
Abstract
Background
Copy number variation (CNV) has been implicated in the genetics of multiple human diseases. Spinal muscular atrophy (SMA) and 22q11.2 deletion syndrome (22q11.2DS) are two of the most common diseases which are caused by DNA copy number variations. Genetic diagnostics for these conditions would be enhanced by more accurate and efficient methods to detect the relevant CNVs.
Methods
Competitive PCR with limited deoxynucleotide triphosphates (dNTPs) and high-resolution melting (HRM) analysis was used to detect 22q11.2DS, SMA and SMA carrier status. For SMA, we focused on the copy number of SMN1 gene. For 22q11.2DS, we analyzed CNV for 3 genes (CLTCL1, KLHL22, and PI4KA) which are located between different region-specific low copy repeats. CFTR was used as internal reference gene for all targets. Short PCR products with separated Tms were designed by uMelt software.
Results
One hundred three clinical patient samples were pretested for possible SMN1 CNV, including carrier status, using multiplex ligation-dependent probe amplification (MLPA) commercial kit as gold standard. Ninety-nine samples consisting of 56 wild-type and 43 22q11.2DS samples were analyzed for CLTCL1, KLHL22, and PI4KA CNV also using MLPA. These samples were blinded and re-analyzed for the same CNVs using the limited dNTPs PCR with HRM analysis and the results were completely consistent with MLPA.
Conclusions
Limited dNTPs PCR with HRM analysis is an accurate method for detecting SMN1 and 22q11.2 CNVs. This method can be used quickly, reliably, and economically in large population screening for these diseases.
Similar content being viewed by others

Background
Copy number variation (CNV) refers to excess or deficient copies of portions of a genome, from entire chromosomes (e.g., trisomy) to smaller segments. CNVs are pervasive in the human genome and have been reported to be associated with many genetic disorders [1]. Methods for CNV detection are crucial for identifying genetic risk factors for disease, and carrier status. Several techniques such as comparative genomic hybridization (CGH) microarrays, MLPA, and fluorescent in situ hybridization (FISH) are currently utilized to detect copy number alterations [2, 3]. CGH microarrays are efficient to scan for copy number variation on a large (e.g., genome-wide) scale. FISH is routinely used in the clinical laboratory as a gold standard for detection of CNV. MLPA has been widely used to detect specific CNVs associated with genetic disease in the clinical laboratory [2, 3]. These methods are time-consuming and rely on expensive and specialized instrumentation for operation and analysis.
Competitive PCR with restricted dNTPs and high resolution melting (HRM) is a simple, fast, and economical approach with high accuracy that can be used for targeted evaluation of relative copy number [4]. The necessary multiplex PCR assays can be easily designed using uMELT melting curve prediction software. Spinal muscular atrophy (SMA) and 22q11.2 deletion syndrome (22q11.2DS) are two of the most common genetic diseases which are caused by genomic copy number variations [5, 6]. For affected patients, a definite diagnosis largely relies on genetic diagnosis by the copy number assessment of the SMN1 gene and the 22q11.2 region, respectively. In this paper we demonstrate the effectiveness of limited dNTPs PCR with HRM analysis for SMA and 22q11.2DS detection.
Methods
DNA samples
One hundred three clinical DNA samples from Shanghai Children’s Medical Center exhibiting SMA-related symptoms were analyzed for copy number of SMN1. Another 99 clinical DNA samples obtained from Shanghai Children’s Medical Center, 43 having 22q11.2DS and the remaining 56 not having 22q11.2DS were tested for copy number of the 22q11.2 region. All identifying data and copy number related data were removed to perform the comparison study of CNV analysis methods in a blinded fashion. Written consent was obtained from parents or guardians of all patients. This study was approved by the Ethics Committee of Shanghai Children’s Medical Center.
DNA extraction
Peripheral blood samples of all cases were obtained and genomic DNA was extracted by using QIAmp DNA Blood Mini Kit (QIAGEN GmbH, Germany) with standard protocols. DNA was quality tested by optical density 260/280 nm ratios, quantified by UV spectrophotometry, and stored at − 20 °C until use. The DNA concentration of the samples ranged from 30 ng/μl to 214 ng/μl.
Primer design
Primers for target and reference genes were designed with Primer3 online software (http://bioinfo.ut.ee/primer3-0.4.0/primer3/). Primer specificity was checked on the UCSC in silico PCR (http://genome.ucsc.edu/cgi-bin/hgPcr). The PCR fragment should be unique in human genome, and have at most only rare SNPs or other sequence variants. Human genome databases NCBI (http://www.ncbi.nlm.nih.gov) and UCSC (http://genome.ucsc.edu) were used to check the uniqueness and SNP frequency of PCR sequences. uMelt software (https://dna.utah.edu/umelt/umelt.html) was used to predict the melting temperatures (Tms) of reference and target PCR products, and their melting domain. The reference and target products were well distinguished by melting temperatures. PCR products having only one melting domain were selected. The ΔTm between selected reference and one or more targets were in the necessary range of between 2 °C to 10 °C [4].
Selection of target genes
SMA is most commonly caused by the homozygous deletion of SMN1 (0 copies of SMN1). SMN1 and SMN2 are highly homologous, having only 5 different nucleotides, that occur on intron 6, exon 7 and on noncoding exon 8. The only coding nucleotide difference is SMN1 c.840C > T in exon 7 [7]. In the duplex PCR to determine the copy number of SMN1, an allele specific primer was used to amplify only SMN1 c.840C, but not the SMN2 homolog [4]. Another primer pair amplified a reference segment of CFTR exon 7. For SMA, each assay contained 3 control samples, having 2, 1, and 0 copies of SMN1, respectively. They were obtained from an SMA non-carrier, an SMA carrier and an affected patient with SMA which had been confirmed by MLPA in our previously study.
22q11.2DS is caused by a hemizygous deletion in 22q11.2 region. The deletions start from the LCR22-A region and about 87% extend to LCR22-D, though in some cases they extend only as far as LCR22-B, or LCR22-C [8, 9]. Three genes, from between each pair of adjacent regions, CLTCL1 from between LCR22-A and LCR22-B, KLHL22 from between LCR22-B and LCR22-C, and PI4KA from between LCR22-C and LCR22-D (Fig. 1) were chosen as targets to detect copy number and determine deletion extent of 22q11.2. For each 22q11.2DS CNV analysis, one duplex PCR and one triplex PCR were performed, both using CFTR as copy number reference. The duplex PCR quantified CLTCL1 copy number, and the triplex PCR simultaneously quantified the copy numbers of KLHL22 and PI4KA genes. The reference and targets primers’ chromosome location, gene name, sequences, Tms and sizes of PCR amplicons are listed in Table 1. All the primers were synthetized by the Beijing Genomics Institute (Beijing, China). For 22q11.2 CNV detection, each assay contained 2 controls, 1 normal sample and 1 sample from a patient having the most frequent 22q11.2 hemizygous deletion, from LCR22-A to LCR22-D.

The distribution of CLTCL1, KLHL22, and PI4KA genes in the 22q11.2 region. CLTCL1 is located between LCR22-A and LCR22-B. KLHL22 is located between LCR22-B and LCR22-C. The PI4KA gene is located between LCR22-C and LCR22-D
PCR and high resolution melting
PCR was performed in 10 μl volume containing 0.125 μM–1 μM each primers, 10 mM Tris (pH = 8.3), 0.5 mg/ml BSA, 2 mM MgCl2, 0.4 U KlenTaq (Ab Peptides), 6.25 μM dNTPs, 1X LCGreen® Plus dye (BioFire Defense) and 30 ng to 214 ng genomic DNA. Primers concentration varied between reactions to optimize analytical resolution. In duplex PCR, the concentrations of each primer (forward and reverse) were 0.25 μM CFTR and 0.5 μM SMN1; 0.125 μM CFTR and 0.5 μM CLTCL1. In the triplex PCR, the concentrations each pair of primers were 0.125 μM CFTR, 1 μM KLHL22 and 0.5 μM PI4KA.
PCR and high resolution melting were performed on the Rotor-Gene Q thermocycler (Qiagen GmbH, Germany). PCR conditions were: Denature at 95 °C for 60s, followed by 35 cycles of 95 °C for 10s and 64 °C for 30s. Following amplification, the samples were melted at a 0.3 °C/s melting rate from 65 °C to 95 °C and high-resolution melting data were acquired. To determine the reproducibility of the new method, the PCR and HRM analyses were performed in three independent runs on all samples.
Analysis
High-resolution melting data were exported and analyzed by MeltWizard 5.1 software [4]. Analysis steps began with exponential background subtraction to remove the fluorescence background, and optional normalization was performed to eliminate the fluorescence differences between samples and correspond to total helicity. Next, melting curves were temperature shifted so that shared intrinsic features were optimally overlaid, so as to remove minor temperature variation between wells. The negative derivative was calculated by degree 2 Savitzky-Golay differentiation. To properly quantify the copy number differences, the reference peaks of all derivative plots were normalized both vertically (to the geometric mean reference peak amplitude) and horizontally (scaling and shifting to the mean temperatures of each peak). After performing unbiased hierarchical clustering in the uniform metric, different copy numbers of the target were visualized and quantified.
MLPA
MLPA is a copy number quantification method that may be performed using commercially available kits (MRC-Holland, The Netherlands). The P060-B2 kit was used to determine the copy number of SMN1. The P250-B2 kit was used to determine the copy number of 22q11.2. All procedures were performed according to the manufacturer’s protocol. After the MLPA reaction, including denaturation, hybridization, ligation and the PCR amplification performing on C1000™ Thermal Cycler (Bio-Rad, USA), the amplified products were separated by electrophoresis on ABI 3130 genetic analyzer (Applied Biosystems, USA). Electrophoresis data were visualized and analyzed with GeneMaker software (SoftGenetics, LLC, USA).
Results
To verify the effectiveness of limited dNTPs PCR and HRM, 103 clinical samples were tested for SMA, and 99 clinical samples were tested for 22q11.2DS, using a commercial MLPA kit as a gold standard control. Both methods were used to quantify copy numbers of various genes associated with SMA and 22q11.2DS. SMA will be manifested when there are zero copies (homozygous deletion) of the SMN1 gene. An individual will be classified to be a SMA carrier when there is one copy (heterozygous deletion) of SMN1. 22q11.2 deletion syndrome is diagnosed when there are hemizygous deletions of various regions of 22q11.2 from LCR22-A to LCR22-D. Approximately 87% of 22q11.2 deletions span about 3 Mb from LCR22-A to LCR22-D; approximately 8% of deletion span about 1.5 Mb from LCR22-A to LCR22-B; the remaining deletions, approximately 5%, span about 2 Mb from LCR22-A to LCR22-C or about 1.5 Mb LCR22-B to LCR22-D [9]. The 3 genes analyzed were chosen to distinguish these possibilities.
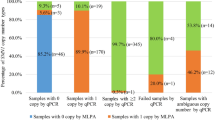
Among the 103 potential SMA samples tested for copy number of SMN1 exon 7, the MLPA method detected 23 samples having 2 copies (normal), 1 sample having 3 copies (no SMA), 43 samples having 1 copy (SMA carrier) and 36 samples having 0 copies (SMA patient). Among the 99 potential 22q11.2 deletion samples, the MLPA method detected 56 samples having 2 copies (normal), 38 cases having deletions from LCR22-A to LCR22-D; 2 samples having deletions from LCR22-A to LCR22-C; and 3 cases having deletions from LCR22-A to LCR22B.
All samples that were pretested using MLPA for SMA and 22q11.2 associated CNV were blinded prior to performing limited dNTPs PCR and HRM. Copy numbers were quantified by target melting peak heights.
For SMA, the melting peak heights obtained following duplex PCR easily distinguished 2, 1 and 0 copies of SMN1 (Fig. 2). In 103 blinded potential SMA samples, limited dNTPs PCR and HRM detected 23 samples having 2 copies (normal); 1 sample having more than 2 copies (normal); 43 samples having one copy (carrier, heterozygous deletion) and 36 samples having zero copies (SMA patients, homozygous deletion). Not only were the total number of samples with each copy number the same, but also all individual samples had corresponding copy numbers. For example, the sample exhibiting more than 2 copies by limited dNTPs and HRM was the sample that exhibited 3 copies according to MLPA. There was complete concordance of the results of limited dNTPs PCR and HRM with the results of MLPA (Table 2a, Additional file 1).

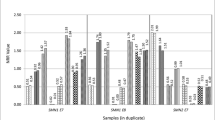
Duplex PCR and HRM result for SMN1 exon7 copy number assessment. Using the restricted dNTPs and duplex PCR, samples with 3 copies (green), 2 copies (blue), 1 copy (black) and 0 copy (red) of SMN1 exon7 were well distinguished after normalization against CFTR gene. −dF/dT, negative first derivative of fluorescence with respect to temperature
For 22q11.2DS, the melting peak heights obtained following the triplex and duplex PCRs easily distinguished 2 vs. 1 copies of the 3 target genes CLTCL1, KLHL22, and PI4KA spread across 22q11.2 from LCR22-A to LCR22-D (Fig. 3). In 56 samples 2 copies of the CLTCL1 gene were found in the duplex reaction (no CNV related 22q11.2DS). In 3 samples, 1 copy of the CLTCL1 gene was found in the duplex reaction, while 2 copies of the KLHL22 and PI4KA genes were found in the triplex reaction (deletion from LCR22-A to LCR22-B). In 2 samples, 1 copy of the CLTCL1 gene was found in the duplex reaction, 1 copy of the KLHL22 gene and 2 copies of the PI4KA gene were found in the triplex reaction (deletion from LCR22-A to LCR22-C). In 38 samples, 1 copy of the CLTCL1 gene was found in the duplex reaction, and 1 copy of the KLHL22 and PI4KA genes were found in the triplex reaction (deletion from LCR22-A to LCR22-D). Not only were the total number of samples with each copy number the same, but also all individual samples had corresponding copy numbers. For example, the 2 samples exhibiting 1 copy of the KLHL22 gene and 2 copies of the PI4KA gene by limited dNTPs and HRM were the same 2 samples having these CNVs according to MLPA. There was complete concordance of the results of limited dNTPs PCR and HRM with the results of MLPA (Table 2b, Additional file 2).

22q11.2 region copy number determination by restricted dNTPs and multiplex PCR. Blinded tests for a CLTCL1, b PI4KA/KLHL22 are shown. Samples were normalized to the CFTR gene. Two copies (Black) and one copy (red) of target genes were well distinguished after normalization against the reference. The green signal in Fig. 3b showed a sample with two copies of PI4KA gene and one copy of KLHL22 gene. −dF/dT, signifies the negative first derivative of fluorescence with respect to temperature
Discussion
As more CNV associated diseases are being investigated, there is a growing demand for widely applicable, accurate, rapid and economical approaches to discern variations in copy number [10]. Here, we proposed a detection method based on amplification with limited dNTPs and HRM analysis that can be easily adapted to any genomic targets of interest. Careful optimization of primers, conditions, and concentrations are essential so that PCR yields the most informative melting peak data [11]. Besides conventional primer design criteria, additional considerations are required. First, one must aim to design short PCR products in order to prevent the occurrence of multiple melting domains and avoid internal sequence variation. Simultaneously, amplicon sequence differences must result in sufficiently distinct melting temperatures so their peaks can be clearly distinguished in duplex or triplex reactions. The implementation of DNA melting analysis also relies on the instrument resolution. A heating rate of 0.1–1 °C/s is common for high resolution melting devices [12]. For all samples that will be compared in one test, the DNA isolation and purification should be standardized so that they can be dissolved in the same buffer solution. Variable concentrations of salt and different ionic strength will affect the relative peak height of products dramatically.
Above all, the determination of appropriate dNTPs concentration is the most crucial factor for precise relative quantification. When the dNTPs concentration is too low (lower than 1.56 μM), the multiplex PCR may only amplify one target and the shortest amplicon will have the competitive advantage. Above a certain dNTPs concentration (greater than 25 μM), quantification of initial relative reference and target copies are lost in the plateau of primer limitation and no CNV information remains [4]. For each particular CNV assay, an optimal dNTPs concentration can be found at which the amplification is still limited but each copy number is clearly recognizable. Based on these design strategies, we developed assays that illustrate the effectiveness and superior time and cost efficiency of the method in screening for two common CNV diseases, SMA and 22q11.2 deletion syndrome.
SMA is characterized by symmetrical and progressive proximal muscular atrophy owing to the degeneration of α-motor neurons in the anterior horns of spinal cord [13]. Among autosomal recessive disorders, SMA is the most frequent inherited cause of infant and early childhood mortality [14]. Meanwhile, SMA has a high carrier frequency of 1:47 to 1:72 in the general population [15,16,17]. Most people won’t know their carrier status until they have an affected child [13]. Considering the severity of the disease and the relatively high carrier frequency, timely diagnosis by molecular detection and prevention of new cases by carrier screening are the best ways to help SMA families [18].
SMN1 produces a full-length SMN protein and is the SMA-determining gene [19]. The homozygous absence of the SMN1 has been observed in the majority of patients. Direct analysis of SMN1 gene deletion is valuable in both SMA molecular diagnosis and carrier screening [14, 20]. The quantitative analysis and clinical implications of SMN1 gene dosage are somewhat complicated by the presence of a highly homologous gene SMN2 that only differs in five base pairs. Among these differences, a single nucleotide variation of c.840C > T substitution in the coding sequence of exon 7 is pivotal [21]. This site can affect RNA splicing in SMN2 and produce an unstable and nonfunctional spliced isoform. It is commonly used to differentiate SMN1 from SMN2. In our research, by using allele-specific duplex PCR with limited dNTPs, we can distinguish between the SMN1 and SMN2 genes and evaluate the copy number of SMN1 exon7. Deficient SMN2 copy number does not cause SMA, so the copy number analysis of SMN2 it is not routinely performed within the setting of diagnostic or carrier testing for SMA [22]. However, additional SMN2 copies, that can in rare cases arise in conjunction with missing SMN1 copies, can partially mitigate the most severe effects of SMA [14, 23, 24]. Followup testing of SMN2 copy number could be beneficial when screening finds zero copies of SMN1.
Our technique can identify the majority SMA patients with homozygous absence of SMN1 gene and provide universal carrier detection for population based screening. However, this method has limitations. For SMA carrier screening, the silent carriers having two copies of SMN1 on one chromosome but no one on the other, as well as SMN1 intragenic mutations could be missed [25]. In this aspect of clinical diagnosis, approximately 5% of affected SMA patients with intragenic mutations in the SMN1 gene will not be detected by deletion testing methods, including our method and MLPA [26]. Limited dNTPs amplification with HRM is a dosage analysis of the SMN1 gene copies, while sequence analysis of the SMN1 gene can be especially useful for the patient who possesses a single copy of SMN1 yet exhibits an SMA-like phenotype.
The 22q11.2 deletion is the most frequent interstitial deletion in humans with an incidence of 1 in 4000 live births [27]. This region is known to have eight low copy number repeats which make it particularly susceptible to suffer unequal meiotic exchange. The majority of patients have a typical deletion region spanning LCR22-A to LCR22-D, while a smaller portion of patients have a smaller proximal nested deletion extending from LCR22-A to LCR22-B or LCR22-A to LCR22-C [28]. For the detection of 22q11.2 deletion syndrome, two competitive PCRs were performed to obtain a rapid determination of the patient genotype. The use of 3 genes, CLTCL1, KLHL22 and PI4KA, located within the deleted regions allowed for discrimination between the various extents of deletion from LCR22-A to LCR22-B, LCR22-C, or LCR22-D. Children with 22q11.2DS display a wide variability in clinical phenotype and experience a host of medical difficulties during early life [29, 30]. When the patients have an identified cause, evaluation of the disease-related features and prevention of certain medical sequelae is feasible and recommendable. Based on previous study, the association of congenital heart disease (CHD) with 22q11.2DS has been well established and it is usually the first presenting symptom in such patients [31]. Molecular screening for 22q11.2 in CHD patients is of great importance in diagnosis and prognosis of disease.
The effectiveness of limited dNTPs amplification with HRM copy number analysis for SMA and 22q11.2DS was validated on clinical samples. Gene dosage results for all cases were in 100% concordance with the results of MLPA, which demonstrated it could be a reliable method for copy number assessment for these patients. In addition, the limited dNTPs method exhibits promising advantages on rapidity, low cost and simple implementation. Firstly, the entire process requires only PCR, melting acquisition and analysis, enabling results to be obtained within 1 h. Secondly, only saturating (high-resolution) fluorescent dye, primers and common PCR components are needed, allowing low reagent cost. Meanwhile, as a closed-tube system, our system requires no additional processing or separation steps, saving considerable labor for operators and greatly reducing the risk of laboratory contamination. Finally, the fluorescence signal can be detected by instrumentation which is readily available in most diagnostic platforms, democratizing the molecular genetic analysis in all kinds of platforms. MLPA has been commonly used for clinical CNV analysis and up to 50 loci can be tested simultaneously in the process. However, MLPA requires at least 2 days for DNA denaturation, probe hybridization and ligation, PCR amplification and data analysis. It demands customized oligonucleotide probes and expensive electrophoresis instruments, both of which increase the cost.
Conclusions
As a relatively common lethal autosomal recessive disorder with high carrier frequency, SMA copy number testing is an ideal candidate for population-based screening. Considering the high incidence of 22q11.2DS and its close association with congenital heart defects, screening for 22q11.2 deletion is also advisable for all infants with CHD. The rapid and cost-effective detection of CNV in SMN1 and 22q11.2DS related genes which can be performed and interpreted in all levels of diagnostic platforms are invaluable. Our research demonstrates that limited dNTPs amplification with HRM could be an ideal method for prompt detection of SMA and 22q11.2DS and for SMA carrier screening.
Abbreviations
- 22q11.2DS:
-
22q11.2 deletion syndrome
- CGH:
-
Comparative genomic hybridization arrays
- CHD:
-
Congenital heart diseases
- CNV:
-
Copy number variation
- dNTPs:
-
Deoxynucleotide triphosphates
- FISH:
-
Fluorescent in situ hybridization
- HRM:
-
High-resolution melting
- LCR:
-
Low copy repeat
- MLPA:
-
Multiplex ligation-dependent probe amplification
- SMA:
-
Spinal muscular atrophy
- Tm:
-
Melting temperature
References
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–54.
Carter NP. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat Genet. 2007;39:S16–21.
Shen Y, Wu B-L. Designing a simple multiplex ligation-dependent probe amplification (MLPA) assay for rapid detection of copy number variants in the genome. J Genet Genomics. 2009;36(4):257–65.
Zhou L, Palais RA, Paxton CN, Geiersbach KB, Wittwer CT. Copy number assessment by competitive PCR with limiting deoxynucleotide triphosphates and high-resolution melting. Clin Chem. 2015;61(5):724–33.
Merikangas AK, Corvin AP, Gallagher L. Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends Genet. 2009;25(12):536–44.
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071.
Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, JD MP. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–83.
Zhang X, Xu Y, Liu D, Geng J, Chen S, Jiang Z, Fu Q, Sun K. A modified multiplex ligation-dependent probe amplification method for the detection of 22q11.2 copy number variations in patients with congenital heart disease. BMC Genomics. 2015;16:364.
Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, et al. Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9(4):489–501.
Marcinkowska-Swojak MUB, Figlerowicz M, Kozlowski P. An MLPA-based strategy for discrete CNV genotyping: CNV-miRNAs as an example. Hum Mutat. 2013;34(5):763–73.
Erali M, Wittwer CT. High resolution melting analysis for gene scanning. Methods. 2010;50(4):250–61.
Zhou L, Wang L, Palais R, Pryor R, Wittwer CT. High-resolution DNA melting analysis for simultaneous mutation scanning and genotyping in solution. Clin Chem. 2005;51(10):1770–7.
Smith M, Calabro V, Chong B, Gardiner N, Cowie S, du Sart D. Population screening and cascade testing for carriers of SMA. Eur J Hum Genet. 2007;15(7):759–66.
Prior TW, Nagan N. Spinal Muscular Atrophy: Overview of Molecular Diagnostic Approaches. Curr Protoc Hum Genet. 2016;88:Unit 9.27.
Hendrickson BC, Donohoe C, Akmaev VR, Sugarman EA, Labrousse P, Boguslavskiy L, Flynn K, Rohlfs EM, Walker A, Allitto B, et al. Differences in SMN1 allele frequencies among ethnic groups within North America. J Med Genet. 2009;46(9):641–4.
Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, Flynn K, Hendrickson BC, Scholl T, Sirko-Osadsa DA, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. 2012;20(1):27–32.
Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in the autosomal recessive spinal muscular atrophy (SMA). Hum Mutat. 2000;15:228–37.
Ben-Shachar S, Orr-Urtreger A, Bardugo E, Shomrat R, Yaron Y. Large-scale population screening for spinal muscular atrophy: clinical implications. Genet Med. 2011;13(2):110–4.
Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65.
Miskovic M, Lalic T, Radivojevic D, Cirkovic S, Ostojic S, Guc-Scekic M. Ten years of experience in molecular prenatal diagnosis and carrier testing for spinal muscular atrophy among families from Serbia. Int J Gynaecol Obstet. 2014;124(1):55–8.
Lorson CLHE, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–11.
Prior TW, Nagan N, Sugarman EA, Batish SD, Braastad C. Technical standards and guidelines for spinal muscular atrophy testing. Genet Med. 2011;13(7):686–94.
Feldkötter MSV, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–68.
Calucho M, Bernal S, Alias L, March F, Vencesla A, Rodriguez-Alvarez FJ, Aller E, Fernandez RM, Borrego S, Millan JM, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28(3):208–15.
Ar Rochmah M, Awano H, Awaya T, Harahap NIF, Morisada N, Bouike Y, Saito T, Kubo Y, Saito K, Lai PS, et al. Spinal muscular atrophy carriers with two SMN1 copies. Brain and Development. 2017;39(10):851–60.
Feng Y, Ge X, Meng L, Scull J, Li J, Tian X, Zhang T, Jin W, Cheng H, Wang X, et al. The next generation of population-based spinal muscular atrophy carrier screening: comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet Med. 2017;19(8):936–44.
Oskarsdóttir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child. 2004;89(2):148–51.
Burnside RD. 22q11.21 deletion syndromes: a review of proximal, central, and distal deletions and their associated features. Cytogenet Genome Res. 2015;146(2):89–99.
Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EW, Chung B, Cytrynbaum C, Faghfoury H, Fishman L, et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet Med. 2015;17(8):599–609.
Pretto D, Maar D, Yrigollen CM, Regan J, Tassone F. Screening newborn blood spots for 22q11.2 deletion syndrome using multiplex droplet digital PCR. Clin Chem. 2015;61(1):182–90.
McDonald-McGinn DM, Zackai EH. Genetic counseling for the 22q11.2 deletion. Dev Disabil Res Rev. 2008;14(1):69–74.
Acknowledgments
We thank all the patients for their participating in this study.
Funding
This work was supported by the National Natural Science Foundation of China (No.81371893), Shanghai Sailing Program (18YF1414800), Shen Kang Municipal Hospital Emerging Frontier Technology joint research project (SHDC12017109) and Translational Medicine Collaborative Innovation research project of Shanghai Jiao Tong University School of Medicine (TM201615). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The data generated or analysed during this study are included in this published article and its supplementary information files.
Author information
Authors and Affiliations
Contributions
XZ and BW performed the experiments and drafted the manuscript. LCZ and GY performed the bioinformatics analyses. RP and LMZ helped with the analysis and writing of the manuscript. QF and LMZ conceived of and supervised the overall research. LZ and RP are listed as inventors on patent application WO2014210199A3. All authors read, approved and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Shanghai Children’s Medical Center. Written consent was obtained from parents or guardians of all patients.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Restricted dNTPs /HRM and MLPA results for SMN1 exon7 copy number assessment. Figure S1-S2. SMN1 copy number determination by restricted dNTPs and multiplex PCR. Figure S3. MLPA results for normal control, SMA carrier and SMA samples. Table S1. Comparison of SMN1 detection results of Limited dNTPs/HRM and MLPA. (PDF 1052 kb)
Additional file 2:
22q11.2 region copy number determination by restricted dNTPs/HRM and MLPA. Figure S4. CLTCL1 copy number determination by restricted dNTPs and multiplex PCR. Figure S5. PI4KA/KLHL22 copy number determination by restricted dNTPs and multiplex PCR. Figure S6. MLPA results for normal control and 22q11.2 deletion samples. Table S2. Comparison of 22q11.2 detection results of Limited dNTPs/HRM and MLPA. (PDF 1098 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, X., Wang, B., Zhang, L. et al. Accurate diagnosis of spinal muscular atrophy and 22q11.2 deletion syndrome using limited deoxynucleotide triphosphates and high-resolution melting. BMC Genomics 19, 485 (2018). https://doi.org/10.1186/s12864-018-4833-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4833-4