
Abstract
Background
Identification of genomic regions that have been targets of selection may shed light on the genetic history of livestock populations and help to identify variation controlling commercially important phenotypes. The Azeri and Kuzestani buffalos are the most common indigenous Iranian breeds which have been subjected to divergent selection and are well adapted to completely different regions. Examining the genetic structure of these populations may identify genomic regions associated with adaptation to the different environments and production goals.
Results
A set of 385 water buffalo samples from Azeri (N = 262) and Khuzestani (N = 123) breeds were genotyped using the Axiom® Buffalo Genotyping 90 K Array. The unbiased fixation index method (FST) was used to detect signatures of selection. In total, 13 regions with outlier FST values (0.1%) were identified. Annotation of these regions using the UMD3.1 Bos taurus Genome Assembly was performed to find putative candidate genes and QTLs within the selected regions. Putative candidate genes identified include FBXO9, NDFIP1, ACTR3, ARHGAP26, SERPINF2, BOLA-DRB3, BOLA-DQB, CLN8, and MYOM2.
Conclusions
Candidate genes identified in regions potentially under selection were associated with physiological pathways including milk production, cytoskeleton organization, growth, metabolic function, apoptosis and domestication-related changes include immune and nervous system development. The QTL identified are involved in economically important traits in buffalo related to milk composition, udder structure, somatic cell count, meat quality, and carcass and body weight.
Similar content being viewed by others

Background
The water buffalo (Bubablus bubalis) is an important livestock resource in many regions of the world, particularly in tropical and subtropical countries. Water buffalo produce milk, meat and are used as draught animal in developing countries [1, 2]. There are two types of domestic water buffalo: the river buffalo that originated in the Indian sub-continent and are now spread widely from India to Europe, and the swamp buffalo, that originated in N. Thailand or Laos and are the most common buffalo in Asia from India to the Philippines [2, 3]. The current world population of buffalo is about 200 million head compared with 1.49 billion cattle, 1.17 billion sheep and 1 billion goats [4]. Although, water buffalo represents only 11.8% of the world bovinae population, a large proportion of the world’s population depend on the domestic water buffalo [1, 5]. Unlike other domesticated bovids, whose populations are declining, the water buffalo population worldwide has increased constantly at a rate of 1.65% per year during the last five decades. However, the potential of buffalo has not been fully exploited. Water buffalo breeders and farmers face many challenges, such as poor reproductive efficiency, suboptimal production potential, and low rates of calf survival [6]. Improvement of these traits will support increasing buffalo production, particularly in poorer communities.
Iranian buffalo breeds originated from the Indian sub-continent and have been farmed in the Lorestan province of Iran since the ninth Century B.C. [7]. In the 1930s there were 1.5 million head of buffalo in Iran [7]. In contrast to the world trend, the number of Iranian buffaloes has dramatically decreased to ~ 204,000 head today [AGRI, 2014].
There are three main buffalo breeds in Iran: Azeri, Khuzestani and Mezandrani, with 119,000, 81,000 and 4000 individuals, respectively. These three breeds are reared in three different geographical areas of the country: the Azeri is widespread in the north-west and north of Iran, the Khuzestani is focused in the west and south-west (mainly in Khuzestan province), and the Mazandrani in the north of the country, mainly in the Mazandran province. These breeds experience dramatically different climatic conditions: in the northwest, Azeri buffalo are exposed to cold, sub-zero winters with heavy snowfall and hot, dry summers a with temperatures reaching 35 °C. The climate of Khuzestan in the South-west is generally very hot and occasionally humid with summertime temperatures routinely exceeding 45 °C degrees while in the winter, it can drop below freezing.
The buffalo farming system in Iran is based on smallholders (99%) and the median herd size is five animals, with a small number of herds of between 20 and 50 buffaloes and only a very few herds with 200 buffaloes (AGRI, 2010). Farming systems differ between breeds. Khuzestan buffaloes are raised outdoors throughout the year, while Azeri and Mazandrani breeds are housed in autumn and winter seasons [7]. Water buffalo provide about 239 thousand tons (2.8% of Iran's total milk production) and 24.7 thousand tons (2.5%) of meat [AGRI, 2010]. Average milk production of Iranian buffaloes in a 202-day lactation ranges from, 1141Kg in W. Azarbaijan, to 2017 Kg in Khuzestan [8]. Azeri and Khuzestani are different in milk production, body size and weight. Milk production in 210-day lactation period was 1865 and 1200 kg for Khuzestani and Azeri, respectively. Height-at-wither is 133 cm and body weight is varied 400 to 600 kg for Azeri adult female. Height-at-wither is 141 cm and body weight is 600 kg for Khuzestani adult female [7].
In recent years, the development of high-density SNP platforms has boosted genomics research in many livestock species [9]. A de novo assembly of the water buffalo genome from mixed Illumina and 454 data, and an Affymetrix panel of SNP markers has been created for water buffalo [10]. These new buffalo genomic resources open an unprecedented range of research possibilities for this species: from genome wide association studies to identifying genomic regions controlling target traits to genome assisted genetic selection [11,12,13,14].
Identifying signatures of recent selection in domesticated animals could provide information on the genomic response to domestication, climatic adaptation and selection for production traits [15]. This information may assist the design of more efficient selection schemes [16]. During the domestication process, livestock have been selected for desired morphological characteristics, physiology, increased yield, behavior and adaptation to particular environments [17, 18].
Genetic selection changes the frequency of beneficial variants and neutral variations in neighboring regions, leaving patterns in genome that can be distinguished between the population [19]. These patterns, referred to as signatures of selection, are detectable in genomic datasets as (i) changes in the ratio of non-synonymous to synonymous variations in the open reading frames (ii) a deficiency in heterozygosity compared with the rest of the genome, (iii) deviation in the Site Frequency Spectrum (SFS), (iv) differences in the allele frequencies among populations, (v) LD persistency, and (vi) unusual long-range haplotypes [20, 21]. Several statistical methods have been developed to identify selection signatures in genomic data [22,23,24,25,26,27,28,29,30,31,32,33]. To date, several studies have identified loci and genomic regions subject to positive selection in different domestic animals [18, 27, 34,35,36]. Once identified, these signals of selection can be used to search for the genes involved in adaptation or which are under selection [18].
The study presented here used 90,000 SNP genotypes for Azeri and Khuzestani buffalo breeds to search for selection signatures and explore putative candidate genes under the selection signatures identified.
Results
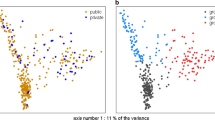
Least square means and standard deviations of some morphometric traits in Iranian Azari and Khuzestani water buffalo breeds are shown in Additional file 1. For the SNPs analyzed in this study, the average MAF for Azeri and Khuzestani buffalo breeds were 0.317 (SD = 0.118) and 0.308 (SD = 0.122), respectively. The Azeri and Kuzestani buffalo breeds are genetically distinct, as seen by the principal components analysis [37]. Assuming two ancestral populations the structure plots show that the two breeds are distinct, but with a moderate levels of admixture (Fig. 1). The relationships among the studied animals were revealed by relationship matrix and heat map plot (see Additional files 2 and 3).

Principal components and population structure diagrams of studied buffalo populations. (a) Principal components diagram provided based on the genomic kinship coefficients between individuals. The first two principal components (PC) and the variance explained by each component are shown on the corresponding axis. (b) Population structure of studied buffalo populations provided by Admixture software with k = 2
Averaged Weir&Cockerham’s unbiased FST values obtained for sliding windows of 500 kb across the genome gave an average of unbiased between population FST of 0.0178 (SD = 0.027), as shown in Fig. 2. Evidence of selection was found in 13 regions which contained 0.1% of windows with the highest FST values over 0.1 included in 65 significant windows. These regions were located on chromosomes 2 (65,490–66,490 and 111,415–112,415 kb), 3 (56,750–57,750 and 114,861–115,861 kb), 4(26,287–26,287 kb), 7 (55,042–56,042 kb), 9 (54,934–55,934 kb), 10 (22,554–23,554 kb), 19 (22,916–23,916 kb), 21 (59,937–60,937 kb), 23 (24,776–25,776 kb), 27 (51.5–344 kb) and X (97,516–98,516 kb). Statistics for linkage disequilibrium, including r2 and D’ were calculated for regions that were selected using FST as selection signature (See Additional files 4 and 5).

Genome-wide distribution of pairwise unbiased FST (Theta) between Azari and Khuzestani buffalo breeds. Overlapping windows of 300 kb across the genome were used to identify putative signatures of selection. The threshold determined based on the 0.1% of the empirical genome-wide distribution is shown by the blue line
The iHS and XP-EHH values were determined across the genome (see Additional files 6, 7, 8 and 9). The results revealed that the selected regions using the FST method on chromosomes 2 (2 regions), 3 (2 regions), 10, 19 and 23 were also determined as selected regions by iHS and XP-EHH methods (Additional files 6, 7, 8 and 9). The haplotype base analyses was carried out to verify selected regions from FST method. Only regions determined as signature of selection from FST were used for gene and QTL detection.
The 1 Mbp genomic regions flanking the putative signature of selection regions were searched for genes that may be have been the subjects of selection. In a total, 13 Mb representing 0.45% of the genome were included and 59 genes and 18 QTLs were identified from the annotation of the cattle genome (UMD3.1 Bos Taurus). The results are shown in Table 1 and Additional file 10.
Discussion
Among the three Iranian buffalo breeds, Azeri and Kuzestani are the most common indigenous breeds that are well adapted to different regions. The Khuzestani have high milk, meat and growth in comparison with other breeds in Iran. The Azeri and Khuzestani breeds have differences in behavior, milk production and body size and are adapted to different environments and rearing methods. In this study, the population structure of these two buffalo breeds was analyzed. Principal component analysis (PCA) of the genotype data formed two distinct clusters with no overlap between them, each containing one of the two breeds showing that the breeds are genetically distinct (Fig. 1). Further analysis of the population structures showed that there was significant admixture (Fig. 1). The mean genomic FST value across all SNPs was 0.0178, indicating low genetic differentiation (FST ranged less than 0.05) according to Wright’s classification [38].
Iranian buffalo are exposed to extreme heat stress which is known to reduce an animal’s performance in tropical, sub-tropical and arid areas [39]. Compared with other farm animals, buffaloes have poor heat tolerance capacity and are more sensitive to heat because of scarcely distributed sweat glands and dark body color [40]. The extent of heat stress depends on the individual animal’s genetics which can alter a number of physiological and behavioral responses [41]. Adaptation to heat stress requires the physiological integration of many organs and systems including endocrine, cardiorespiratory and immune system [42]. Thus, genetic variation could be selected for better adaptation [43]. Here, 59 genes and 18 QTLs were found within the regions of high FST. Some of these loci may be related to environmental adaptation such as the cytosketal organization and immune function, while others affect production traits including milk production, and growth (Table 1 and Additional file 10).
Natural selection is expected to act strongly on immunity genes through disease exposure and response to stress [18]. The highly polymorphic major histocompatibility complex (MHC) has been implicated in the resistance and susceptibility to a broad range of diseases [44], differences in milk production, growth rate, reproductive performance and immune response [45]. The bovine BoLA locus is located on BTA23 between 25.3 and 25.6 Mb which was identified as a region of high FST in the comparison of the two buffalo breeds studied here (Table 1). Other genes in this region are also involved in immune responses, specifically FBXO9. Also, NDFIP1 (located on BTA7 between 54.9 and 55.9 Mb) is another gene detected in a region under selection and is involved in immune response.
Candidates genes involved in cytoskeleton organization within high FST regions included ACTR3, ARHGAP26 and CLN8. Genes in this category within high FST have been implicated in muscle development, including MYOM2 [46] which has a role in protein synthesis and modification of skeletal muscle [46], and, ARHGAP26 and ACTR3 involved in actin filament polymerization and organization (NCBI). MOGAT1 and AC8 L3 have key role in lipid and Fatty Acyl-CoA biosynthesis. GPR39 (located on BTA2 between 65 and 66 Mb) is involved in the control of growth hormone release [47].
Apoptotic pathways participate in growth, proliferation, development, immunity and stress responses. Genes involved in apoptosis within high FST regions included TMEM14A (Trans membrane protein 14A) which stabilizing mitochondrial membrane potential [48] that may be affected by heat stress. The CLN8 [36] has a negative regulatory function in the apoptotic process. DNAJB2 encodes a heat shock binding protein which has anti-apoptosis function and has been implicated in meat tenderness. SERPINF2 is involved in regulation of proteolysis, which is a response to heat stress (NCBI). SERPINF2 is involved in the Wnt signaling pathway which associated with apoptosis response, but is also involved in mammary gland alveolus development, possibly related with milk production traits [49].
The QTLs that were associated with regions of high FST are shown in Table 1 and are involve traits such as fatty acid content, milk protein percentage, milk protein yield, somatic cell score, teat placement, udder attachment, subcutaneous fat, meat percentage, marbling score (EBV), shear force, carcass and body weight in mature, somatic cell count and residual feed intake traits.
Conclusions
In this study, a genomic scan was performed on two distinct Iranian buffalo breeds and was analyzed using a population differentiation index approach. A total of 13 regions with outlier FST were detected, indicating greater than genome average divergence between the two Iranian buffalo breeds in these regions where natural or breeding selection may have been acting. A total of 59 genes were identified within these regions. Many of these genes are involved with physiological pathways including milk production, cytoskeleton organization and growth, metabolic and apoptosis processes, immune function. Hence, these genes may be considered as candidates for genes under selection. However, from this large number of candidate genes and very wide range of functions it will be necessary to refine the study to identify those under selection and variants that are beneficial for production nor climate adaptation traits.
Methods
Animal samples and phenotype data
Blood and hair root samples were collected from 159 milk-recorded herds, including 112 herd for Azeri (AZI) breed and 47 herd for Khuzestani (KHU) breeds, respectively, which participate in the registration and recording system of the Animal Breeding Centre of Iran. Sample collection from studied animals was performed in accordance with animal ethics and approved by the Animal Use Committee in University of Tehran and Animal Breeding Center of Iran (ABCI). Fewer than 5 animals were selected from each herd. Individuals from each herd were selected based on the lowest possible pedigree-based relationship. Production records and type traits were considered to assess the diversity of the each breed. Extensive sampling was carried out to cover a large proportion of each breed. The AZI breed samples were collected from East-Azarbaijan, West-Azarbaijan, Ardebil and Gilan provinces, located in north and north-western part of Iran (37.02° – 38.78° N, 44.81° - 49.52° E), whereas the KHU breed samples were collected from Khuzestan (30.68–32.55° N, 48.02°- 48.97° E) and Kermanshah provinces (34.54°N, 45.60°E), located in the south-western part of Iran (Fig. 3). In total 510 samples were collected from which 385 were selected for genotyping. Selected animals had milk production records and type traits including height-at-withers and chest girth. The data, normally distributed in each breed, were analyzed by SAS software (SAS 2014, SAS Institute Inc., Cary, NC, USA) using the GLM procedure. The results showed that breed had significant effect on traits (P < 0.001) and KHU had higher height-at-withers (143.63 vs 138.73), chest girth (195.37 vs 183.89) and milk production (10.92 vs 6.98) than AZI.

Distributions of the two Iranian Buffalo breeds used in this study. (a) The Iran country in Middle-East (south-west of Asia). (b) More than 99% of AZI and KHU buffalo breeds distribute in red and green highlighted areas, respectively. The red highlighted area (located in north and north-western part of Iran) consist of East-Azarbaijan, west-Azarbaijan, Ardebil and Gilan provinces, and the green highlighted area (west and south-western part of Iran) consist of Khuzestan and Kermanshah provinces
Genotyping and data quality control
Genomic DNA was extracted from blood by the modified salting out method [50] and from hair samples as described by Alberts et al. [51]. The quality and concentration of extracted DNA were assessed by visualizing on 1.2% agarose gel and spectrophotometrically based on absorbance at (260 nm /280 nm). DNA samples were diluted to 50 ng/ul for genotyping.
The set of 385 water buffalo samples from AZI (N = 262) and KHU (N = 123) breeds with milk production and type records were genotyped using the Axiom® Buffalo Genotyping 90 K Array (Affymetrix). SNP genotypes were extracted from raw data using the AffyPipe workflow [52] and applying default thresholds (dish-quality control < 0.82 and individual call rate < 0.97). Primary quality control and filtering, was carried out and genotypes exported in PLINK format. In total, 4 animals were removed because they failed the quality standards. Furthermore, 5501 (6.1%) and 9857 (11.7%) SNP were discarded because the call rate was below the threshold and low quality genotypes, respectively. A total of 73,935 SNP passed the quality control which had an average sample call rate of 99.68%. Genotype repeatability assessed from 5 replicate samples was 99.96%, demonstrating a high quality of the genotyping results. A total of 64,339 (71.6%) probes were high-quality polymorphic (PolyHighResolution class), 7924 (8.8%) showed high-quality monomorphic signals (MonoHighResolution) and 1672 (1.8%) had one homozygous genotypes class missing (NoMinorHom). The latter three classes were retained for further analyses. Therefore, the quality-edited dataset has a total of 383 animals, AZI n = 260 and KHU n = 123 individuals and 73,935 SNP genotypes.
A second QC procedure was performed breed-wise, using PLINK software [53], retaining SNPs on autosomal and X chromosomes, minor allele frequency (MAF) > 0.02%, divergence from Hardy-Weinberg Equilibrium (HWE) (P-value >10e-6) and SNP Call rate (CRSNP) > 0.95%. After this quality control, individuals with call rate (CRIND) below 0.95% were excluded from further analysis (Table 2 and Additional file 11). This procedure yielded 371 individuals (253 AZI and 118 KHU) and 64,866 SNPs with average distances between 2 adjacent SNPs about 40 kb based on the bovine genome UMB 3.1, which were used for further analyses (Table 2).
Population structure and FST estimation
PCA analysis based on SNPs which passed quality control, was carried out using the identical by state (IBS) matrix generated with GenABEL [54] by converting the calculated genomic kinship coefficients to squared Euclidean distances that capture the differences between individuals via classical multidimensional scaling [55]. Individuals located outside the expected breed cluster were excluded from further analysis. As the selection of individuals was based on pedigree, the identity-by-state (IBS) relationship matrix was used to remove closely related animals, as proposed by Leutenegger et al. [56]. The IBS matrix was estimated using GenABEL R package IBS function [54] and individuals with an IBS > 0.8 were removed from further analysis. Genetic structure of the population was tested using ADMIXTURE software [57]. The r2 statistics between adjacent SNP pairs were calculated for both of the studied populations for all marker pairs, using SnppldHD software (Sargolzaei M, University of Guelph, Canada).
The unbiased Fixation index (Theta) estimator proposed by Weir and Cockerham was calculated (Additional file 12) to detect signatures of selection [32, 58] in R (the R project website, http://www.r-project.org/). The FST outlier method was used to detect signatures of selection [59] where adjacent SNPs show outlier FST values [60]. A modified sliding window (SW) approach (referred to as a “Creeping Window”: CW) was used to scan the entire genome for evidence of selective sweeps, using a one SNP step [61]. The optimal size of the window depends on time since the occurrence of the selection sweep, as LD breaks down with time [35]. An arbitrary 300Kbp window size was chosen in this study (Additional file 13). In total, 13 regions exceeding the 0.1% threshold of the empirical outlier window-wise unbiased FST values. Overall unbiased FST was calculated using Weir and Cockerham method [32] in R as population differentiation index. To verify selected regions from unbiased FST results and appropriately identify selection signatures, two haplotype base methods integrated Haplotype Homozygosity score (iHS) [31] and Cross Population Extended Haplotype Homozygosity (XP-EHH) were applied using rehh package [62] in R. Imputation of missing data and haplotype phasing were carried out by fastPHASE software [63] for use in haplotype base analysis.
Annotation of the outlier regions
The 13 outlier genomic regions were surveyed to find genes within 1 Mb of the outlier region peaks. In total 59 genes were extracted from the corresponding areas in UMD3.1 Bos Taurus Genome Assembly using Biomart. DAVID [64] was used to perform a gene ontology analysis and to identify putative biological networks including the genes found in outlier regions. Finally, the Enrichment Map Cytoscape plug-in was used to construct networks [65].
Abbreviations
- AZI:
-
Azeri
- CRIND :
-
Individuals Call Rate
- CRSNP :
-
SNP Call Rate
- CW:
-
Creeping Window
- FST :
-
Population differentiation index
- HWE:
-
Hardy-Weinberg Equilibrium
- IBS:
-
Identical By State
- KHU:
-
Khuzestani
- MAF:
-
Minor Allele Frequency
- PCA:
-
Principal component analysis
- QC:
-
Quality Control
- SW:
-
Sliding Window
References
Kierstein G, Vallinoto M, Silva A, Schneider MP, Iannuzzi L, Brenig B. Analysis of mitochondrial D-loop region casts new light on domestic water buffalo (Bubalus bubalis) phylogeny. Mol Phylogenet Evol. 2004;30(2):308–24.
Yindee M, Vlamings B, Wajjwalku W, Techakumphu M, Lohachit C, Sirivaidyapong S, Thitaram C, Amarasinghe A, Alexander P, Colenbrander B. Y-chromosomal variation confirms independent domestications of swamp and river buffalo. Anim Genet. 2010;41(4):433–5.
Kumar S, Nagarajan M, Sandhu J, Kumar N, Behl V, Nishanth G. Mitochondrial DNA analyses of Indian water buffalo support a distinct genetic origin of river and swamp buffalo. Anim Genet. 2007;38(3):227–32.
FAO: statistics website. 2014:http://www.fao.org/statistics/en/.
FAO: The state of the world’s animal genetics resources for food and agriculture. 2000. http://www.fao.org/statistics/en/.
Michelizzi VN, Dodson MV, Pan Z, Amaral MEJ, Michal JJ, McLean DJ, Womack JE, Jiang Z. Water buffalo genome science comes of age. Int J Biol Sci. 2010;6(4):333.
Borghese A: Buffalo Production and Research. 2005, FAO Ed. REU Technical Series 67:1–316.
Sanjabi M, Alemi F, Naderfard H. Estimated breeding value in Iranian buffaloes. J Agric Sci Technol. 2011;A 1:570–4.
Cole J, VanRaden P, O’Connell J, Van Tassell C, Sonstegard T, Schnabel R, Taylor J, Wiggans G. Distribution and location of genetic effects for dairy traits. J Dairy Sci. 2009;92(6):2931–46.
Iamartino D, Nicolazzi EL, Tassell CV, Reecy JM, Fritz-Waters ER, Koltes JE, Biffani S, Sonstegard T, Schroeder S, Ajmone-Marsan P, et al. Design and validation of a 90K SNP genotyping assay for the water buffalo (Bubalus bubalis). PLoS One. 2017;12:e0185220.
Saatchi M, Schnabel RD, Rolf MM, Taylor JF, Garrick DJ. Accuracy of direct genomic breeding values for nationally evaluated traits in US Limousin and Simmental beef cattle. Genet Sel Evol. 2012;44:38.
Hayes BJ, Pryce J, Chamberlain AJ, Bowman PJ, Goddard ME. Genetic architecture of complex traits and accuracy of genomic prediction: coat colour, milk-fat percentage, and type in Holstein cattle as contrasting model traits. PLoS Genet. 2010;6(9):e1001139.
Peters S, Kizilkaya K, Garrick D, Fernando R, Reecy J, Weaber R, Silver G, Thomas M. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in Brangus heifers. J Anim Sci. 2012;90(10):3398–409.
McClure M, Ramey H, Rolf M, McKay S, Decker J, Chapple R, Kim J, Taxis T, Weaber R, Schnabel R. Genome-wide association analysis for quantitative trait loci influencing Warner–Bratzler shear force in five taurine cattle breeds. Anim Genet. 2012;43(6):662–73.
Wright S. The genetical structure of populations. Ann Eugenics. 1949;15(1):323–54.
Simianer H. Statistical problems in livestock population genomics. In: 10th World Congress on Genetics Applied to Livestock Production: 2014: Asas; 2014.
Gregory TR. Artificial selection and domestication: modern lessons from Darwin’s enduring analogy. Evolution Educ Outreach. 2009;2(1):5–27.
Qanbari S, Pausch H, Jansen S, Somel M, Strom TM, Fries R, Nielsen R, Simianer H. Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 2014;10(2):e1004148.
Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419(6909):832–7.
Andersen KG, Shylakhter I, Tabrizi S, Grossman SR, Happi CT, Sabeti PC. Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever. Philos Trans R Soc Lond Ser B Biol Sci. 2012;367(1590):868–77.
Utsunomiya YT, Perez O'Brien AM, Sonstegard TS, Van Tassell CP, do Carmo AS, Meszaros G, Solkner J, Garcia JF. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS One. 2013;8(5):e64280.
Fay JC, Wu C-I. Hitchhiking under positive Darwinian selection. Genetics. 2000;155(3):1405–13.
Fu Y-X, Li W-H. Statistical tests of neutrality of mutations. Genetics. 1993;133(3):693–709.
Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148(3):929–36.
Oleksyk TK, Smith MW, O'Brien SJ. Genome-wide scans for footprints of natural selection. Philos Trans R Soc Lond Ser B Biol Sci. 2010;365(1537):185–205.
Ong RT-H, Teo Y-Y. varLD: a program for quantifying variation in linkage disequilibrium patterns between populations. Bioinformatics. 2010;26(9):1269–70.
Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, Jiang L, Ingman M, Sharpe T, Ka S, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464(7288):587–91.
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449(7164):913.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95.
Tang K, Thornton KR, Stoneking M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007;5(7):e171.
Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4(3):e72.
Weir BS, Cockerham CC: Estimating F-statistics for the analysis of population structure. Evolution 1984:1358–1370.
Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, Nielsen R. Localizing recent adaptive evolution in the human genome. PLoS Genet. 2007;3(6):e90.
Mokhber M, Moradi Shahrbabak M, Sadegi M, Moradi Shahrbabak H, Williams J. Genome-wide survey of signature of positive selection in Khuzestani and Mazandrani buffalo breeds. Iran J Anim Sci. 2015;46(2):119–31.
Moradi MH, Nejati-Javaremi A, Moradi-Shahrbabak M, Dodds KG, McEwan JC. Genomic scan of selective sweeps in thin and fat tail sheep breeds for identifying of candidate regions associated with fat deposition. BMC Genet. 2012;13:10.
O’Brien AMP, Utsunomiya YT, Mészáros G, Bickhart DM, Liu GE, Van Tassell CP, Sonstegard TS, Da Silva MV, Garcia JF, Sölkner J. Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet Sel Evol. 2014;46(1):19.
Marsan P, Milanesi M, Vajana E, Iamartino D, Bomba L, Nicolazzi E, AHMED S EL-DIN, HERRERA J, Cruz L, Zhang S. Diversity and origins of domestic buffalo. In: International plant and animal genome conference XXIII: 2015; 2015.
Wright S. Evolution and the genetics of population: variability within and among natural populations. Univ Chicago Press. 1978;4
Silanikove N, Maltz E, Halevi A, Shinder D. Metabolism of water, sodium, potassium, and chlorine by high yielding dairy cows at the onset of lactation 1, 2. J Dairy Sci. 1997;80(5):949–56.
Das S, Upadhyay R, Madan M. Heat stress in Murrah buffalo calves. Livest Prod Sci. 1999;61(1):71–8.
Freeman B. The stress syndrome. World’s Poultry Sci J. 1987;43(01):15–9.
Altan Ö, Pabuçcuoğlu A, Altan A, Konyalioğlu S, Bayraktar H. Effect of heat stress on oxidative stress, lipid peroxidation and some stress parameters in broilers. Br Poult Sci. 2003;44(4):545–50.
Kimothi S, Ghosh C: Strategies for ameliorating heat stress in dairy animals. Dairy Year book 2005:371–377.
Rupp R, Hernandez A, Mallard B. Association of bovine leukocyte antigen (BoLA) DRB3. 2 with immune response, mastitis, and production and type traits in Canadian Holsteins. J Dairy Sci. 2007;90(2):1029–38.
Sharif S, Mallard B, Wilkie B, Sargeant J, Scott H, Dekkers J, Leslie K. Associations of the bovine major histocompatibility complex DRB3 (BoLA-DRB3) alleles with occurrence of disease and milk somatic cell score in Canadian dairy cattle. Anim Genet. 1998;29(3):185–93.
Moreno-Sánchez N, Rueda J, Carabaño MJ, Reverter A, McWilliam S, González C, Díaz C. Skeletal muscle specific genes networks in cattle. Funct Integr Genomics. 2010;10(4):609–18.
McKee KK, Tan CP, Palyha OC, Liu J, Feighner SD, Hreniuk DL, Smith RG, Howard AD, Van der Ploeg LH. Cloning and characterization of two human G protein-coupled receptor genes (GPR38 and GPR39) related to the growth hormone secretagogue and neurotensin receptors. Genomics. 1997;46(3):426–34.
Jin H, Kang ES, Kim HJ, Lee JH, Chang KC, Park J-Y, Choi WS, Seo HG. TMEM14A inhibits N-(4-hydroxyphenyl) retinamide-induced apoptosis through the stabilization of mitochondrial membrane potential. Cancer Lett. 2011;309(2):190–8.
Raven LA, Cocks BG, Goddard ME, Pryce JE, Hayes BJ. Genetic variants in mammary development, prolactin signalling and involution pathways explain considerable variation in bovine milk production and milk composition. Genet Sel Evol. 2014;46:29.
Grimberg J, Nawoschik S, Belluscio L, McKee R, Turck A, Eisenberg A. A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res. 1989;17(20):8390.
Alberts CC, Ribeiro-Paes J, Aranda-Selverio G, Cursino-Santos J, Moreno-Cotulio V, Oliveira A, Porchia B, Santos W, EBd S. DNA extraction from hair shafts of wild Brazilian felids and canids. Genet Mol Res. 2010:2429–35.
Nicolazzi EL, Iamartino D, Williams JL. AffyPipe: an open-source pipeline for Affymetrix axiom genotyping workflow. Bioinformatics. 2014;30(21):3118–9.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23(10):1294–6.
Mardia K. Some properties of classical multi-dimensional scaling. Commun Stat A-Theor. 1978;A7:1233–41.
Leutenegger A-L, Prum B, Génin E, Verny C, Lemainque A, Clerget-Darpoux F, Thompson EA. Estimation of the inbreeding coefficient through use of genomic data. Am J Hum Genet. 2003;73(3):516–23.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64.
Weir BS, Hill WG. Estimating F-statistics. Annu Rev Genet. 2002;36(1):721–50.
Karlsson S, Moen T. The power to detect artificial selection acting on single loci in recently domesticated species. BMC Research Notes. 2010;3(1):1.
Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12(12):1805–14.
Qanbari S, Strom TM, Haberer G, Weigend S, Gheyas AA, Turner F, Burt DW, Preisinger R, Gianola D, Simianer H. A high resolution genome-wide scan for significant selective sweeps: an application to pooled sequence data in laying chickens. PLoS One. 2012;7(11):e49525.
Gautier M, Vitalis R. Rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics. 2012;28(8):1176–7.
Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;78(4):629–44.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One. 2010;5(11):e13984.
Bibi F, Vrba ES. Unraveling bovin phylogeny: accomplishments and challenges. BMC Biol. 2010;8(1):50.
Sherman E, Nkrumah J, Li C, Bartusiak R, Murdoch B, Moore S. Fine mapping quantitative trait loci for feed intake and feed efficiency in beef cattle. J Anim Sci. 2009;87(1):37–45.
Longeri M, Polli M, Strillacci M, Samore A, Zanotti M. Quantitative trait loci affecting the somatic cell score on chromosomes 4 and 26 in Italian Holstein cattle. J Dairy Sci. 2006;89(8):3175–7.
Mosig MO, Lipkin E, Khutoreskaya G, Tchourzyna E, Soller M, Friedmann A. A whole genome scan for quantitative trait loci affecting milk protein percentage in Israeli-Holstein cattle, by means of selective milk DNA pooling in a daughter design, using an adjusted false discovery rate criterion. Genetics. 2001;157(4):1683–98.
Schnabel R, Sonstegard T, Taylor J, Ashwell M. Whole-genome scan to detect QTL for milk production, conformation, fertility and functional traits in two US holstein families. Anim Genet. 2005;36(5):408–16.
Holmbeg M, Andersson-Eklund L. Quantitative trait loci affecting health traits in Swedish dairy cattle. J Dairy Sci. 2004;87(8):2653–9.
Schrooten C, Bovenhuis H, Coppieters W, Van Arendonk J. Whole genome scan to detect quantitative trait loci for conformation and functional traits in dairy cattle. J Dairy Sci. 2000;83(4):795–806.
Gutiérrez-Gil B, Williams J, Homer D, Burton D, Haley C, Wiener P. Search for quantitative trait loci affecting growth and carcass traits in a cross population of beef and dairy cattle. J Anim Sci. 2009;87(1):24–36.
Kühn C, Bennewitz J, Reinsch N, Xu N, Thomsen H, Looft C, Brockmann G, Schwerin M, Weimann C, Hiendleder S. Quantitative trait loci mapping of functional traits in the German Holstein cattle population. J Dairy Sci. 2003;86(1):360–8.
Takasuga A, Watanabe T, Mizoguchi Y, Hirano T, Ihara N, Takano A, Yokouchi K, Fujikawa A, Chiba K, Kobayashi N. Identification of bovine QTL for growth and carcass traits in Japanese black cattle by replication and identical-by-descent mapping. Mamm Genome. 2007;18(2):125–36.
McClure M, Morsci N, Schnabel R, Kim J, Yao P, Rolf M, McKay S, Gregg S, Chapple R, Northcutt S. A genome scan for quantitative trait loci influencing carcass, post-natal growth and reproductive traits in commercial Angus cattle. Anim Genet. 2010;41(6):597–607.
Daetwyler HD, Schenkel FS, Sargolzaei M, Robinson JAB. A genome scan to detect quantitative trait loci for economically important traits in Holstein cattle using two methods and a dense single nucleotide polymorphism map. J Dairy Sci. 2008;91(8):3225–36.
Bennewitz J, Reinsch N, Grohs C, Levéziel H, Malafosse A, Thomsen H, Xu N, Looft C, Kühn C, Brockmann GA. Combined analysis of data from two granddaughter designs: a simple strategy for QTL confirmation and increasing experimental power in dairy cattle. Genet Sel Evol. 2003;35(3):1.
Acknowledgements
The authors gratefully acknowledge the Animal Breeding Center of Iran (ABCI) and Towsee Kesht va Dam Noandish Alborz Co (Takdna) for access to the animals and recording. The authors also acknowledge the staff from university of Tehran, Dr. Saber Qanbari, Dr. Mohammad Hossein Moradi, Mr. Parviz Azizi and Mr. Amir Sobhani who helped and supported this research.
Funding
This study was supported by Iranian Buffalo Genomics project with 2014–800/91/20, 2014–888, 2014–1049, 2014–92/487, 2014–289 and 2014–91/120/1771 research grants from Iran’s Ministry of Agriculture-Jahad, and Towsee Kesht va Dam Noandish Alborz Co (Takdna). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this manuscript and its supplementary information files. In addition, more detail data are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
MM MMS HMS MS JR JLW conceived and designed the study, MMS HMS MS MM EN JLW contributed reagents/materials/analysis tools, MM JR created the data, MM and AS analyzed the data and MM JLW wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Sample collection from studied animals were performed in accordance with animal ethics and approved by the Animal Use Committee in University of Tehran and Animal Breeding Center of Iran (ABCI). In addition, all the samples were collected from commercial farms and the animal owners agreed to be involved in the project through their respective associations with Animal Breeding Center of Iran.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Least-square means and standard deviations of some morphometric and body size traits in Iranian Azeri and Khuzestani water buffalo breeds. (DOCX 19 kb)
Additional file 2:
Genomic matrix of studied Iranian Azeri and Khuzestani buffalo breeds. (XLS 1750 kb)
Additional file 3:
HeatMap of studied Iranian Azeri and Khuzestani buffalo breeds. (PDF 235 kb)
Additional file 4:
Haploview LD graph for selected regions. (PDF 904 kb)
Additional file 5:
Adjecent r2 information. (XLS 67 kb)
Additional file 6:
Genome wide distribution of iHS and XP-EHH plot. (PDF 349 kb)
Additional file 7:
Genome wide distribution of iHS from Azeri buffalo breed values. (TXT 2913 kb)
Additional file 8:
Genome wide distribution of iHS from Khuzestani buffalo breed values. (TXT 2864 kb)
Additional file 9:
Genome wide distribution of XP-EHH from Azeri and Khuzestani buffalo breed values. (TXT 2963 kb)
Additional file 10:
Genes included in selected regions (Biomart input & output). (XLS 105 kb)
Additional file 11:
Map file after QC. (TXT 1809 kb)
Additional file 12:
FST output file. (XLS 9242 kb)
Additional file 13:
Windowed FST output file. (XLS 6816 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mokhber, M., Moradi-Shahrbabak, M., Sadeghi, M. et al. A genome-wide scan for signatures of selection in Azeri and Khuzestani buffalo breeds. BMC Genomics 19, 449 (2018). https://doi.org/10.1186/s12864-018-4759-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4759-x