wang YM, et al. Genome-wide discovery of novel and conserved microRNAs in white shrimp (Litopenaeus vannamei). Mol Biol Rep. 2015;42(1):61–9.
Yang YH, Dudoit S, Luu P, et al. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 2002;30(4):e15.
Ye H, Tao Y, Wang GZ, et al. Experimental nursery culture of the mud crab Scylla paramamosain (Estampador) in China. Aquac Int. 2011;19:313–321.
Ye J, Fang L, Zheng HK, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Web Server issue):W293–7.
Yuan J, Xiao G, Peng G, et al. MiRNA-125a-5p inhibits glioblastoma cell proliferation and promotes cell differentiation by targeting TAZ. Biochem Biophys Res Commun. 2015;457(2):171–6.
Zheng Q, Wang XJ. GOEAST: a web-based software toolkit for gene ontology enrichment analysis. Nucleic Acids Res. 2008;36(Web Server issue):358–63.
Zhou Z, Wang L, Song L, et al. The identification and characteristics of immune-related micro RNAs in haemocytes of oyster Crassostrea gigas. PLoS One. 2014;9(2):e88397.
Acknowledgments
Aside from funding support, we also thank BGI (BGI, Shenzhen, China) for sequencing consultation and support.
Funding
This work was supported by the Major Sci & Tech Special Project of Zhejiang Province (no: 2016C02055-8), China Agriculture Research System-48 (no: CARS-48), Research Fund of Ningbo University (no: XYL18013), and the K. C. Wong Magna Fund in Ningbo University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
Raw sequences were deposited into the NCBI Sequence Read Archive (SRA) database with accession numbers SRP143427.
Author information
Authors and Affiliations
Contributions
CW and HWang conceived and designed the study. HWang and JL performed the cultivation of experimental animals, HWang, JL, LT, HWei, and CM performed and analyzed all the other experiments. HWang and HWei wrote the manuscript with support from all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal subjects used in the present study are crabs, which are invertebrates and are exempt from this requirement. S. paramamosain is not an endangered or protected species. All animal work has been conducted according to the relevant national and international guidelines. No specific permissions are required to work with invertebrates in China. Similarly, no specific permissions are required for the collection of S. paramamosain from sample sites because they were not collected from protected areas of land.
Consent for publication
“Not applicable”
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Differentially expressed miRNAs. Table S2. Gene expression of 18 miRNAs for the comparison of CK and LS groups according to qPCR analysis. Table S3. KEGG pathway annotation and classification. (DOCX 60 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, H., Wei, H., Tang, L. et al. Identification and characterization of miRNAs in the gills of the mud crab (Scylla paramamosain) in response to a sudden drop in salinity. BMC Genomics 19, 609 (2018). https://doi.org/10.1186/s12864-018-4981-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4981-6