
Abstract
Background
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorders in the world. Studying PD from systems biology perspective involving genes and their regulators might provide deeper insights into the complex molecular interactions associated with this disease.
Result
We have studied gene co-expression network obtained from a PD-specific microarray data. The co-expression network identified 11 hub genes, of which eight genes are not previously known to be associated with PD. Further study on the functionality of these eight novel hub genes revealed that these genes play important roles in several neurodegenerative diseases. Furthermore, we have studied the tissue-specific expression and histone modification patterns of the novel hub genes. Most of these genes possess several histone modification sites those are already known to be associated with neurodegenerative diseases. Regulatory network namely mTF-miRNA-gene-gTF involves microRNA Transcription Factor (mTF), microRNA (miRNA), gene and gene Transcription Factor (gTF). Whereas long noncoding RNA (lncRNA) mediated regulatory network involves miRNA, gene, mTF and lncRNA. mTF-miRNA-gene-gTF regulatory network identified a novel feed-forward loop. lncRNA-mediated regulatory network identified novel lncRNAs of PD and revealed the two-way regulatory pattern of PD-specific miRNAs where miRNAs can be regulated by both the TFs and lncRNAs. SNP analysis of the most significant genes of the co-expression network identified 20 SNPs. These SNPs are present in the 3′ UTR of known PD genes and are controlled by those miRNAs which are also involved in PD.
Conclusion
Our study identified eight novel hub genes which can be considered as possible candidates for future biomarker identification studies for PD. The two regulatory networks studied in our work provide a detailed overview of the cellular regulatory mechanisms where the non-coding RNAs namely miRNA and lncRNA, can act as epigenetic regulators of PD. SNPs identified in our study can be helpful for identifying PD at an earlier stage. Overall, this study may impart a better comprehension of the complex molecular interactions associated with PD from systems biology perspective.
Similar content being viewed by others
Background
Parkinson’s disease (PD) is one of the well-reported neurodegenerative disorders, only second to the Alzheimer’s disease (AD), throughout the world [1]. The primary pathology of PD is the loss of dopaminergic neurons in the substantia nigra with Lewy bodies (intracytoplasmic inclusion deposits of aggregated alpha-synuclein and ubiquitin protein, and damaged nerve cells) [2, 3].
A good number of studies have been performed to identify the causative factors and molecular markers of PD. Several previous studies have pointed out the role of different genes in this disease [4]. Gene expression profiling analysis has identified differentially expressed genes in PD [5]. Besides, differential expression of several microRNAs (miRNAs) has also been associated with the pathophysiology of several neurodegenerative diseases [6, 7] including PD [8]. Study of gene regulatory networks has emerged as an important approach for computational analyses of diseases [9]. However, limited previous studies have tried to understand the association of both of these (miRNAs and mRNAs) PD markers in the context of biological networks. In order to gain a proper understanding of this disease, one needs to study the detailed regulatory network involving genes, miRNAs and transcription factors (TFs). A thorough examination of regulatory networks can help us to identify the key genes or miRNAs as well as different network motifs associated with a disease. These network motifs, in turn, provide us several important aspects of a disease progression.
Previous studies have indicated the role of epigenetic modifications in the development of neurodegenerative diseases including Parkinson’s Disease and Alzheimer’s Disease (AD) [10, 11]. Epigenetics refers to the meiotically and mitotically heritable changes in gene expression that does not involve changes to the DNA sequence [12]. Interpretation of epigenetic profiling leads to the identification of changes in gene expression responsible for the disease progression. There are three distinct yet highly interrelated mechanisms of epigenetic regulation - DNA methylation, Histone modifications and non-coding RNA-based mechanisms [13]. Epigenetic changes can be influenced by several factors including age, environment, lifestyle and disease state [13]. Recent systematic review on neurodegenerative disease, investigated epigenetic marks in PD and identified the most consistently reported methylation genes and histone modifications associated with PD [14].
Studies have revealed that non-coding RNAs such as miRNAs (~22 nt long) and long non-coding RNAs (lncRNAs) (>200 nt long), play crucial roles in epigenetic pathways and gene silencing. The function of miRNAs involves binding to a specific sequence in the 3′ UTR of a gene and inhibiting the expression of that gene. Thus, miRNAs act as cellular post-transcriptional regulators. The miRNA profiling of PD samples offers insight into the molecular mechanism of PD progression and several miRNAs have been implicated in PD pathogenesis [8, 15, 16].
The function of lncRNAs involves diverse cellular processes, such as chromatin remodeling, cell cycle regulation and several developmental processes [17]. It can influence the post-transcriptional regulation by interfering with the miRNA pathways, by acting as competing endogenous RNAs (ceRNAs) [18]. lncRNAs possess miRNA response elements (MRE) or miRNA binding sites in them. This enables lncRNAs to act as miRNA sponges to control the availability of endogenous miRNA for binding to their target mRNAs and subsequently reducing the repression of these target mRNAs [18]. lncRNAs are implicated in neurodegenerative processes, including AD and Huntington’s disease (HD) [19, 20]. However, very little is known about the association of lncRNAs in PD [21].
Single base alteration in the gene sequence or single nucleotide polymorphism (SNP) can affect the phenotypes either by altering the amount of protein produced or by changing the type of protein produced [22]. SNPs are believed to cause differences between individuals, such as susceptibility to diseases [23]. There are numerous SNPs present in the human genome [24]. These are considered as invaluable markers and potentially powerful tools for both genetic research and applications in practice [25]. Several studies have identified SNPs associated with complex diseases, which in turn serve as a potential marker for diagnosis [26]. A recent miRNA-related SNP analysis study identified SNPs as independent prognostic markers for the survival in small cell lung cancer patients [27]. However, very few such studies have been performed for PD. A recent genome-wide association study identified significant association between bone marrow stromal cell antigen 1 SNP and increased risk of PD which is enhanced by environmental factors [28]. SNP analysis or genotyping of PD patients can be helpful to identify this disease at an earlier state. Besides single base alteration in the miRNA binding sites can give us important information about the mode of regulation of regulatory factors in this disease. It is believed that more and more genetic studies combined with machine learning and statistical methods will be required in near future to investigate the underlying molecular signature of a disease [29].
With the increase of transcriptomic data, novel system biological approaches are required that can explore the complex molecular interactions associated with a disease. In this study, we have analyzed gene co-expression network based on a PD microarray dataset. Two regulatory networks were built from the highly co-expressed genes. The mTF-miRNA-gene-gTF regulatory network involves microRNA Transcription Factor (mTF), microRNA (miRNA), gene and gene Transcription Factor (gTF) whereas, long noncoding RNA (lncRNA) mediated regulatory network involves miRNA, gene, mTF and lncRNA. mTF-miRNA-gene-gTF regulatory network identified a novel feed-forward loop. lncRNA-mediated regulatory network identified novel lncRNAs of PD and revealed the two-way regulatory pattern of PD-specific miRNAs where miRNAs can be regulated by both the TFs and lncRNAs. SNP analysis of the most significant genes of the co-expression network identified 20 SNPs. Thus, our study provides important insight into the epigenetic mechanism (lncRNA, miRNA, histone modification) associated with PD. Moreover, SNPs identified in our study can be helpful for identifying PD at an earlier stage.
Results
Figure 1 depicts the workflow of our analysis.

Workflow of the methodology used in our study
Differentially expressed gene selection
SAM
We identified the differentially expressed (DE) genes between PD and control patients by applying the Significance Analysis of Microarray (SAM) [30]. In chip A, SAM identified 1518 DE genes at FDR value 0.19% and tail strength 44.1%. Among the 1518 DE genes 293 genes were positive (upregulated) and 1225 were negative (downregulated). In chip B, SAM identified 673 DE genes at FDR value 0.11% and tail strength 37.6%. Among the 673 differentially expressed genes, 91 genes were positive (upregulated) and 582 were negative (downregulated).
t-test
With the t-test analysis, a much higher number of DE genes were found than with SAM. Results identified 4797 and 3120 DE genes in chip A and chip B respectively at p-value 0.05 or 95% confidence level.
The common DE genes found by both SAM and t-test were considered as the most significant DE gene sets and these were used for further study. In chip A, 521 genes were found to be commonly DE in both SAM and t-test whereas, in chip B, 130 genes were found to be commonly DE in both SAM and t-test (Table 1). Out of the 521 and 130 genes from chip A and chip B only 458 genes in chip A and 105 genes in chip B were annotated.
Enrichment analysis of the DE genes
The DE genes found from SAM and t-test were both annotated via EASE (Expression Analysis Systematic Explorer) [31]. The shared 458 DE genes of chip A obtained from SAM and t-test were then subjected to enrichment analysis in FatiGO (Table 2) [32]. Results of the enrichment analysis of identified several neurodegenerative disease pathways as the most significant over representative KEGG pathways such as Parkinson’s disease pathway (hsa05012), Huntington disease (hsa05016) and Alzheimer’s disease (hsa05010) (Table 3). This also signifies the importance of this gene set in the context of the PD-specific study. The 105 DE genes in chip B were not associated with any significant terms in FatiGo. Therefore DE genes of chip B were not considered for further analysis. The 458 DE genes of chip A were considered as the significant gene set for further study and were termed as common DE gene set of chip A.
Co-expression network construction and analysis
On the basis of the co-expression pattern, WGCNA (please refer to the methods section) divided the 458 common DE genes into six modules (turquoise, blue, brown, yellow, green and red containing 266, 56, 43, 42, 25 and 25 mRNAs). FatiGO analysis revealed that out of the six WGCNA modules Turquoise module was the most significant co-expressed module (Additional file 1: Table S1A and B).
Topological analysis of the WGCNA module and identification of hub genes
We analyzed two centrality measures - degree and betweenness centrality (BC) in tYNA [33]. We sorted the 266 genes according to their degree or connectivity. The degree represents the number of connections or edges of a particular node [34], whereas BC quantifies the flow of information through a node in the network. It specifies how a node influences the communication among other nodes [35]. In our study, the 266 genes exhibited a varied degree distribution with the highest degree of 262 and lowest degree of 1. The average degree value was found to be 217.63 with standard deviation 52.74. We found that highest BC value was 391.50 and the lowest was 0 with an average of 24.95 and standard deviation 33.71. We chose the top 8 nodes (i.e. top 3% of the total nodes) with highest degree value as High Connectivity (HC) hub nodes. AP3B2, MAGED1, NSF, STXBP1, CYB561, AF1Q, C14ORF78 and GASP were identified as HC hub genes (Additional file 2: Table S2). Interestingly these 8 HC nodes were assigned with low BC values. Surprisingly we found three nodes with high BC values but low degree value. Although these nodes have low connectivity, they might be important regarding information flow. Therefore, we identified these as High BC low connectivity (HBLC) hub nodes. HNRPC, MAN1C1 and HSPA1A were identified as HBLC hub genes (Additional file 2: Table S2). Figure 2 shows the gene co-expression network of the turquoise module with the 11 hub genes. Out of the 11 hubs, three hubs (NSF, HSPA1A and CYB561) were already found to be associated with PD. The remaining eight novel hub genes (MAGED1, AP3B2, STXBP1, AF1Q, GASP, C14ORF78, MAN1C1, HNRPC) were further studied for their association in PD.

Gene co-expression Network of the most significant co-expressed module (Turquoise module) obtained from WGCNA. Green nodes represent genes and edges represent co-expression relationship. 11 Hub genes are represented by larger node size
Epigenetic regulation of the hub genes
In order to identify the probable epigenetic regulation of the hub genes, histone modification data for eight hub genes (Table 4) were retrieved from HHMD [36]. Table 4 shows that all the eight hub genes were associated with several histone modification sites. Further study identified the experimentally validated non-coding RNA mediated regulation of hub genes (Table 5) [37,38,39,40]. It was found that four out of the eight hub genes were associated with miRNAs already known in PD. Moreover, most of these hub gene associated miRs were in turn regulated by lncRNAs.
Regulatory network construction and analysis
mTF-miRNA-gene-gTF regulatory network
In order to get a view of the regulatory pattern of the turquoise module, we built a regulatory network comprising genes of the turquoise model and the TFs associated with these genes (gTFs) [41]. 160 gTFs were found to be associated with 81 genes of the turquoise module. It was found that PSME3 and PTBP1 are the genes, which are regulated by maximum gTFs, 31 and 25 gTFs respectively. Both of these genes were found to be involved in several cancers [42, 43]. PSME3 was found to be involved in Huntington’s Disease [44]. Besides, we found gTFs for hub gene, HSPA1A, MAGED1 and NSF.
two hundred twenty-six genes of the turquoise module were found to be associated with 51 experimentally validated miRNAs. ATP6V1C1, CBFB and PSME3 are the genes, which are regulated by a maximum number of miRNAs i.e. 7, 6 and 6 miRNAs, respectively. These 51 experimentally validated miRNAs were associated with 117 mTFs as obtained from TransmiR database [45]. By combing all these regulatory information, we constructed a mTF-miRNA-gene-gTF regulatory network (Fig. 3) which represents the four layers of complex regulatory interactions taking place within the most significant WGCNA module.

The four layered mTF-miRNA-gene-gTF Regulatory Network of the turquoise module. In this network, blue rectangular nodes represent miRNAs, green circular nodes represent genes, green circular node with black border represents gene that can regulate other genes as TF, diamond shaped magenta nodes represent mTFs, diamond shaped orange nodes represent gTFs, diamond shaped pink nodes with cyan borders represent the common TFs regulating both miRNAs and genes. The Feed-Forward Loop involving hsa-miRNA-375, gene PAFAH1B1 and TF ASH1L is also shown in the network
lncRNA-mediated gene regulatory network
Fifty-one miRNAs that were previously found to be associated with the genes of the turquoise module were further searched in the lncbase module of DIANA tools [40] to acquire information on the miRNA-lncRNA pair. Thirteen out of the 51 miRNAs were found to be PD-specific which were associated with 57 lncRNAs. These 13 miRs control 29 genes of the turquoise module and the 13 miRNAs are in turn controlled by 44 mTF. By combining all these regulatory information, we constructed a regulatory network involving the mTFs, lncRNA and genes (Table 6, Figure not Shown).
SNP analysis of the most significant co-expressed module
Selection of biologically significant SNPs
SNPs corresponding to the 266 genes of the turquoise module were obtained from the online database SCAN [46]. We studied for those SNPs that are present at the 3’UTR of these genes. Using MirSNP database [47] (miRNAs usually bind at the 3’UTR of the target gene and controls the expression of that gene). 1525 miRNAs were found to be associated with these SNPs. Of these 1525 miRNAs, 82 miRNAs were found to be PD related. It was found that 140 SNPs were associated with these 82 miRNAs (p-value ≤10−05) (Additional file 3: Table S3). These 140 SNPs were then searched in the dbSNP database (http://www.ncbi.nlm.nih.gov/snp/) [48] for SNP sequence, chromosome locus, and gene corresponding to each of the SNPs (data not shown). It was found that these 140 SNPs were associated with 157 genes (out of the 266) of the turquoise module.
Discussion
In our study, Co-expression network analyses revealed that out of the six WGCNA modules Turquoise module was the most significant co-expressed module. Enrichment analysis revealed that Parkinson disease pathway (hsa05012) is one of the over-representative pathways associated with this module (Additional file 1: Table S1A). Moreover, Epithelial cell signaling in Helicobacter pylori infection (hsa05120) appeared as one of the most significant KEGG pathways (Additional file 1: Table S1A). Previous studies have already reported that H. pylori infection is associated with PD [49]. Additional file 1: Table S1B depicts the highly Significant GO Biological Processes such as microtubule-based process (GO:0007017), synaptic transmission (GO:0007268), intracellular transport (GO:0046907), etc. associated with the genes of turquoise module.
Co-expression network yielded 11 hub genes based on their topological significance. Out of the 11 hubs, three hubs (NSF, HSPA1A and CYB561) were already found to be associated with PD. The remaining eight novel hub genes were further studied for their association in PD.
Differential expression pattern of the hub genes
Table 7 represents the differential expression pattern of the eight novel hub genes obtained from the co-expression network. Most of the novel hub genes (MAGED1, AP3B2, STXBP1, AF1Q, GASP, C14ORF78, and MAN1C1) were down regulated in PD with respect to control, whereas, one hub gene (HNRPC) was up-regulated in PD with respect to control.
Biological significance of hub genes
We further studied the biological significance of these eight novel hub genes. Table 8 represents the GO biological processes associated with the eight novel hub genes which shows the involvement of these genes in several PD-related processes such as protein transport, neurotransmitter release, synaptic transmission, etc. We found that a recent study has pointed out the role of MAGED1 in the central nervous system in both developmental and adult stages [50]. Studies have found reporting on the vesicle coat protein complex AP3B2 to have some neuron-specific functions such as neurotransmitter release [51, 52]. STXBP1 was found to be listed as an AD-specific marker in Genotator [53], Polysearch [54] and Pescador [55]. AF1Q is a retinoic acid target gene and reported to have an association with ovarian cancer disease [56]. GASP was found as a potential tumor marker for several cancers [57]. C14ORF78 was reported to be associated with calcium channel proteins of cardiomyocytes [58]. MAN1C1 was identified as a differentially expressed gene in PD blood samples. Transcriptome changes related to epigenetic modifications including chromatin remodeling and methylation was also studied for this gene in PD [59]. Protein products of HNRPC gene are associated with pre-mRNA processing and other aspects of mRNA metabolism and transport [60]. All these information validate our finding regarding the association of these genes as hubs in PD.
Epigenetic regulation of hub genes
The epigenetic regulations of hub genes are shown in Tables 4 and 5. We have studied the association of experimentally validated miRNAs and lncRNAs with eight hub genes. It was found that four (STXBP1, AF1Q, C14ORF78, MAN1C1) out of the eight hub genes were regulated by PD-specific miRNAs. Interestingly, AF1Q was identified to be regulated by a maximum number of PD-specific miRNAs (10). It is evident from Tables 5 and 6 that four lncRNAs namely (XIST, n406658, n338391, n410211) are regulating both PD-specific miRNAs and miRNAs not previously known in PD.
We have studied the histone modification patterns of hub genes. Histone modification refers to the posttranslational modifications of the amino-terminal tails of histone proteins which upon modification affect the downstream molecular interactions, hence regulates the gene expression. Interestingly, we found several histone modification sites those are already known to be associated with several neurodegenerative diseases [61] present within these eight hub genes (Table 4).
Identification of feed forward loop from mTF-miRNA-gene-gTF regulatory network
Analysis of regulatory network revealed the presence of an interesting FFL, where a TF regulates a miRNA and they both regulate a target gene (Fig. 3). We found such a FFL between the gene PAFAH1B1, hsa-miR-375 and TF ASH1L. TransmiR data indicated that hsa-miR-375 is activated by TF ASH1L. By combing the TransmiR and TarBase data, we found that ASH1L and hsa-miR-375 both regulate the expression of its target gene PAFAH1B1. Studies have found that ASH1L activates hsa-miR-375 and hsa-miR-375 inhibits its target PAFAH1B1. Interestingly, however, ASH1L has been found to be over-expressed in neuroblastoma cell line transfected with normal or mutated alpha-synuclein [62]. This indicates a possibility of higher expression of this TF in brain tissues of PD patients. Besides, studies have identified the association of miR-375 in gastric cancer, breast cancer, cervical cancer [63,64,65]. A recent study with AD patients has identified higher expression of this miRNA (has-miR-375) in patients than controls [66]. This information provides a link to the finding of upregulation of hsa-miR-375 by the TF ASH1L. It is possible that up-regulation of this miRNA in PD patients is responsible for the aberrant production of downstream target genes involved in the pathogenesis. Moreover, the FFL gene PAFAH1B1 has been listed in Genotator database as a responsible candidate gene in AD. PAFAH1B1 was found to be associated with epilepsy, schizophrenia, neuronal migration disorders, cerebellar hypoplasia, etc. nerve related diseases in GeneCards database (http://www.genecards.org/). Therefore, this can be considered as a validation of our findings in PD. Further study on this novel FFL can help us to understand the molecular biology of PD progression.
Significance of lncRNA-mediated gene regulatory network
This network depicts an interesting functional module where a PD-specific miRNA is being regulated by both mTF (either activation or repression) and lncRNA, and this regulatory information is then conveyed to the gene in terms of post-transcriptional repression. Modes of regulations of 44 mTFs associated with 13 PD-specific miRNAs (out of 51 miRNA of the turquoise module) indicated that most of these interactions were ‘activation’ (Table 6). The regulation of hsa-miR-103a-3p of this network is noteworthy. It is not associated with mTFs but has a maximum number of lncRNAs (14 lncRNAs) associated with it (Table 6). This miRNA represses two genes, namely, KPNA1 and NSF. NSF is known to be involved in PD [53] whereas KPNA1 is known to be involved in several neurological disorders including autism and schizophrenia [67]. In contrast to the above findings, hsa-let-7a-5p and hsa-miR-9-5p each has one identified lncRNA (Table 6). These two miRNAs are in turn repress one gene each namely HRAS and OPTN. These two genes are known to be involved in PD [68, 69]. However, both of the miRNAs are associated with 11 and 12 mTFs respectively (Table 6). Since all the 57 lncRNAs of this regulatory network are associated with known PD-specific miRNAs, they might be important epigenetic regulators in PD that were not identified by previous studies. Moreover, the conservation scores of 57 lncRNAs indicate high conservations that strengthen the association of these lncRNAs with PD (Additional file 4: Table S4).
Final screening and selection of 20 most significant SNPs associated with PD
One hundred forty SNPs were identified from the 157 co-expressed genes of the turquoise module. Out of these 157 genes, 18 genes were already known in PD. 20 SNPs are identified to be associated with 18 genes which in turn controlled by PD-specific miRNAs. This strengthens the association of these 20 SNPs in PD (Table 9). In order to find out the functional role of these 20 SNPs, we further analyzed them in F-SNP database (http://compbio.Cs.Queensu.Ca/F-SNP/) [70]. Table 10 Describes the functional category, allele and region of each SNPs. Interestingly 3 SNPs namely, rs535860, rs3814309 and rs3766286 are found to be classified as the ‘conserved’ functional category (predicted by PhastCons_8way and PhastCons_17way within F-SNP database) signifying a conserved functional role of these variations throughout the evolution. Furthermore, our study identified several SNPs associated with hsa-miR-375 involved in FFL of the regulatory network. Among them, SNP rs193223230 is present in the locus of an already known PD-related gene (YWHAZ) (Table 11). Therefore, hsa-miR-375 can be an important PD epigenetic biomarker in our study.
Conclusion
In this study, we have analyzed gene co-expression network, gene regulatory network, and lncRNA-mediated regulatory network based on a PD microarray dataset. The co-expression network, generated through WGCNA, identified eight novel hub genes based on their topological significance in the network. The biological significance and epigenetic regulations of hub genes indicated their involvement in PD-related processes. Analysis of the gene regulatory network (mTF-miRNA-gene-gTF) resulted in the identification of a novel FFL, the regulators of which are unidentified in PD. The lncRNA-mediated regulatory network provided important insight into the lncRNA-mediated regulation of known PD miRNAs. These lncRNAs might be important epigenetic regulators in PD those were not identified by previous studies. Moreover, 57 lncRNAs obtained from lncRNA-mediated regulatory network indicate high conservations that strengthen the association of these lncRNAs with PD. Four lncRNAs (XIST, n406658, n338391, n410211) were identified to be regulating both PD-specific miRNAs and miRNAs not previously known in PD. Moreover, SNP analysis identified 20 significant SNPs along with their associated genes and regulatory miRNAs. These SNPs can be considered as potential risk factors upon further validation. Out of these 20 SNPs, 3 SNPs, namely rs535860, rs3814309 and rs3766286 have conserved functional role throughout the evolution. Thus, findings of our study will be helpful for further PD clinical research and diagnostic purposes.
Methods
Figure 1 depicts the workflow of our analysis.
Microarray data collection
Microarray data generated by Affymetrix HG_U133 array sets (A and B chips) was downloaded from GEO Dataset Browser for data set GDS3128 and series GSE 8397 (from the link http://www.ncbi.nlm.nih.gov/gds/?term=GDS3128) [71]. The microarray data contains 94 samples (47samples from chipA and 47 samples from ChipB) taken from three brain regions Frontal Cerebral Cortex (FCC), Lateral Substantia Nigra (LSN) and Medial Substantia Niagra (MSN). A total of 15 samples were taken from MSN, 9 from LSN, 5 samples from frontal cerebral cortex. 8 medial nigra control samples and 7 lateral nigra control samples and 3 frontal cerebral cortex control samples were considered. The whole dataset was normalized with GCRMA (Gene Chip Robust Multi-Array Averaging) which performs background correction, probe level intensity calculation and summarization [72].
Analysis of differential gene expression
To identify the most significant DE gene set from the microarray data, we performed both the SAM and t-test analysis. When we performed t-test and SAM, we did not get any differentially expressed genes for frontal cerebral cortex.
SAM
Significance Analysis of Microarray (SAM) [30] was used to identify the differentially expressed (DE) genes that are positively and negatively regulated genes among the control and disease samples. The test statistic of SAM is given by:
Where d i is the relative difference in gene expression, r is the linear regression coefficient of gene i, s i is the standard error of r and s o is a constant chosen to minimize the coefficient of variation of d i . Thus, SAM assigns a score to each gene on the basis of change in gene expression relative to the standard deviation of repeated measurements. In chip A, SAM identified 1518 DE genes at FDR value 0.19%. In chip B, SAM identified 673 DE genes at FDR value 0.11%.
t-test
We further performed paired two sample t-test to identify differentially expressed genes in chip A and chip B. 2-tailed t-test is a measure of the statistical significance of the dataset, in terms of a test statistic t, which is given by:
Where \( \overline{x} \) and \( \overline{y} \) are the sample means, s x and s y are the sample standard deviations, n and m are the sample sizes for two samples, x and y. Under the null hypothesis, this test returns the probability (p-value) of observing a value as extreme or more extreme of the test statistic. Probes corresponding to a portion of the genes showed significant changes in signal intensities in disease sample groups, as compared to the control. These genes were selected as DE genes. t-test analysis identified 4797 and 3120 DE genes in chip A and chip B respectively at p-value 0.05 or 95% confidence level.
Construction of the gene co-expression network
The 458 common DE genes from the chip A were subjected to Weighted Gene co-expression Network Analysis (WGCNA) [73]. This correlation networking method deals with genes differentially expressed over two different conditions (control and disease). In this method, highly correlated nodes are placed into a single module or cluster which are thought to be regulated by the same kind of transcription factors. Therefore, identification of the hub genes of the most significant module can provide insight into the biological significance of that module [74]. Figure 2 describes the gene co-expression network of the most significant co-expressed module (Turquoise module) obtained from WGCNA.
Identification and further analysis of the hub genes
In order to find out the hub nodes, we analyzed the topological properties of the most significant turquoise module using tYNA (http://tyna.gersteinlab.org/) web interface [33]. Degree and Betweenness Centrality (BC) were selected as the criteria for hub gene selection. Nodes with high degree-low BC value (HC nodes) and nodes with high BC-low degree value (HBLC nodes) were considered as hub nodes. Histone modification data for the hub genes were retrieved from human histone modification database (HHMD, http://202.97.205.78/hhmd/index.jsp) [36]. DIANA-Tarbase [37], miRWalk database [38] and TargetScan database [39] were used to study experimentally validated non-coding miRNA-mediated regulation of hub genes. DIANA-LncBase [40] was used to study lncRNAs associated with these miRNAs. The lncRNAs, which are both experimentally validated and computationally predicted (prediction score ≥ 0.70) are considered in our study. The tissue-specific expression data of eight hub genes were collected from GNF Gene Atlas (http://biogps.org/).
Construction of regulatory networks
To get a detailed view of the regulatory pattern of the turquoise module, we built a regulatory network comprising genes of the turquoise module, TFs and miRNAs associated with these genes and TFs associated with the miRNAs. The gene-TF information was obtained from TRANSFAC [41]. Information about the miRNAs associated with the genes of the turquoise module was identified from the DIANA-TarBase database [37]. Information about the TFs regulating the transcription of these miRNAs was obtained from the TransmiR database [45]. By combing all these regulatory information, we constructed a TF-miRNA-gene-TF regulatory network (Fig. 3) which represents the four layers of complex regulatory interactions taking place within the most significant WGCNA module. The network was generated by using Cytoscape software [75].
To identify possible lncRNA-mediated regulation of the miRNAs associated with the genes of the turquoise module, we constructed a lncRNA-mediated regulatory network (Figure not shown). The PD-specific miRNAs that were previously found to be associated with the genes of the turquoise module were searched in the lncbase module of DIANA-LncBase [40] to acquire information on the miRNA-lncRNA pair. This database contains experimentally verified and computationally predicted miRNA targets on lncRNAs. The lncRNAs which are both experimentally validated and computationally predicted (prediction score ≥ 0.70) are considered in our study. In order to identify the regulation of these PD-specific miRNAs present in the turquoise module, we constructed a regulatory network involving the TFs, lncRNA and genes associated with these 13 miRNAs (Figure not shown). The TF-miR-lncRNA-gene regulatory network consisted of 44 TFs, 57 lncRNA, 13 miRNAs and 29 genes of the turquoise module (Table 6). The network was generated by using Cytoscape software [75].
SNP analysis of the highly significant WGCNA module
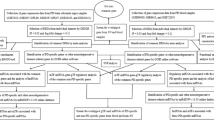
Figure 4 depicts the flowchart for SNP analysis performed in our study. In order to gain insight about the PD-associated SNPs, the 266 genes of the turquoise module were subjected to SNP analysis. SNPs corresponding to these genes were obtained from the online database SCAN (SNP and Copy number ANnotation database; http://www.scandb.org/) [46]. The expression data served in SCAN has been assayed in HapMap (87 CEU and 89 YRI) [76]. CEU represents the human samples of European descent from Utah and YRI represents the Yoruban samples from Ibadan Nigeria. Genes were queried to retrieve information about the relationship between SNPs and genes at user-specified p-value thresholds [77]. We chose the SNPs that predict gene expression with p-values less than ≤10−05 and frequency greater than 0.10. We obtained a huge number of SNPs corresponding to these genes. To identify only the biologically significant SNPs from this huge number of SNPs we sought the SNPs in MirSNP database (http://202.38.126.151/hmdd/mirsnp/search/) [47]. This database identifies SNPs present in the 3′ UTR of miRNA target sites. We obtained 1525 miRNAs corresponding to the SNPs of 266 genes. These 1525 miRNAs were compared with a list of 92 PD-related miRNAs that was obtained through text mining in PubMed and Human MicroRNA Disease Database (HMDD) [78]. We found 82 miRNAs, related to these 92 miRNAs already known in PD. 140 SNPs associated with these 82 miRNAs were considered as the most relevant SNPs in our study, and these were used for further screening.

Flowchart for SNP analysis performed in our study
Abbreviations
- BC:
-
Betweenness centrality
- DE:
-
Differentially expressed
- FFL:
-
Feed Forward Loop
- gTF:
-
gene Transcription Factor
- HBLC:
-
High Betweenness Low Connectivity
- HC:
-
High Connectivity
- lncRNA:
-
Long non-coding RNA
- miRNA:
-
microRNA
- mTF:
-
microRNA Transcription Factor
- PD:
-
Parkinson’s disease
- SAM:
-
Significance Analysis of Microarray
- SNP:
-
Single nucleotide polymorphism
- TF:
-
Transcription factor
- WGCNA:
-
Weighted Gene Coexpression Network
References
Fitzgerald JC, Plun-Favreau H. Emerging pathways in genetic Parkinson’s disease: autosomal-recessive genes in Parkinson’s disease—a common pathway? FEBS J. 2008;275(23):5758–66.
Jankovic J. Parkinson’s Disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry. 2008;79(4):368–76.
Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson's disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology. 2007;27(5):494–506.
Liao XY, Wang WW, Yang ZH, Wang J, Lin H, Wang QS, Wu YX, Liu Y. Microarray analysis of transcriptome of medulla identifies potential biomarkers for Parkinson's disease. Int J Genomics. 2013;2013:606919.
Gao L, Gao H, Zhou H, Xu Y. Gene expression profiling analysis of the putamen for the investigation of compensatory mechanisms in Parkinson's disease. BMC Neurol. 2013;13:181.
Zovoilis A, Agbemenyah HY, Agis-Balboa RC, Stilling RM, Edbauer D, et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011;30:4299–308.
Gaughwin PM, Ciesla M, Lahiri N, Tabrizi SJ, Brundin P, et al. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum Mol Genet. 2011;20:2225–37.
Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, et al. A microRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):1220–4.
Rankin SA, Zorn AM. Gene regulatory networks governing lung specification. J Cell Biochem. 2014;115(8):1343–50.
Lardenoije R, Iatrou A, Kenis G, Kompotis K, Steinbusch HW, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. 2015;131:21–64.
Devall M, Roubroeks J, Mill J, Weedon M, Lunnon K. Epigenetic regulation of mitochondrial function in neurodegenerative disease: new insights from advances in genomic technologies. Neurosci Lett. 2016;625:47–55.
Weinhold B. Epigenetics: the science of change. Environ Health Perspect. 2006;114(3):A160–7.
Peschansky VJ, Wahlestedt C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics. 2014;9(1):3–12.
Wen KX, Milic J, El-khodor B, Dhana K, Nano J, et al. The role of DNA methylation and histone modifications in neurodegenerative diseases: a systematic review. PLoS One. 2016;11(12):e0167201.
Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466(7306):637–41.
Martins M, Rosa A, Guedes LC, Fonseca BV, Gotovac K, et al. Convergence of miRNA expression profiling, α-synuclein interaction and GWAS in Parkinson’s disease. PLoS One. 2011;6:e25443.
Guttman M, Amit I, Garber M, French C, Lin MF, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7.
Das S, Ghosal S, Sen R, Chakrabarti J. lnCeDB: database of human long noncoding RNA acting as competing endogenous RNA. PLoS One. 2014;9(6):e98965.
Wu P, Zuo X, Deng H, Liu X, Liu L, Ji A. Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res Bull. 2013;97:69–80.
Johnson R. Long non-coding RNAs in Huntington's disease neurodegeneration. Neurobiol Dis. 2012;46(2):245–54.
Wang S, Zhang X, Guo Y, Rong H, Liu T. The long noncoding RNA HOTAIR promotes Parkinson’s disease by upregulating LRRK2 expression. Oncotarget. 2017;8(15):24449–56.
Brookes AJ. The essence of SNPs. Gene. 1999;234:177–86.
Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7.
Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, et al. Characterization of single nucleotide polymorphisms in coding regions of human genes. Nature Genet. 1999;22:231–8.
Schork NJ, Fallin D, Lanchbury JS. Single nucleotide polymorphisms and the future of genetic epidemiology. Clin Genet. 2000;58:250–64.
Huang GL, Lu Y, Pu XX, He YX, Chen ML, et al. Association study between miR-149 gene polymorphism and nasopharyngeal carcinoma. Biomed Rep. 2013;1(4):599–603.
Guo Z, Wang H, Li Y, Li B, Li C, et al. A microRNA-related single nucleotide polymorphism of the XPO5 gene is associated with survival of small cell lung cancer patients. Biomed Rep. 2013;1(4):545–8.
Chen ML, Lin CH, Lee MJ, Wu RM. BST1 rs11724635 Interacts with environmental factors to increase the risk of Parkinson's disease in a Taiwanese population. Parkinsonism Relat Disord. 2014;20(3):280–3.
Liu Y, Ng M. Shrunken methodology to genome wide SNPs selection and construction of SNPs networks. BMC Syst Biol. 2010;4(Suppl 2):S5.
Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21.
Hosack DA, Dennis G, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4(6):4.
Al-Shahrour F, Minguez P, Tarraga J, Medina I, Alloza E, et al. FatiGO + : a functional profiling tool for genomic data. Integration of functional annotation, regulatory motifs and interaction data with microarray experiments. Nucleic Acids Res. 2007;35:W91–6.
Yip KY, Yu H, Kim PM, Schultz M, Gerstein M. The tYNA platform for comparative interactomics: a web tool for managing, comparing and mining multiple networks. Bioinformatics. 2006;22(23):2968–70.
Barabási AL, Albert R. Emergence of scaling in random networks. Science. 1999;286(5439):509–12.
Barabási AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101–13.
Zhang Y, Lv J, Liu H, Zhu J, Su J, et al. HHMD: the human histone modification database. Nucleic Acids Res. 2010;38(Database issue):D149–54.
Vergoulis T, Vlachos IS, Alexiou P, Georgakilas G, Maragkakis M, et al. Tarbase 6.0: Capturing the exponential growth of miRNA targets with experimental support. Nucl. Acids Res. 2012;40(D1):D222–9.
Dweep H, Sticht C, Pandey P, Gretz N. miRWalk - database: prediction of possible miRNA binding sites by "walking" the genes of 3 genomes. J Biomed Inform. 2011;44:839–7.
Agarwal V, Bell GW, Nam J, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. elife. 2015;4:e05005.
Paraskevopoulou MD, Georgakilas G, Kostoulas N, Reczko M, Maragkakis M, et al. DIANA-LncBase: experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013;41(Database issue):D239–45.
Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nuc Ac Res. 2006;34(Database issue):D108–10.
Roessler M, Rollinger W, Mantovani-Endl L, Hagmann ML, Palme S, et al. Identification of PSME3 as a novel serum tumor marker for colorectal cancer by combining two-dimensional polyacrylamide gel electrophoresis with a strictly mass spectrometry-based approach for data analysis. Mol Cell Proteomics. 2006;5(11):2092–101.
He X, Arslan AD, Ho TT, Yuan C, Stampfer MR, et al. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogene. 2014;3:e84.
Seo H, Sonntag KC, Kim W, Cattaneo E, Isacson O. Proteasome activator enhances survival of Huntington's disease neuronal model cells. PLoS One. 2007;2(2):e238.
Wang J, Lu M, Qiu C, Cui Q. TransmiR : a transcription factor-microRNA regulation database. Nucleic Acids Res. 2009;38:D119–22.
Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner E, et al. SCAN: SNP and copy number annotation. Bioinformatics Advance Access. 2009; doi:10.1093/bioinformatics/btp644 PMID 19933162.
Liu C, Zhang F, Li T, Lu M, Wang L, et al. (2012) MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPsand eQTLs. BMC Genomics. 2012;13:661.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11.
Suwarnalata G, Tan AH, Isa H, Gudimella R, Anwar A, et al. Augmentation of autoantibodies by helicobacter pylori in Parkinson's disease patients may be linked to greater severity. PLoS One. 2016;11(4):e0153725.
Mouri A, Noda Y, Watanabe K, Nabeshima T. The roles of MAGE-D1 in the neuronal functions and pathology of the central nervous system. Rev Neurosci. 2013;24(1):61–70.
Faúndez V, Horng JT, Kelly RB. A function for the AP3 coat complex in synaptic vesicle formation from endosomes. Cell. 1998;93(3):423–32.
Grabner CP, Price SD, Lysakowski A, Cahill AL, Fox AP. Regulation of large dense-core vesicle volume and neurotransmitter content mediated by adaptor protein 3. Proc Natl Acad Sci U S A. 2006;103(26):10035–40.
Wall DP, Pivovarov R, Tong M, Jung J-Y, Fusaro VA, et al. Genotator: a disease-agnostic tool for genetic annotation of disease. BMC Med Genet. 2010;3:50.
Cheng D, Knox C, Young N, Stothard P, Damaraju S, et al. PolySearch: a web-based text mining system for extracting relationships between human diseases, genes, mutations, drugs and metabolites. Nucleic Acids Research. 2008;36(Web Server issue):W399–405.
Barbosa-Silva A, Fontaine JF, Donnard ER, Stussi F, Ortega JM, et al. PESCADOR, a web-based tool to assist text-mining of biointeractions extracted from PubMed queries. BMC Bioinformatics. 2011;12:435.
Tiberio P, Cavadini E, Callari M, Daidone MG, Appierto V. AF1q: A novel mediator of basal and 4-HPR-induced apoptosis in ovarian cancer cells. PLoS One. 2012;7(6):e39968.
Zheng X, Chang F, Zhang X, Rothman VL, Tuszynski GP. G-protein coupled receptor-associated sorting protein 1 (GASP-1), a ubiquitous tumor marker. Exp Mol Pathol. 2012;93(1):111–5.
Komuro A, Masuda Y, Kobayashi K, Babbitt R, Gunel M, et al. The AHNAKs are a class of giant propeller-like proteins that associate with calcium channel proteins of cardiomyocytes and other cells. Proc Natl Acad Sci U S A. 2004;101(12):4053–8.
Calligaris R, Banica M, Roncaglia P, Robotti E, Finaurini S, et al. Blood transcriptomics of drug-naïve sporadic Parkinson’s disease patients. BMC Genomics. 2015;16:876.
McCloskey A, Taniguchi I, Shinmyozu K, Ohno M. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science. 2012;335(6076):1643–6.
Pattaroni C, Jacob C. Histone methylation in the nervous system: functions and dysfunctions. Mol Neurobiol. 2013;47(2):740–56.
Baptista MJ, O'Farrell C, Daya S, Ahmad R, Miller DW, et al. co-ordinate transcriptional regulation of dopamine synthesis genes by alpha-synuclein in human neuroblastomacell lines. J Neurochem. 2003;85(4):957–68.
Zhang X, Yan Z, Zhang J, Gong L, Li W, et al. Combination of hsa-miR-375 and hsa-miR-142-5p as a predictor for recurrence risk in gastric cancer patients following surgical resection. Ann Oncol. 2011;22(10):2257–66.
Giricz O, Reynolds PA, Ramnauth A, Liu C, Wang T, et al. Hsa-miR-375 is differentially expressed during breast lobular neoplasia and promotes loss of mammary acinar polarity. J Pathol. 2012;226(1):108–19.
Bierkens M, Krijgsman O, Wilting SM, Bosch L, Jaspers A, et al. Focal aberrations indicate EYA2 and hsa-miR-375 as oncogene and tumor suppressor in cervical carcinogenesis. Genes Chromosomes Cancer. 2013;52(1):56–68.
Villa C, Ridolfi E, Fenoglio C, Ghezzi L, Vimercati R, et al. Expression of the transcription factor Sp1 and its regulatory hsa-miR-29b in peripheral blood mononuclear cells from patients with Alzheimer's disease. J Alzheimers Dis. 2013;35(3):487–94.
Jouan L, Girard SL, Dobrzeniecka S, Ambalavanan A, Krebs MO, et al. Investigation of rare variants in LRP1, KPNA1, ALS2CL and ZNF480 genes in schizophrenia patients reflects genetic heterogeneity of the disease. Behav Brain Funct. 2013;9:9.
Tanaka H, Ishikawa A, Ginns EI, Miyatake T, Tsuji S. Linkage analysis of juvenile parkinsonism to tyrosine hydroxylase gene locus on chromosome 11. Neurology. 1991;41(5):719–22.
Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, et al. Ubiquitin independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126(Pt 2):580–92.
Lee PH, Shatkay H. F- SNP: computationally predicted functional SNPs for disease association studies. Nucleic Acids Res. 2008;36(Database issue):D820–4.
Moran LB, Duke DC, Deprez M, Dexter DT, Pearce RK, et al. Whole genome expression profiling of the medial and lateral substantia nigra in Parkinson's disease. Neurogenetics. 2006;7(1):1–11.
Wu Z, Irizarry RA, Gentleman R, Martinez Murillo F, Spencer F. A model based background Adjustement for oligonucleotide expression arrays. J Amer Stat Assoc. 2003;99:909–17.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Clarke C, Madden SF, Doolan P, Aherne ST, Joyce H, et al. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis. 2013;34(10):2300–8.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Gibbs RA, Belmont JW, Hardenbol TD, Willis P, Yu FL, et al. The international HapMap project. Nature. 2003;426:789–96.
Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6(4):e1000888.
Lu M, Zhang Q, Deng M, Miao J, Guo Y, et al. An analysis of human MicroRNA and disease associations. PLoS One. 2008;3(10):e3420.
Acknowledgements
The authors would like to thank the Department of Biophysics, Bose Institute, IIEST, Shibpur and the Indian Statistical Institute (ISI) for their co-operation and support.
Funding
The authors have no funding to report.
Availability of data and materials
All the data supporting the findings of this study are contained in the manuscript and the corresponding supplementary files.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: DR. Performed the experiments: PC. Analyzed the data: PC, DR, MB, SB. Wrote the paper: PC, DR, MB, SB. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors have declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
FatiGO analysis of the turquoise module. S1A - Highly Significant KEGG pathways associated with the turquoise module. S1B - Highly Significant GO Biological Process associated with the turquoise module. (DOCX 12 kb)
Additional file 2: Table S2.
Topological properties of the hub genes obtained from the turquoise module. (DOCX 11 kb)
Additional file 3: Table S3.
140 PD-related SNPs identified in our study with their associated chromosome number and PD-related miRNAs. (XLSX 20 kb)
Additional file 4: Table S4.
List of lncRNAs associated with the PD-specific miRNAs with their binding type, transcript position, conservation, and MRE sequence. (XLSX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chatterjee, P., Roy, D., Bhattacharyya, M. et al. Biological networks in Parkinson’s disease: an insight into the epigenetic mechanisms associated with this disease. BMC Genomics 18, 721 (2017). https://doi.org/10.1186/s12864-017-4098-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4098-3