
Abstract
Background
Bovine leukemia virus (BLV) infection is omnipresent in dairy herds causing direct economic losses due to trade restrictions and lymphosarcoma-related deaths. Milk production drops and increase in the culling rate are also relevant and usually neglected. The BLV provirus persists throughout a lifetime and an inter-individual variation is observed in the level of infection (LI) in vivo. High LI is strongly correlated with disease progression and BLV transmission among herd mates. In a context of high prevalence, classical control strategies are economically prohibitive. Alternatively, host genomics studies aiming to dissect loci associated with LI are potentially useful tools for genetic selection programs tending to abrogate the viral spreading. The LI was measured through the proviral load (PVL) set–point and white blood cells (WBC) counts. The goals of this work were to gain insight into the contribution of SNPs (bovine 50KSNP panel) on LI variability and to identify genomics regions underlying this trait.
Results
We quantified anti–p24 response and total leukocytes count in peripheral blood from 1800 cows and used these to select 800 individuals with extreme phenotypes in WBCs and PVL. Two case-control genomic association studies using linear mixed models (LMMs) considering population stratification were performed. The proportion of the variance captured by all QC-passed SNPs represented 0.63 (SE ± 0.14) of the phenotypic variance for PVL and 0.56 (SE ± 0.15) for WBCs. Overall, significant associations (Bonferroni’s corrected -log10p > 5.94) were shared for both phenotypes by 24 SNPs within the Bovine MHC. Founder haplotypes were used to measure the linkage disequilibrium (LD) extent (r2 = 0.22 ± 0.27 at inter-SNP distance of 25−50 kb). The SNPs and LD blocks indicated genes potentially associated with LI in infected cows: i.e. relevant immune response related genes (DQA1, DRB3, BOLA-A, LTA, LTB, TNF, IER3, GRP111, CRISP1), several genes involved in cell cytoskeletal reorganization (CD2AP, PKHD1, FLOT1, TUBB5) and modelling of the extracellular matrix (TRAM2, TNXB). Host transcription factors (TFs) were also highlighted (TFAP2D; ABT1, GCM1, PRRC2A).
Conclusions
Data obtained represent a step forward to understand the biology of BLV–bovine interaction, and provide genetic information potentially applicable to selective breeding programs.
Similar content being viewed by others


Background
The Bovine leukemia virus (BLV) is an oncogenic δ-retrovirus that persistently infects cattle [1]. BLV infection elapses without noticeable symptoms in most of the animals while 30–40% of them evolve into a sub-clinical persistent lymphocytosis (PL) characterized by an expansion of proviral infected B-lymphocytes in peripheral blood increasing white blood cells (WBC) counts (leukocytosis). Less than 10% of infected animals will develop the severe form of the disease leading to lethal lymphomas or lymphosarcoma in various organs [2]. The BLV is transmitted among cows through direct exposure to blood, body fluids or consumption of contaminated colostrum/milk with proviral DNA [3, 4]. In the absence of effective measures controlling BLV transmission, the virus is massively propagated reaching high levels of endemicity. BLV is world-wide distributed. In North America BLV’s prevalence reached 70–85% at farm level [5,6,7], and in Argentina more than 80% of the lactating cows in the main dairy production area are infected [8]. International trade restrictions of livestock products from affected herds [9] impact on the regional economy, but the total economic loss is significantly higher if we consider milk production dropping and faster culling of asymptomatic BLV carriers compared to BLV free herds [10, 11]. These conditions are emphasised in lymphocytotics animals and also in those with high anti-BLV humoral response [12,13,14,15,16]. Traditional control strategy implies segregating/eliminating infected cows after BLV testing, but this is not economically affordable when the regional prevalence is high [17]. A suitable long term approach to contain endemic livestock diseases, especially those where the therapeutics or preventives treatments failed, is DNA-based breeding for host resistance to infection [18]. Mastitis incidence is currently being incorporated in genomic evaluations for dairy cattle ([19, 20]). A key component to set up host genetic studies is properly defining and recording a consistent phenotype [21, 22]. The high PVL has been associated with progression of the BLV pathogenesis [23, 24], and with enhanced viral dissemination [25]. Previous evidence pointed to DNA polymorphisms in candidates genes (in particular, alleles from the bovine MHC class II DRB gene) as contributors to the inter-individual variation in the PVL and PL [26,27,28,29,30,31,32,33,34]. However, genetic variation for resistance/susceptibility to diseases in cattle is usually polygenic, i.e. susceptibility to paratuberculosis [35, 36], mastitis [37, 38] bovine respiratory disease complex (BRDC) [39] and recently observed in a genome-wide mapping for genetics determinants of BLV incidence [40, 41].
With the final objective in mind of developing (implementing) genomics selection programs to control BLV dissemination it is essential to understand the biological mechanism implicated. In an attempt to address this questions we utilized antibodies anti-p24 as cost-effective predictors of the level of PVL in blood [8] on over 1800 animals, characterized with WBC counts, in order to recruit a population of Holstein and Holstein x Jersey crossbred dairy cows representing extreme phenotypes of level of infection (LI) in vivo, and therefore, with different individual risk for viral transmission. Afterwards, we integrate these phenotypes in a case-control genome-wide association study (GWAS) using the Illumina BovineSNP50v2 BeadChip to investigate genomic regions associated with BLV infection.
Methods
Study population
Blood samples were obtained from 1800 Holstein and Holstein x Jersey cows representing 25 half-sisters families from 16 dairy herds located in the central region of Argentina. Farm owners’ consent was obtained before animal sampling. All animals have full pedigree and were over 3 years old at the time of sampling and between 1st and 9th lactation (78.7% concentrated between 2nd and 4th lactation). The procedures followed for extraction and handling of samples were approved by the Institutional Committee for Care and Use of Experimental Animals of the National Institute of Agricultural Technology (CICUAE-INTA) under protocol number 35/2010, and followed the guidelines described in the institutional Manual. Blood samples were collected in heparinized tubes. Plasma and fresh blood were frozen at − 20 °C until used.
Quality and concentration of genomic DNA extracted from whole blood samples using a commercial kit (Blood Genomic DNA AxyPrep™, Axygen Biosciences, Union City, USA) was assessed using a micro−volume spectrophotometer (NanoDrop™ Technologies, Inc. Wilmington, USA). DNA samples were normalized to a minimum concentration of 20 ng/μl.
Phenotyping
The anti-p24 ELISA assay was used to discriminate BLV infected animals [42], followed by total WBC counts. In a previous study we showed that the individual anti-BLV humoral response is a cost effective indirect measure of infection in vivo [8]. Therefore, animals were first screened by anti-p24 ELISA and then divided into two groups of contrasting phenotypes on the basis of their antibodies level. The LI in selected animals was then corroborated by determining blood PVL set-point.
Serology: The recombinant viral core-p24 protein was used to detect plasma anti-BLV antibodies for each sample as described in [42] Briefly, a ratio sample to positive (S/P), called % of reactivity (% R) is obtained: S/P = [(ODSample - ODWS-)/(ODWS + 1/7 - ODWS-)] × 100, where OD = optical density, WS- = negative serum, WS + 1/7 = serum international standard weak positive control (diluted 1:7 in negative serum) used as reference value. The % R of all samples tested was indicated as “low” (LR), for records between a cut-off value (25%) [42] and the weak positive serum value (100%); and as “high” (HR) in those samples with measurements higher than 200%.
PVL relative quantification: PVL determination was performed over LR and HR animals by real-time PCR using TaqMan technology as described in [42, 43]. A fragment of BLV pol gene (target) was amplified simultaneously with the reference gene 18S. Fetal Lamb Kidney cells (FLK) containing 4 copies/cell of BLV proviral DNA, in a final concentration of 1% in BLV-free peripheral blood mononuclear cells (PBMCs) were used as internal control. This proportion of BLV-infected cells corresponds to a low level of natural infection [42], proved to be 5% in aleukemics animals [44]. The relative PVL was defined according to the equation described by Pfaffl et al. [45]:
where E = reaction efficiency; Ct = threshold cycle. The control PVL was indicated as 1 and all samples tested were referred to this. The detection limit was 1 infected cell /2000 uninfected cells. The PVL was qualified as “No Detectable” (ND) if a Ct value could not be obtained (below the detection limit), as “low” if R < 1 (LPVL) and as “high” if R > 1 (HPVL).
WBC counts: Total leukocyte count was performed manually using a haemocytometer. Fresh blood was diluted 1:20 in a 1% Gentian Violet and 2% acetic acid solution. Normal WBC counts in BLV-seronegative adult cattle are in the range of 2100 to 11,500 cells/μl (mean: 6800 ± 2 SD) [46].
Genotyping with the medium density SNPs chip
A total of 970 cows with PVL determination (74% Holstein and 26% Holstein x Jersey), and 29 bulls (24 Holstein and 5 Jersey) where chosen for genotyping. SNP genotyping was performed by GeneSeek (Neogen Corporation Company, Lincoln, NE, USA) using the BovineSNP50 v2 BeadChip. The panel tests 54,609 SNPs spaced 49.4 kb on average (median spacing of 37 kb) [47]. SNPs were mapped to the bovine assembly UMD 3.1 [48]. The SNPs identification was reassigned to its corresponding “rs” catalogued in dbSNP [49] using the SNPchiMp v.3 tool [50].
Quality control (QC) of the genotypic data was carried out using a set of tools and routines provided by PLINK v1.07 [51]. Individuals with a genotype call rate (CRIND) < 90% were excluded from further analysis. Mendelian inheritance discordances were checked and set as missing. SNPs with a call rate (CRSNPs) < 90%, a deviation from HWE p < 1.10− 08 and Minor Allele Frequency (MAF) < 0.01 were removed from the study. After the QC, the data set was composed of 44,174 SNPs and 925 individuals (898 cows and 27 bulls).
Case-control definitions
The case-control phenotypes to conduct GWAS for LI in vivo considered 396 cases (HPVL) and 373 controls (ND/LPVL). The alternate phenotype was defined by the number of circulating white blood cells (WBCs), which is indicative of the presence of PL, a sub-clinical symptom of BLV infection [52], clustering 283 cases (WBC ≥ 11,500 cells/μl) and 611 control cows (WBC < 11,500 cells/μl).
Linkage disequilibrium
We performed chromosomal haplotypic phase assignment using the software DualPHASEv1.1 included in the PHASEBOOK 2.3 package [53] using the default settings. To calculate the extent of linkage disequilibrium (LD) only maternal founders chromosomes were used. Phasing algorithms allowed the imputation of not assigned genotypes [54] so that the missing genotypes or those set as missing due to Mendelian inconsistencies were imputed. Measure of LD extent was addressed by calculating the correlation of alleles in two loci (r2 and D’) for each chromosome with Haploview v4.2 [55]. The calculations were performed on all pairs of syntenic SNPs with an inter-marker distance < 1 Mb. The r2 measure was then averaged at intervals of 10 kb and plotted versus the inter-SNPs distance.
Population stratification
We inspected the genealogy of bulls in the sampled families (http://www.sra.org.ar/rrgg/) evidencing a strong co-ancestry among them (Additional file 1: Figure S1). In order to prevent spurious associations due to population substructure we performed a multi-dimensional scaling (MDS) analysis on identity by state (IBS) pairwise based distances matrix as implemented in PLINKv1.07 [51]: 1 − (IBS2 + 0.5IBS1)/N, where IBS2 is the number of SNPs in which two individuals share two IBS alleles; IBS1 is the number of SNPs in which only one allele is shared IBS and N is the number of markers used. Finally, the distance matrix was partitioned into a number of uncorrelated variables (or Principal Components, PC). The first two PCs were used to visualize differences in ancestry among individuals (Additional file 2: Figure S2).
Association analysis
The GWAS were performed using LMMs as implemented in GCTA v1.24 [56]. The general model in the observed scale can be written as:
where y is a vector n × 1 that contains binaries values for PVL or WBC; b is a vector that contains the fixed effects of the SNP i to be tested for association (considering an additive genetic effect) and the co-variables Age (A j ) (j = 3 to 12); Herd (H k ) (k = 1 to 16); Lactation Number (L l ) (l = 1 to 9); Percentage of Holstein breed (PH m ) (m = 0, 0.125, 0.25, 0.50, 0.75 and 1); Bull (B n ) (n = 1 to 25) and PC derived from the co-ancestry analysis (PC1 – PC8). The random effects included the additive polygenic effect u of the animal; and the vector e of residuals effects, X and Z stand for the incidence matrix that relate the values in y to the fixed effects in b and the random effects in u, respectively. A liability threshold model l = Xb + g + e was utilized in order to relate observations on the observed scale to liabilities on the unobserved continuous scale. Liability phenotypes are distributed as ~N(0,1), On the liability scale, the random effects included the additive polygenic effect g of the animal, distributed ~N(0, σ\( {}_g{}^2G \)), where G represents the genetic relationship matrix (GRM) calculated from the proportion of shared alleles among individuals for all QC-passed SNPs; and the vector e of residuals effects, distributed ~N(0, σ\( {}_e{}^2I \))), where I is the identity matrix. Other terms are the same as in the linear model but on the liability scale [57, 58]. The heritability on the scale observed \( \left({h}^2=\frac{\sigma_u^2}{\sigma_u^2+{\sigma}_{\varepsilon}^2}\right) \) was transformed to that on the liability scale [58]: \( {h}_l^2=\frac{h_0^2\ K\left(1-K\right)}{z^2} \); where \( {h}_l^2 \) is the heritability on the underlying scale; \( {h}_o^2 \) is the heritability on the observed scale; K is the disease prevalence and zis the height of the density function of the standard normal probability at the threshold value (K).
This model was utilized to calculate the genetic correlation between PVL and WBC. According to Lee et al. [59] the genetic correlation was defined as\( \mathit{\operatorname{cov}}\left({g}_1,{g}_2\right)\overset{\sim }{=}{z}_1\frac{P_1\left(1-{P}_1\right)}{K_1\left(\ 1-{K}_1\right)}{z}_2\frac{P_2\left(1-{P}_2\right)}{K_2\left(\ 1-{K}_2\right)} \) \( \mathit{\operatorname{cov}}\left({g}_1^{\ast },{g}_2^{\ast}\right) \); where, gis the genetic value on the observed scale, g∗ is the genetic value on the underlying scale, z is the height of the density function of the standard normal probability at the threshold value (K), P is the proportion of cases in the sample and K is the prevalence for features 1 (PVL) and 2 (WBC), respectively.
The association analysis implemented in GCTA v1.24 uses the restricted maximum likelihood method (REML) to solve the equations of LMMs [56, 57].
The genetic variance for PVL and WBCs traits was calculated on the 29 individual autosomes and on the X bovine sexual chromosome. The genetic variance was decomposed into separate chromosomal segments following:
where, g i is a random genetic effects vector corresponding to the complete genome or a particular chromosome. The phenotypic variance was partitioned so that \( \mathit{\operatorname{var}}(y)=\sum \limits_{i=1}^r \)\( {\sigma}_i^2G \) + \( {\sigma}_i^2I \) where \( {\sigma}_i^2 \)is the variance of the i-th genetic factor with its related GRM.
The family-wise error rate was controlled by Bonferroni’s correction α′ ≈ α/n, where α is the significance level of 5% and n is the number of SNPs tested. The α′value represents the threshold that the p-value must reach for each test to be considered significant.
Gene content of the genomic regions identified in the association studies
Gene annotation for BTA 23 (UMD3.1 bovine assembly, [48]) was retrieved using BioMart tools [60]. Firstly, SNPs were assigned to a particular gene if they were located on exons, introns or untranslated regions (UTRs). Secondly, SNPs lying within a 20 kb window upstream and downstream from gene boundaries, aiming to capture promoter/expression regulatory regions [61]. Finally, remaining SNPs were assigned to the closest gene from 20 to 30 kb, within the range of useful average LD values found (r2 > 0.2) [62,63,64,65]. Gene to SNPs mapping was performed using BedTools package [66]. Gene functional classification based in Gene Ontology terms (GO) was performed using the PANTHER classification system website (http://www.pantherdb.org/) [67].
Results
Genome-wide distributed SNPs were tested for association with both PVL and WBCs traits using logistic regressions assuming an additive model that incorporated as covariates 8 PCs and other confounding factors Age (A); Herd (H); Lactation number (L); Percentage of Holstein (PH) and Bull (B). The observed statistical values of association were ordered from lowest to highest and plotted against their expected values and genomic inflation factor (λ) was calculated using PLINK. Both for PVL (Additional file 3: Figure S3, λ = 1.21) and WBCs (Additional file 4: Figure S4, λ = 1.30), the data was best fitted with models considering PCs1–8 + A_H_L_PH_B. Given that kinship and/or cryptic relationship might be contributing sources of distortion in the case-control associations we decided to use LMMs as implemented in GCTA v1.24 [56] that incorporates each of QC-passed SNP as a fixed effects and the polygenic animal effect as random. QQ plots showed a proper fit for stratification and a pronounced deviation to the right, which is suggestive of true associations (Fig. 1).

QQ plots. pobs vs pexp obtained from the association mapping using LMMs of 44,174 SNPs for a) PVL and b) WBCs
In parallel, genome-wide regression methods incorporating GRM allowed to estimate the fraction of phenotypic variance captured by the common SNPs in the genotyping panel. The heritability (h2) for PVL was estimated at 0.63 (SE ± 0.14) on the scale of liability, with a prevalence of 40% of cows with high PVL. A h2lia = 0.56 (SE ± 0.15) to equal prevalence was estimated for a phenotype corresponding to a high WBCs count (> 11,500 cells/μl). Polymorphisms in only three chromosomes explained more than 5% of the phenotypic variation for PVL: Bos taurus autosome (BTA) 10 7.1% (SE ± 5.0%), BTA 11 5.8% (SE ± 4.1%) and BTA 23 30.6% (SE ± 7.8%). Similarly, when considering the percentage of variation in WBCs, BTA 11 12.4% (SE ± 6.5%), BTA 21 5.4% (SE ± 5.0%) and BTA 23 24.0% (SE ± 7.2%) autosomes accounted for 74% of the total variance explained by all SNPs (58%).
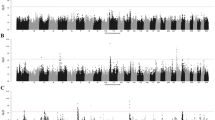
The Manhattan plots revealed significantly associated SNPs with HPVL in peripheral blood (Fig. 2) and with a high number of circulating WBCs (Fig. 3).

Manhattan plot depicting GWAS results of BLV level of infection using LMMs for PVL. The -log10(p) values for each SNP association is represented for each chromosome (BTA) and location within it. The SNPs exceeding the significance threshold according to Bonferroni’s correction (−log10(p) > 5.94, blue horizontal line) are highlighted in green

Manhattan plot depicting GWAS results of BLV level of infection using LMMs for WBCs. The -log10(p) values for each SNP association is represented by chromosome (BTA) and location within it. The SNPs exceeding the significance threshold according to Bonferroni’s correction (−log10(p) > 5.94, blue horizontal line) are highlighted in green
All SNPs exceeding the significance threshold after Bonferroni’s correction (−log10p > 5.94) are located on BTA 23. Notably, SNPs mapping on BTA 23 largely captured most of the genetic variation for both traits, influencing BLV levels of infection. Overall, a group of 24 SNPs were significantly associated with high PVL, whilst 11 SNPs were associated to high WBCs counts. Of these, only 1 SNP (rs110277740) is not shared with those associated with PVL (Table 1). PVL and WBCs are correlated (~ 0.81 (SE ± 0.10)). Joint significant positions for PVL and WBCs taken together delimit a region on BTA 23 that is located within the bovine MHC, approximately from the beginning of bovine leukocyte antigen (BoLA) class IIa region (~ 20.6 Mb) extending to the Histone cluster I which mark off the end of the complex (~ 33.3 Mb). Only a small fraction of phenotypic variance is explained by SNPs located outside this MHC region, so we decided to repeat the GWAS for PVL but excluding the SNPs located between ~ 20–36 Mb of BTA 23, which could be masking the SNPs effects in non-MHC regions. As result, we did not find any significantly associated SNP (Additional file 5: Table S1).
Unless a significant SNP is itself the causal variant, it will capture the effects of other variants in LD responsible for the phenotype. The clustering of genes with related functions in the linkage disequilibrium pattern in the bovine MHC region makes it difficult to detect effects of individual loci. We characterized the LD extent (Additional file 6: Figure S5) in our Holstein and Holstein x Jersey population. At distances ≤30 kb, the average correlation value r2 was 0.26 ± 0.30 (34% of pairwise SNPs showed r2 > 0.25). This value rapidly decays to 0.14 ± 0.21 when the inter-markers distance was between 50−100 kb. About 60% of the inter-SNPs distances in this work is in the range of 25–50 kb with an average r2 = 0.22 ± 0.27, indicating that causal genetics variants effects could be retrieved from SNPs hits.
We found five significantly associated SNPs pairs having a strong allelic correlation (r2 > 0.80). The rs110579760 (BTA23: 25,507,676 bp) was in LD with rs109343703 (r2 = 0.95; BTA23: 24,699,202 bp) and rs110499907 (r2 = 0.88; BTA23: 24,727,613 bp). The r2 between rs109343703 and rs110499907 was 0.90. The remaining pairs were rs41255514 (BTA23: 27,305,227 bp) and rs17872223 (BTA23: 27,306,795 bp) with r2 = 0.96, and the pair rs110794231 (BTA23: 27,887,914 bp) and rs41587536 (BTA23: 27,923,154 bp) had the same r2 value. Figure 4 shows the gene context and the LD map in the BTA23: 24,872,758–29,535,762 bp region.

Gene context and LD map of the bovine chromosome 23 containing the SNPs significantly associated in the GWAS. A) BTA23:24,872,758–29,535,762 bp. ■ PVL significant SNPs; red square symbol: WBCs significant SNPs; ----- log10(p)_PVL; red broken line: log10(p)_WBC; MAF: Minimum Allele Frequency. Local LD map estimates (D´) based on 235 SNPs located within the cattle MHC region in founder maternal chromosomes. Triangles delimited with black lines identify haplotype blocks derived from Haploview [55]. The gene content of the region according to the bovine genome assembly UMD3.1 was obtained from Ensembl Genome Browser (http://www.ensembl.org). Red vertical bars indicate SNPs of the SNP50K chip located in the region
SNPs were assigned to putative biological function if they fell within a coding, intronic, or regulatory region. For those SNPs mapping outside genic regions we scrutinised the closest gene located at a distance where a moderate LD was kept. A total of 44 genes were identified using the criteria described above. Table 2 details the genomic context of each SNP (position and related genes). Half of significant SNPs mapped to putative regulatory regions, 24% to intergenic regions, 12% to introns and 12% of SNPs to exons. Only one SNP (rs41255514) is located in a 3′-untranslated region (3’-UTR). The SNP with the lowest p-value for both conditions was rs110525467 (pPVL = 5.32 × 10− 16 and pWBC = 5.90 × 10− 10) introducing a synonymous substitution in the MHC class II gene BoLA-DQA1 (ENSBTAG00000037605) located in BTA23: 25,426,330–25,430,097 bp. The remaining 10 positions associated with a high number of peripheral blood leukocyte counts included an intronic SNP located in the Polycystic kidney and hepatic disease 1 gen, (PKHD1 - ENSBTAG00000011237) and a synonymous substitution in the Proline-rich coiled-coil 2A gene (PRRC2A - ENSBTAG00000019682). In addition, six SNPs significantly associated with this condition were located in regulatory regions of the GPR111 (ENSBTAG00000037687), CD2AP (ENSBTAG00000000322), TFAP2D (ENSBTAG00000020425), C23H6ORF47 (ENSBTAG00000023628), ABHD16A (ENSBTAG00000000578), LY6G5C (ENSBTAG00000025449), LY6G5B (ENSBTAG00000039740), CSNK2B (ENSBTAG00000008837), BAT4 (ENSBTAG00000008835), APOM (ENSBTAG00000008833), BAG6 (ENSBTAG00000019685), TUBB5 (ENSBTAG00000006969), IER3 (ENSBTAG00000011358), FLOT1 (ENSBTAG00000009960), TRIM31 (ENSBTAG00000004490), TRIM40 (ENSBTAG00000037381) and ABT1 (ENSBTAG00000010784) genes (Table 2). The two remaining SNPs, rs41587216 and rs110473048, were at 39.3 kb of CRISP1 (ENSBTAG00000008085) and 21.7 kb GCM-1 (ENSBTAG00000008148), respectively.
The PVL phenotype association study included all significant SNPs for the GWAS considering WBCs -except the SNP assigned to Activator of Transcription Basal 1 gene (ABT1, rs110277740) - and other 14 significant SNPs. The functional annotation resulted in a synonymous substitution in TNXB (ENSBTAG00000001444) and a SNP located in the 3’-UTR region of SCL44A4 (ENSBTAG00000005675). Also, there are two SNPs located in intronic regions of NEU1 and TRAM2 (ENSBTAG00000009112 and ENSBTAG00000005674, respectively). The variant rs110260956 is 20 kb upstream or downstream of the following genes: LTA (ENSBTAG00000000016), NFKBIL1 (ENSBTAG00000014492), ATP6V1G2 (ENSBTAG00000014491), LTB (ENSBTAG00000020674) and TNF (ENSBTAG00000025471), while rs110350951 meets the same condition in U6 snRNA (ENSBTAG00000043004) and Ig-like MHCI genes (ENSBTAG00000007075), and rs109754326 in NRSN1gene (ENSBTAG00000009180). Three SNPs significantly associated with high PVL are present in putative regulatory regions of 7 genes encoding Odorant receptors (Olfactory receptor). Finally, rs110579760, rs110794231 and rs41587536 polymorphisms are at 30.7, 20.3 and 22.3 kb of BoLA-DRB3 (ENSBTAG00000013919) BoLA-A (ENSBTAG00000022590) and SFTA2 (ENSBTAG00000038810), respectively. Genes represented by the SNPs were assigned to Gene Ontology (GO) functional terms and the PANTHER (Protein Class, PC) (http://pantherdb.org/) classification system. The analysis was performed for all genes represented by significantly associated SNPs with any of the two traits. A total of 11 PC “parents” terms were identified being the three most common categories “protein defense/immunity”, (PC00090) (21,7%), “signalling molecule” (PC00207) (21,7%) and cytoskeletal protein (PC00085) (8,7%). In Additional file 7: Table S2 a description of each PC term is displayed along with their “child” terms providing specificity to the functional description. Notably, a single gene might be contained in more than one PC class. Afterwards, we classified genes under the category GO “Biological Processes” (BP) wherein each gene identification is assigned according to their function within a network of proteins that collectively carry out a particular process within the cell. Figure 5 presents the distribution of 12 “parent” terms found according to GO BP classification, these represent general cellular processes being the top five terms “immune system process” (16%), “response to stimulus” (16%), “metabolic process” (14%), “cellular process” (12%) and “localization” (11%). The search for GO terms and/or KEGG pathways overrepresented on the gene set identified vs those present in whole bovine gene set using FatiGO method [68] did not produce significant results (data not shown).

Distribution of the GO “Biological Process” (BP) categories for genes represented by the significantly associated SNPs in GWAS for PVL and WBC counts
Discussion
The humoral immune response plays a crucial role in the control of viral infections in cattle and humans, and the level of response varies from individual to individual, being this variability highly heritable [69, 70]. In cattle, we showed that the humoral response to BLV infections indeed varies among animals and, interestingly, it can be used to infer the circulating PVL in an infected animal [8]. In this study, we use the % of Reactivity (anti-p24) to screen a population of BLV naturally infected animals aiming to select cows most likely to have extreme values of PVL set-point, which was subsequently confirmed by qPCR. The predictive ability of serological markers to detect high PVL conditions was good being the ROC area under the curve > 0.80 (data not shown). We then performed a case-control study setting levels of infection (LI) as phenotype aiming to estimate (a) the proportion of phenotypic variation that can be attributed to genetic components and (b) the cattle genome mapping positions of genetic polymorphisms underlying this variation. A positive genetic correlation between PVL and WBCs of magnitude ~ 0.81 (SE ± 0.10) supports the idea of shared or genomic regions influencing both traits.
We used kinship captured from QC pruning variants to assess the additive genetic variance of LI produced by BLV in the population. The percentage of variation reached was 63% (h2GRM_PVL = 0.63) and 56% (h2GRM_WBC = 0.56) on the risk scale or liability. On this scale, the phenotypic variances do not depend on the disease prevalence, so h2lia values allow comparisons between populations and environments where incidence varies. When partitioned, the proportion of the phenotypic variance explained by markers for both phenotypes was distributed on different chromosomes. In a pedigree-based study by Abdalla et al. [71], the contribution of genetic components to the incidence of BLV in Holsteins and Jersey herds (approximately 30% of herd level prevalence) was estimated to be 8%, showing that environmental factors could play a major role over genetics in susceptibility to BLV occurrence. Paradoxically, the implementation of good veterinary practices and proper management in order to reduce iatrogenic issues, considered one of the main factors of infection among cows within herds [44], failed to reduce the prevalence ([7, 43]). However, we focused on phenotypes representing the BLV level of infection in cows. The LI is directly related to the cows’ viral transmission ability to BLV free herd mates. Methods incorporating GRM as used in this study, have been successfully tested in selective breeding of animals and plants for complex traits [72, 73] and proposed to predict human disease phenotypes [74,75,76]. The high heritability values obtained for LI have remarkable practical implications in genetic improvement programs aiming to reduce the dissemination of BLV in heavily infected populations. Sources of non-additive genetic variance, such as dominance or epistasis among markers, were not considered in this study; however, the evidence indicates that the additive genetic variance is the major contributor to the phenotypic variance in complex characters [77,78,79]. To our knowledge, this is the first report that estimates the level of phenotypic variation in PVL and WBC explained by SNPs markers in BLV infected animals.
The molecular basis influencing the inter-individual difference in LI in vivo during aleukemic phase remains unexplained. The mechanisms of BLV infection involve an infectious cycle after a primary infection, where the virus replicates and spreads within the host [80] by cell-to-cell virion transference, but this process is promptly abrogated by the anti-BLV host immune response [81]. Subsequently, the PVL is maintained by polyclonal expansion of surviving infected B-cells elapsed by mitotic division where viral factors hijack host cellular physiological pathways.
To evaluate which genes might be influencing the set-point of PVL and WBC counts, we identified genomic regions harbouring significantly associated SNPs (p < 1.13 × 10− 6). These SNPs were located in the MHC region on BTA 23, the most relevant gene cluster of the immune sub-genome with high gene density and exceptional diversity.
Polymorphisms in the MHC loci −featured by an intricate structure of linkage disequilibrium [82,83,84,85]− are associated with viral infections outcomes, such as HIV-1, HBV, HCV and HPV [86,87,88]. Fellay et al. [89, 90] confirmed through GWAS that polymorphisms in HLA are leading host genetic determinants on the viral load set-point in HIV−1 infected individuals. To investigate whether effects in others loci were masked by the BoLA region, we have also performed the same genome-wide analysis with the PVL level but excluding SNPs within the bovine MHC. No tested SNP reached significance level (Additional file 5: Table S1). However, when BLV incidence (based on ELISA test) was considered as phenotype for GWAS in US Holstein and Poland Holstein-Frisian populations, many genes outside of the MHC appears to be modulating this trait [40, 41]. The phenotype evaluated in these studies (BLV-seronegatives vs BLV-seropositives cows) indicated novel potentially associated genomic regions, however they do not corroborate previous finding suggesting the MHC as the main genomic region harbouring genes influencing BLV infection [28, 31, 33].
Genomic positions highlighted by GWAS emphasised the potential influence of polymorphic variants in MHC class I and II genes related to their central role in cell-mediated immunity. Four SNPs shared the functional term antigen processing and presentation (GO:0019882). The SNP with the lowest p-value for both conditions studied was rs110525467 (pPVL = 5.32 × 10− 16 and pWBC = 5.90 × 10− 10) producing a synonymous substitution in the MHC class II gene BoLA-DQA1. The rs110579760 (pPVL = 3.66 × 10− 09) located 30.7 kb downstream BoLA-DRB3 was significantly associated with PVL, but not with WBC (Table 1). After infection, a strong cytotoxic T-cell response is developed in peripheral blood against specific viral epitopes [91]. In this regard, two significantly associated SNPs in strong LD (r2 = 0.96) were functionally related with BoLA−A and SFTA2 (Surfactant Associated 2) genes. The magnitude of the immune response to viruses exerted by CD8+ T cells and its association with polymorphisms in HLA class I molecules is widely described in the literature [92,93,94]. In the case of BLV infections in cattle, previous studies have identified associations between polymorphisms in MHC class I genes and resistance to PL in different breeds [26, 27, 95, 96]. The closest SNP (rs110794231) to BoLA-A is 35.2 Kb away corresponding to the SFTA2 gene. The SFTA2 gene encodes a secreted hydrophilic protein with surfactant properties with a role in host defence pathogen recognition patterns (bacteria, viruses and fungi) [97, 98].
The SNP rs41587216 (pPVL = 5.06 × 10− 07 and pWBC = 4.95 × 10− 07) was positioned in the secreted protein CRISP1 (Cysteine-rich secretory protein 1), expressed in skeletal muscle, salivary/lacrimal glands and during lymphoblast development [99] potentially interfering in innate and adaptive immunity (GO:0002376 “immune system process”). Other GWAS hit, the SNP rs109856572, is involved in biological processes that are associated with anti-retroviral innate immunity. It is located in a transcriptional regulatory region of TRIM31 and TRIM40 genes that pertain to the E3 ubiquitin ligases protein family [100,101,102,103]. Perhaps the most studied TRIM mechanism is the restriction of infections caused by HIV−1 exerted by TRIM5α [104,105,106].
A particular haplotype block located in the MHC Class III region, encompasses a number of genes with a plethora of biological effects that may explain the associations detected (Fig. 4). These genes play relevant roles in the immune response, specifically in the regulation of apoptosis and proteasome degradation (LTA, TNF, BAG6) [107], HIV−1 progression (BAT1–5, LY6) [86], T cells response to interferon and viral shedding prevention during innate response to infections (LY6) [108, 109].
Tumor necrosis factor (TNF) and LTA are pro-inflammatory cytokines classified to the GO BP “apoptotic process” (GO:0006915). Polymorphisms in TNF have been reported to contribute partially to the development of lymphosarcoma and PVL in BLV infected cattle [30, 110] and early elimination of BLV in experimentally infected sheep [111].
Several of the targeted genomic regions in the GWAS might be involved in viral intra-host spreading after primary infection according to mechanisms proposed in [81]. For example, in the case of cells-cells fusions the SNP rs110473048 (pPVL = 1.78 × 10− 12 and pWBC = 9.97 × 10− 08) is at 7.21 kb from the Transcription Factor (TF) GCM1 (Glial Cells Missing 1) gene that regulates the expression of the ENV gene (located in the endogenous retrovirus HERV-W) [112] encoding for a fusogenic protein called Syncytin-1. Recently, it has been postulated that infection by an exogenous virus can trans˗activate ENV ectopically by increasing the expression of GCM1 [113] and promoting syncytia formation in breast cancer and endometrial carcinoma [114, 115].
Two significantly associated SNPs mapped within and close to TRAM2 (Translocation-Associated Membrane Protein 2) gene, involved in the biosynthesis and excretion of type I collagen [116] that constitutes the extracellular matrix components [117] where virions are stored. The SNP rs109343703 causes an intronic substitution and the rs110499907 SNP is located 24.7 kb upstream of it. Both SNPs were associated with PVL and not with WBCs, and the nearest SNP in BoLA-DRB3 was in strong LD (r2 > 0.85) with them. Another associated polymorphism (rs110836188) produces a synonymous substitution in the TNXB gene (Tenascin-X), which encodes an extracellular matrix protein involved in matrix-cell adhesion (GO:0007160) and organization of collagen fibres (GO:0030199). Ultimately, analogous to “immunological synapses” some viruses subvert cellular processes to disseminate in the host through “virological synapses” [118, 119]. In this process, cytoskeletal proteins are required for polarization of cells involved in the synapses [120, 121]. Crucially, the SNP rs110155623 associated with both phenotypes is located in the putative regulatory region of CD2AP (CD2-associated protein). CD2AP organizes the cytoskeleton in polarization sites (GO:2,000,249) interacting directly with the Actin protein [122]. Two other GWAS hit variants could capture the effects of genes related to regulation and polymerization of the cytoskeleton structures. A substitution in an intron in the polycystic kidney and hepatic disease 1 (PKHD1) gene (rs41641297) and a SNP (rs110742604) positioned in regulatory regions of TUBB5 and FLOT1 genes. The TUBB5 gene encodes β5-tubulin which belongs to the β-tubulin family.
Finally, four SNPs were assigned to TFs (Table 2) regulating transcriptional processes in promoters depending on RNA polymerase II (GO:0006357). TF genes are candidates for host determinants controlling BLV intra−host spreading, due to a tight mechanism of transcriptional regulation exerted by the trans-activating protein Tax, modulating several host signalling pathways such as AP-1, NF-κB and CREB to induce oncogenic transformation [123].
Altogether, in addition to classical immune response related genes, in this study we pointed novel genes/mechanisms as possibly involved in PVL control and hence BLV dissemination.
Despite our results do not conclusively identify causal genomic variants; we have successfully contracted the landscape of genes potentially related to BLV infections to a short list of very specific candidate genes suitable for experimental validations by gene-expression assays. These results also encourage fine mapping and validation studies. Furthermore, functional studies integrating multi-omics data will allow discerning the underlying molecular mechanisms of the BLV−host interaction.
Conclusions
Results obtained in this study revealed relevant genetic variants in the bovine MHC strongly influencing the control of BLV infection in cattle. Functional inspection of genes harbouring statistically significant associated SNPs brought preliminary evidence about possible biological pathways underlying the LI in infected cows. The proviral BLV load is associated with the stage of the disease progression in the infected cows and directly determines its ability to transmit BLV to healthy animals. Therefore, genome wide identification of genetics variants associated with low proviral load BLV infections would be useful not only to design therapeutics and preventive treatments but also as alternative control programs based on selective breeding of animals through genomic selection in dairy cattle.
Abbreviations
- BLV:
-
Bovine Leukemia Virus
- BoLA:
-
Bovine leukocyte antigen
- BTA:
-
Bos taurus autosome
- ELISA:
-
Enzyme-linked Immunosorbent Assay
- GO:
-
Gene ontology terms
- GRM:
-
Genetic relationship matrix
- GWAS:
-
Genome-Wide Association Study
- HLI:
-
High level of infection
- HPVL:
-
High proviral load
- IBS:
-
Identity by State
- LD:
-
Linkage Disequilibrium
- LMMs:
-
Linear Mixed Models
- LPVL:
-
Low proviral load
- MAF:
-
Minor Allele Frequency
- MHC:
-
Major Histocompatibility Complex
- PBMCs:
-
Peripheral Blood Mononuclear Cells
- PCs:
-
Principal Components
- PL:
-
Persistent Lymphocytosis
- PVL:
-
Proviral Load
- QC:
-
Quality Control
- SNPs:
-
Single Nucleotide Polymorphisms
- WBC:
-
White Blood Cells
References
Kettmann R, Burny A, Callebaut Y, Droogmans L, Mammerickx M, Willems L, et al. Bovine leukemia virus. Levy JA, ed The Retroviridae Plenum Press, NY. 1994:39–81.
European Food Safety Authority (EFSA). Panel on Animal Health and Welfare (AHAW). Scientific Opinion on Enzootic Bovine Leukosis. EFSA Journal. 2015;13:4188.
Barez PY, de Brognez A, Carpentier A, Gazon H, Gillet N, Gutiérrez G, et al. Recent advances in BLV research. Viruses. 2015;24:6080–8.
Gutiérrez G, Lomonaco M, Alvarez I, Fernandez F, Trono K. Characterization of colostrum from dams of BLV endemic dairy herds. Vet Microbiol 2015; 12: 366−9.
VanLeeuwen JA, Forsythe L, Tiwari A, Chartier R. Seroprevalence of antibodies against bovine leukemia virus, bovine viral diarrhea virus, Mycobacterium avium subspecies paratuberculosis, and Neospora caninum in dairy cattle in Saskatchewan. Can Vet J. 2005;46:56–8.
VanLeeuwen JA, Tiwari A, Plaizier JC, Whiting TL. Seroprevalences of antibodies against bovine leukemia virus, bovine viral diarrhea virus, Mycobacterium avium subspecies paratuberculosis, and Neospora caninum in beef and dairy cattle in Manitoba. Can Vet J. 2006;47:783–6.
NAHMS, 2007.https://www.aphis.usda.gov/animal_health/nahms/dairy/downloads/dairy07/Dairy07_is_BLV.pdf. Accessed 27 Apr 2016.
Gutierrez G, Carignano H, Alvarez I, Martínez C, Porta N, Politzki R, et al. Bovine leukemia virus p24 antibodies reflect blood proviral load. BMC Vet Res. 2012; https://doi.org/10.1186/1746-6148-8-187.
OIE. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. 7th Ed; 2012.
Erskine RJ, Bartlett PC, Byrem TM, Render CL, Febvay C, Houseman JT. Using a herd profile to determine age-specific prevalence of bovine leukemia virus in Michigan dairy herds. Vet Med Int. 2012a; https://doi.org/10.1155/2012/350374.
Bartlett PC, Norby B, Byrem TM, Parmelee A, Ledergerber JT, Erskine RJ. Bovine leukemia virus and cow longevity in Michigan dairy herds. J Dairy Sci. 2013;96:1591–7.
Norby B, Bartlett PC, Byrem TM, Erskine RJ. Effect of infection with bovine leukemia virus on milk production in Michigan dairy cows. J Dairy Sci 2016; 99:2043−52.
Da Y, Shanks RD, Stewart JA, Lewin HA. Milk and fat yields decline in bovine leukemia virus-infected Holstein cattle with persistent lymphocytosis. Proc Natl Acad Sci US A. 1993;90:6538–41.
Pelzer KD. Economics of bovine leukemia virus infection. Vet. Clin. North Am. Food Anim. Pract. 1997;13:129–41.
Ott SL, Johnson R, Wells SJ. Association between bovine-leukosis virus seroprevalence and herd-level productivity on US dairy farms. Prev Vet Med. 2003;61:249–62.
Erskine RJ, Bartlett PC, Byrem TM, Render CL, Febvay C, Houseman JT. Herd-level determinants of bovine leukaemia virus prevalence in dairy farms. J Dairy Res. 2012b;79:445–50.
Rodríguez SM, Florins A, Gillet N, de Brogniez A, Sánchez-Alcaraz MT, Boxus M, et al. Preventive and therapeutic strategies for bovine leukemia virus: lessons for HTLV. Viruses. 2011;3:1210–48.
Bishop SC. Disease Genetics: Successes, Challenges and Lessons Learnt. Proc. 10th World Congr. Genet. Appl. to Livest. Prod.; 2014.
VanRaden PM, Cole JB. Net merit as a measure of lifetime profit: 2014 revision. AIP Research Report. https://aipl.arsusda.gov/reference/nmcalc-2014.htm. Accessed 22 Apr 2016.
Beavers L, Van Doormaal B. Pro$: Genetic selection for profit. Canadian Dairy Network. https://www.cdn.ca/document.php?id=404. Accessed 20 Mar 2016.
Koeck A, Miglior F, Kelton DF, Schenkel FS. Health recording in Canadian Holsteins: data and genetic parameters. J Dairy Sci. 2012;95:4099–108.
Parker-Gaddis KL, Cole JB, Clay JS, Maltecca C. Genomic selection for producer-recorded health event data in US dairy cattle. J Dairy Sci 2014; 97:3190−9.
Mirsky ML, Olmstead CA, Da Y, Lewin HA. The prevalence of proviral bovine leukemia virus in peripheral blood mononuclear cells at two subclinical stages of infection. J Virol. 1996;70:2178–83.
Jimba M, Takeshima S, Matoba K, Endoh D, Aida Y. BLV-CoCoMo-qPCR: quantitation of bovine leukemia virus proviral load using the CoCoMo algorithm. Retrovirology. 2010;7:91.
Mammerickx M, Portetelle D, de Clercq K, Burny A. Experimental transmission of enzootic bovine leukosis to cattle, sheep and goats: infectious doses of blood and incubation period of the disease. Leuk Res. 1987;11:353–8.
Lewin HA, Bernoco D. Evidence for BoLA-linked resistance and susceptibility to subclinical progression of bovine leukaemia virus infection. Anim Genet. 1986;17:197–207.
Lewin HA, Wu MC, Stewart JA, Nolan TJ. Association between BoLA and subclinical bovine leukemia virus infection in a herd of Holstein-Friesian cows. Immunogenetics. 1988a;27:338–44.
Xu A, van Eijk MJ, Park C, Lewin HA. Polymorphism in BoLA-DRB3 exon 2 correlates with resistance to persistent lymphocytosis caused by bovine leukemia virus. J Immunol. 1993;151:6977–85.
Mirsky ML, Olmstead C, Da Y, Lewin HA. Reduced bovine leukaemia virus proviral load in genetically resistant cattle. Anim Genet. 1998;29:245–52.
Konnai S, Usui T, Ikeda M, Kohara J, Hirata T, Okada K, et al. Tumor necrosis factor-alpha genetic polymorphism may contribute to progression of bovine leukemia virus-infection. Microbes Infect. 2006;8:2163–71.
Juliarena MA, Poli M, Sala L, Ceriani C, Gutierrez S, Dolcini G, et al. Association of BLV infection profiles with alleles of the BoLA-DRB3.2 gene. Anim Genet. 2008;39:432–8.
Jimba M, Takeshima SN, Murakami H, Kohara J, Kobayashi N, Matsuhashi T, et al. BLV-CoCoMo-qPCR: a useful tool for evaluating bovine leukemia virus infection status. BMC Vet Res. 2012;8:167.
Miyasaka T, Takeshima SN, Jimba M, Matsumoto Y, Kobayashi N, Matsuhashi T, et al. Identification of bovine leukocyte antigen class II haplotypes associated with variations in bovine leukemia virus proviral load in Japanese black cattle. Tissue Antigens. 2013;81:72–82.
Carignano HA, Beribe MJ, Caffaro ME, Amadio A, Nani JP, Gutierrez G, et al. BOLA-DRB3 gene polymorphisms influence bovine leukaemia virus infection levels in Holstein and Holstein × Jersey crossbreed dairy cattle. Anim Genet. 2017;48:420–30.
Minozzi G, Williams JL, Stella A, Strozzi F, Luini M, Settles ML, et al. Meta-analysis of two genome-wide association studies of bovine paratuberculosis. PLoS One. 2012;7:e32578.
Bermingham ML, Bishop SC, Woolliams JA, Pong-Wong R, Allen AR, McBride SH. At al. Genome-wide association study identifies novel loci associated with resistance to bovine tuberculosis. Heredity. 2014;112:543–51.
Sahana G, Guldbrandtsen B, Thomsen B, Lund MS. Confirmation and fine-mapping of clinical mastitis and somatic cell score QTL in Nordic Holstein cattle. Anim Genet. 2013;44(6):620.
Wu X, Lund MS, Sahana G, Guldbrandtsen B, Sun D, Zhang Q, Su G. Association analysis for udder health based on SNP-panel and sequence data in Danish Holsteins. Genet Sel Evol. 2015;47:50.
Neibergs HL, Seabury CM, Wojtowicz AJ, Wang Z, Scraggs E, Kiser JN, et al. Susceptibility loci revealed for bovine respiratory disease complex in pre-weaned holstein calves. BMC Genomics. 2014;15:1164.
Abdalla EA, Peñagaricano F, Byrem TM, Weigel KA, Rosa GJ. Genome-wide association mapping and pathway analysis of leukosis incidence in a US Holstein cattle population. Anim Genet. 2016;47:395–407.
Brym P, Bojarojc-Nosowicz B, Olenski K, Hering DM, Rusc A, Kaczmarczyk E, Kaminski S. Genome-wide association study for host response to bovine leukemia virus in Holstein cows. Vet Immunol Immunopathol. 2016;175:24–35.
Gutiérrez G, Alvarez I, Fondevila N, Politzki R, Lomónaco M, Rodríguez S, et al. Detection of bovine leukemia virus specific antibodies using recombinant p24-ELISA. Vet Microbiol. 2009;137:224–34.
Gutiérrez G, Alvarez I, Politzki R, Lomónaco M, Dus Santos MJ, Rondelli F, et al. Natural progression of bovine leukemia virus infection in Argentinean dairy cattle. Vet Microbiol. 2011;151:255–63.
Hopkins SG, DiGiacomo RF. Natural transmission of bovine leukemia virus in dairy and beef cattle. Vet Clin North Am Food Anim Pract. 1997;13:107–28.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45.
Alvarez I, Gutiérrez G, Gammella M, Martínez C, Politzki R, González C, et al. Evaluation of total white blood cell count as a marker for proviral load of bovine leukemia virus in dairy cattle from herds with a high seroprevalence of antibodies against bovine leukemia virus. Am J Vet Res. 2013;74:744–9.
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One. 2009;4:e5350.
Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, et al. A whole genome assembly of the domestic cow, Bos Taurus. Genome Biol. 2009;10:R42.
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, et al. Database resources of the National Center for biotechnology information. Nucleic Acids Res. 2007;36:D13–21.
Nicolazzi EL, Caprera A, Nazzicari N, Cozzi P, Strozzi F, Lawley C, et al. SNPchiMp v.3: integrating and standardizing single nucleotide polymorphism data for livestock species. BMC Genomics. 2015;16:283.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Lewin HA, Wu MC, Nolan TJ, Stewart JA. Peripheral B lymphocyte percentage as an indicator of subclinical progression of bovine leukemia virus infection. J Dairy Sci. 1988b;71:2526–34.
Druet T, Georges M. A hidden Markov model combining linkage and linkage disequilibrium information for haplotype reconstruction and quantitative trait locus fine mapping. Genetics. 2010;184:789–98.
Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–13.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9.
Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88:294–305.
Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28:2540–2.
Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database. 2011; https://doi.org/10.1093/database/bar030.
Veyrieras JB, Kudaravalli S, Kim SY, Dermitzakis ET, Gilad Y, Stephens M, et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4:e1000214.
Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet. 2002;3:299–309.
McKay SD, Schnabel RD, Murdoch BM, Matukumalli LK, Aerts J, Coppieters W, et al. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 2007;8:74.
Khatkar MS, Zenger KR, Hobbs M, Hawken RJ, Cavanagh JA, Barris W, et al. A primary assembly of a bovine haplotype block map based on a 15,036-single-nucleotide polymorphism panel genotyped in holstein-friesian cattle. Genetics. 2007;176:763–72.
Khatkar MS, Nicholas FW, Collins AR, Zenger KR, Cavanagh JA, Barris W, et al. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genomics. 2008;9:187.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Daverman R, et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–41.
Al-Shahrour F, Díaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of gene ontology terms with groups of genes. Bioinformatics. 2004;20:578–80.
Glass EJ, Baxter R, Leach R, Taylor G. Bovine viral diseases – the role of host genetics. In: Bishop SC, Axford R, Owen J, Nicholas F, ed. Breeding for Disease Resistance in Farm Animals. 3rd edition. CAB International; Nosworthy Way, Wallingford OX10 8DE. 2010.
Rubicz R, Leach CT, Kraig E, Dhurandhar NV, Duggirala R, Blangero J, et al. Genetic factors influence serological measures of common infections. Hum Hered. 2011;72:133–41.
Abdalla EA, Rosa GJ, Weigel KA, Byrem T. Genetic analysis of leukosis incidence in United States Holstein and Jersey populations. J Dairy Sci. 2013;96(9):6022.
VanRaden PM, Van Tassell CP, Wiggans GR, Sonstegard TS, Schnabel RD, Taylor JF, et al. Invited review: reliability of genomic predictions for north American Holstein bulls. J Dairy Sci. 2009;92:16–24.
Crossa J, de los Campos G, Pérez P, Gianola D, Burgueño J, Araus JL, et al. Prediction of genetic values of quantitative traits in plant breeding using pedigree and molecular markers. Genetics. 2010;186:713–24.
Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk of complex disease. Curr Opin Genet Dev. 2008;18:257–63.
Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet. 2013;14:507–15.
de los Campos G, Vazquez AI, Fernando R, Klimentidis YC, Sorensen D. Prediction of complex human traits using the genomic best linear unbiased predictor. PLoS Genet. 2013;9:e1003608.
Hill WG, Goddard M, Visscher P. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 2008;4:e1000008.
Hill WG, Mäki-Tanila A. Expected influence of linkage disequilibrium on genetic variance caused by dominance and epistasis on quantitative traits. J Anim Breed Genet. 2015;132:176–86.
Zhu Z, Bakshi A, Vinkhuyzen AA, Hemani G, Lee SH, Nolte IM, et al. Dominance genetic variation contributes little to the missing heritability for human complex traits. Am J Hum Genet. 2015;96:377–85.
Gillet N, Gutiérrez G, Rodriguez SM, de Brogniez A, Renotte N, Alvarez I, et al. Massive depletion of bovine leukemia virus Proviral clones located in genomic transcriptionally active sites during primary infection. PLoS Pathog. 2013;9:e1003687.
Carpentier A, Barez P, Hamaidia M, Gazon H, de Brogniez A, Perike S, et al. Modes of human T cell leukemia virus type 1 transmission, replication and persistence. Viruses. 2015;7:3603–24.
Stewart CA, Horton R, Allcock RJ, Ashurst JL, Atrazhev AM, Coggill P, et al. Complete MHC haplotype sequencing for common disease gene mapping. Genome Res. 2004;14:1176–87.
Miretti MM, Walsh EC, Ke X, Delgado M, Griffiths M, Hunt S, et al. High-resolution linkage-disequilibrium map of the human major histocompatibility complex and first generation of tag single-nucleotide polymorphisms. Am J Hum Genet. 2005;76:634–46.
de Bakker PIW, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet. 2006;38:1166–72.
Traherne J, Horton R, Roberts AN, Miretti MM, Hurles ME, Stewart CA, et al. Genetic analysis of completely sequenced disease-associated MHC haplotypes identifies shuffling of segments in recent human history. PLoS Genet. 2006;2:e9.
Limou S, Le Clerc S, Coulonges C, Carpentier W, Dina C, Delaneau O, et al. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide association study 02). J Infect Dis. 2009;199:419–26.
Trachtenberg E, Bhattacharya T, Ladner M, Phair J, Erlich H, Wolinsky S. The HLA-B/-C haplotype block contains major determinants for host control of HIV. Genes Immun. 2009;10:673–7.
Martin MP, Carrington M. Immunogenetics of HIV disease. Immunol Rev. 2013;254:245–64.
Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–7.
Fellay J, Ge D, Shianna KV, Colombo S, Ledergerber B, Cirulli ET, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5:e1000791.
Lundberg P, Splitter GA. Gammadelta(+) T-lymphocyte cytotoxicity against envelope-expressing target cells is unique to the alymphocytic state of bovine leukemia virus infection in the natural host. J Virol. 2000;74:8299–306.
Goulder PJR, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8:619–30.
Blackwell JM, Jamieson SE, Burgner D. HLA and infectious diseases. Clin Microbiol Rev. 2009;22:370–85.
Fellay J. Host genome influences on HIV-1 disease. Antivir Ther. 2010;14:731–8.
Palmer C, Thurmond M, Picanso J, Brewer AW, Bernoco D. Susceptibility of cattle bovine leukemia virus infection associated with BoLA type. In: Proceedings of the 91st Annual Meeting of United States Animal Health Association, Salk Lake City, USA; 1987. p. 218–28.
Stear MJ, Dimmock CK, Newman MJ, Nicholas FW. BoLA antigens are associated with increased frequency of persistent lymphocytosis in bovine leukaemia virus infected cattle and with increased incidence of antibodies to bovine leukaemia virus. Anim Genet. 1988;19:151–8.
Seaton B, Crouch EC, McCormack FX, Head JF, Hartshorn KL, Mendelsohn R. Structural determinants of pattern recognition by lung collectins. Innate Immun. 2010;16:143–50.
Mittal R, Hammel M, Schwarz J, Heschl KM, Bretschneider N, Flemmer AW, et al. SFTA2—a novel secretory peptide highly expressed in the lung—is modulated by lipopolysaccharide but not Hyperoxia. PLoS One. 2012;7:e40011.
Reddy T, Gibbs GM, Merriner DJ, Kerr JB, O’Bryan MK. Cysteine-rich secretory proteins are not exclusively expressed in the male reproductive tract. Dev Dyn. 2008;237:3313–23.
Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The tripartite motif family identifies cell compartments. EMBO J. 2001;20:2140–51.
Nisole S, Stoye JP, Saïb A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005;3:799–808.
Ozato K, Shin DM, Chang TH, Morse HC. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. 2008;8:849–60.
Uchil PD, Quinlan BD, Chan W, Luna JM, Mothes W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008;4:e16.
Hatziioannou T, Perez-Caballero D, Yang A, Cowan S, Bieniasz PD. Retrovirus Resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc Natl Acad Sci U S A. 2004;101:10774–9.
Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in old world monkeys. Nature. 2004;427:848–53.
Zhang X, Kondo M, Chen J, Miyoshi H, Suzuki H, Ohashi T, et al. Inhibitory effect of human TRIM5alpha on HIV-1 production. Microbes Infect. 2010;12:768–77.
Desmots F, Russell HR, Lee Y, Boyd K, McKinnon PJ. The reaper-binding protein scythe modulates apoptosis and proliferation during mammalian development. Mol Cell Biol. 2005;25:10329–37.
Loeuillet C, Deutsch S, Ciuffi A, Robyr D, Taffé P, Muñoz M, et al. In vitro whole-genome analysis identifies a susceptibility locus for HIV-1. PLoS Biol. 2008;6:e32.
Lee PY, Wang J, Parisini E, Dascher CC, Nigrovic PA. Ly6 family proteins in neutrophil biology. J Leukoc Biol. 2013;94:585–94.
Lendez PA, Passucci JA, Poli MA, Gutierrez SE, Dolcini GL, Ceriani MC. Association of TNF-α gene promoter region polymorphisms in bovine leukemia virus (BLV)-infected cattle with different proviral loads. Arch Virol. 2015;160:2001–7.
Kabeya H, Ohashi K, Oyunbileg N, Nagaoka Y, Aida Y, Sugimoto C, et al. Up-regulation of tumor necrosis factor alpha mRNA is associated with bovine-leukemia virus (BLV) elimination in the early phase of infection. Vet Immunol Immunopathol. 1999;68:255–65.
Yu C, Shen K, Lin M, Chen P, Lin C, Chang GD, et al. GCMa regulates the syncytin-mediated trophoblastic fusion. J Biol Chem. 2002;277:50062–8.
Li F, Nellaker C, Sabunciyan S, Yolken RH, Jones-Brando L, Johansson AS, et al. Transcriptional Derepression of the ERVWE1 locus following influenza a virus infection. J Virol. 2014;88:4328–37.
Bjerregaard B, Holck S, Christensen IJ, Larsson LI. Syncytin is involved in breast cancer-endothelial cell fusions. Cell Mol Life Sci. 2006;63:1906–11.
Strick R, Ackermann S, Langbein M, Swiatek J, Schubert SW, Hashemolhosseini S, et al. Proliferation and cell–cell fusion of endometrial carcinoma are induced by the human endogenous retroviral Syncytin-1 and regulated by TGF-β. J Mol Med. 2006;85:23–38.
Stefanovic B, Stefanovic L, Schnabl B, Bataller R, Brenner D. TRAM2 protein interacts with endoplasmic reticulum Ca2+ pump Serca2b and is necessary for collagen type I synthesis. Mol Cell Biol. 2004;24:1758–68.
Pais-Correia A, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, et al. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med. 2010;16:83–9.
Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–7.
Sattentau QJ. Avoiding the void: animal virus cell-to-cell spread. Nature reviews. Microbiology. 2008;6:815–26.
Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, et al. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science. 2003;299:1713.
Jolly C, Sattentau QJ. Adhesion molecule interactions facilitate human immunodeficiency virus type-1-induced virological synapse formation between T cells. J Virol. 2007;81:13916–21.
Lehtonen S, Zhao F, Lehtonen E. CD2-associated protein directly interacts with the actin cytoskeleton. AJP Ren Physiol. 2002;283:F734–43.
Grassmann R, Aboud M, Jeang KT. Molecular mechanisms of cellular transformation by HTLV-1 tax. Oncogene. 2005;24:5976–85.
Acknowledgements
Authors thank MINCYT, CONICET, INTA and UNaM for continuous support and laboratory setting funds.
Funding
This work was supported by project PAE 37143 MINCyT – FONCyT; INTA PNBIO 1131033 and FAO-IAEA CRP D3.10.28. MM is supported by CONICET.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
HC, KT, MP and MM project design and support; HC, MR, AA, JN sample collection and data processing; HC, GG and IA performed laboratory experiments, HC, DR, MB performed the data analysis and interpreted results; DR, MB, MR, JN, GG, IA, KT, MP participated in discussing and revising the manuscript; HC and MM drafted the manuscript. The final manuscript has been read and approved by all authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The procedures followed for extraction and handling of samples were approved by the Institutional Committee for Care and Use of Experimental Animals of the National Institute of Agricultural Technology (CICUAE-INTA) under protocol number 35/2010, and followed the guidelines described in the institutional Manual.
Farm owners’ consent was obtained before animal sampling.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Pedigree of the Holstein and Jersey crosses cattle population under study. Red lines connect the individual with his father. Blue lines connect the individual with his mother. (DOCX 252 kb).
Additional file 2:
Figure S2. Holstein and Jersey crosses population sub-structure. Each point represents the coordinates of PC1 and PC2 for each individual. The assignment of PCs allowed inferences about the ancestry origin of cows. (0; 12.5; 25; 50; 75 and 100 of Holstein breed percentage). (DOCX 236 kb).
Additional file 3:
Figure S3. QQ plots and genome inflation factor (λ) in GWAS with PVL. The p-values come from a logistic regression under an additive model of association of SNPs with the level of PVL. The genomic inflation factor quantifies the volume of inflation observed. A) A strong deviation in the -log10(p) (black circle) from the line of identity (red line) was observed without considering covariates, λ = 5.31. B) A small adjustment was observed when included the following covariates: Age of the animal (A), Herd (H), Number of lactation (L) Percentage of Holstein (PH) and Bull (B), λ = 4.07. C) A significant correction is manifested including the first 8 Principal Components (PC1-PC8), λ = 1.26. D) When considering all the covariates together, the correction level approaches the CI95% of the null distribution (dotted lines), λ = 1.21. (DOCX 404 kb).
Additional file 4:
Figure S4. QQ plots and genome inflation factor (λ) in GWAS with WBC counts. Analyses were based on the -log10(p) from logistic regression under an additive model of association of SNPs with WBCs. A) A pronounced deviation from the null distribution (red line) is manifested in the model without covariates, λ = 3.99. B) When covariates A_DF_L_PH_B were adjusted in the model λ decreases to 2.76, C) A significant correction is achieved by considering PC1-PC8, λ = 1.31. D) When considering all the covariates together (PC1-PC8_A_H_L_PH_B) the correction level approached the CI 95% of the null distribution (dotted lines), λ = 1.30. (DOCX 429 kb)
Additional file 5:
Table S1. Top 15 SNPs with the lowers p-value from the GWA analysis with PVL excluding SNPs located ~ 20–36 Mb of BTA 23 (DOCX 20 kb).
Additional file 6:
Figure S5. LD decay by distance. LD measures were considered among syntenic SNPs pairs separated < 1 Mb. The r2 values were averaged each 10 Kb and plotted versus inter-SNPs distances. (DOCX 56 kb).
Additional file 7:
Table S2. Description of PC categories identified and genes containing them. (DOCX 22 kb).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Carignano, H.A., Roldan, D.L., Beribe, M.J. et al. Genome-wide scan for commons SNPs affecting bovine leukemia virus infection level in dairy cattle. BMC Genomics 19, 142 (2018). https://doi.org/10.1186/s12864-018-4523-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4523-2