
Abstract
Background
Genome divergence by mobile elements activity and recombination is a continuous process that plays a key role in the evolution of species. Nevertheless, knowledge on retrotransposon-related variability among species belonging to the same genus is still limited. Considering the importance of the genus Helianthus, a model system for studying the ecological genetics of speciation and adaptation, we performed a comparative analysis of the repetitive genome fraction across ten species and one subspecies of sunflower, focusing on long terminal repeat retrotransposons at superfamily, lineage and sublineage levels.
Results
After determining the relative genome size of each species, genomic DNA was isolated and subjected to Illumina sequencing. Then, different assembling and clustering approaches allowed exploring the repetitive component of all genomes. On average, repetitive DNA in Helianthus species represented more than 75% of the genome, being composed mostly by long terminal repeat retrotransposons. Also, the prevalence of Gypsy over Copia superfamily was observed and, among lineages, Chromovirus was by far the most represented. Although nearly all the same sublineages are present in all species, we found considerable variability in the abundance of diverse retrotransposon lineages and sublineages, especially between annual and perennial species.
Conclusions
This large variability should indicate that different events of amplification or loss related to these elements occurred following species separation and should have been involved in species differentiation. Our data allowed us inferring on the extent of interspecific repetitive DNA variation related to LTR-RE abundance, investigating the relationship between changes of LTR-RE abundance and the evolution of the genus, and determining the degree of coevolution of different LTR-RE lineages or sublineages between and within species. Moreover, the data suggested that LTR-RE abundance in a species was affected by the annual or perennial habit of that species.
Similar content being viewed by others

Background
Transposable elements (TEs) play a key role in the evolution of species [1]. They can drive rapid genome remodelling by creating chromosomal rearrangements and new regulatory gene networks, acting as an endogenous force that promotes reproductive isolation [2]. Moreover, the mutagenic action of TEs creates substantial genetic variability [3], novel functions by fine-tuning gene activity [4, 5], and they are also a major driver of genome size evolution [6,7,8].
Although many kinds of TEs have been identified, they generally fall into two major classes based on their transposition mechanisms [9]. Retrotransposons, or Class I elements, transpose by an RNA intermediate which is reverse transcribed from the genomic copy and integrated in a new position elsewhere in the genome [10]. DNA transposons, or Class II elements, can move using an enzymatic method for excision from the chromosome and reintegration in a new location [9].
Retrotransposons (REs) are the most common class of elements, making up the bulk of many genomes [2, 11]. They can be classified into five taxonomic orders [9], among which long terminal repeat (LTR) REs and non-LTR-REs differ in the mechanism of integration. The size of LTR-REs varies from a few hundred base pairs to over 10 kb, in which two identical LTRs side a region containing open reading frames (ORFs) for Gag and for Pol. Pol encodes a polyprotein with protease, reverse transcriptase (RT), RNaseH, and integrase enzyme domains, which are necessary for the replication and the integration of the elements in the host chromosomes [10].
LTR-REs are the most abundant order in plants, especially those belonging to the Copia and Gypsy superfamilies [9] which differ in the position of the integrase domain within the ORFs [10]. Copia and Gypsy superfamilies can be subdivided into several major evolutionary lineages [12, 13]. The main Copia lineages are: AleI/Retrofit/Hopscotch, AleII, Angela, Bianca, Ivana/Oryco, TAR/Tork, and Maximus/SIRE [12]. On the other hand, the most frequent Gypsy lineages are: Ogre/Tat [14], Athila [15], and Chromovirus, a lineage which is especially abundant in centromeric regions and carries a chromodomain at the 5′-end of the coding portion [13, 16]. In certain species, four sublineages (Galadriel, Tekay, CR, and Reina) of Chromoviruses have been distinguished according to the positions of the chromodomain and the polypurine tract, and to the LTR length [17].
Studies on the impact of RE proliferation and loss on genome structure and evolution of plant species have been performed especially in species with small- or medium-sized genomes. In angiosperms, large sized genomes have been studied especially in monocotyledonous species such as maize (2.3 gigabase pairs, [18]) and barley (5.1 gigabase pairs, [19]). For this reason, we decided to study genome size, structure and evolution in a dicotyledonous genus with a large genome, such as Helianthus (for example, H. annuus has a genome size of 3.3 Gbp, [20]).
This genus, which belongs to the Asteraceae family, includes 49 outcrossing species from different habitats and with a remarkable level of variability [21], including differences in phenotypic traits such as reproductive timing, branching patterns, height [22, 23] and especially habitat preferences. The study of Timme et al. [24] provided evidence for multiple, independent hybrid speciation and/or polyploidy events subdividing sunflowers into four different sections: a monophyletic annual section Helianthus, a polyphyletic section Ciliares and the monotypic section Agrestis, all of which were encompassed by a large polyphyletic section, Divaricati.
It has been evident for more than a decade that the sunflower (H. annuus) genome contains many thousands of TEs [25,26,27,28,29]. In particular, the repetitive fraction of the sunflower genome contains more than 81% TEs, 77% of which are LTR-REs [29]. Among LTR-REs, elements belonging to the Gypsy superfamily are 2.3 times more represented than those belonging to the Copia superfamily [27]. Furthermore, massive transposition of Gypsy-like LTR-REs is supposed to have driven a rapid speciation (in less than 60 generations in one case) of three species of the Helianthus section (H. anomalus, H. deserticola and H. paradoxus), derived from the same two parental species (H. annuus and H. petiolaris). The genomes of these hybrid taxa are 50% larger than the genome of parental lines as the result of bursts of transposition. Further analyses of these Helianthus species have shown that RE proliferation has occurred even in relatively recent events [30].
Although many data are available on Helianthus evolution, massively parallel sequencing techniques are providing new possibilities to investigate genome structure and its impact on speciation. The use of these technologies within a computational framework led to the identification of a so called “metagenome” of repetitive elements of the species analysed, allowing us to address many facets of the dynamics of changes of the genomic repetitive component within the largely unexplored genus Helianthus. These include: i) establishing the extent of intrageneric repetitive DNA variation, especially considering LTR-REs, at superfamily, lineage and sublineage levels; ii) studying the relationship between changes of LTR-RE abundance and the evolution of a genus; iii) investigating whether there is a relation between annual or perennial habits of species and LTR-RE abundance; iv) checking whether different LTR-RE lineages or sublineages have coevolved; v) studying variations in the dynamics (amplification, loss, proliferation dating) of specific LTR-REs among species.
Results
Genome characterization of Helianthus species
In order to classify repetitive sequences and identify their homologous groups in individual genomes, 10 species and one subspecies out of 49 Helianthus species were selected (see Table 1). Of these, two species belong to the section Helianthus (H. annuus and H. petiolaris, with two subspecies: H. petiolaris ssp. petiolaris, and H. petiolaris ssp. fallax), one represents the monotypic annual section Agrestis (H. agrestis), another annual species (H. porteri) and six perennial species belong to the section Divaricati (according to [24]). Concerning the six perennial species of the section Divaricati, two diploid species (H. divaricatus and H. giganteus), three tetraploid species (H. hirsutus, H. californicus and H. laevigatus) and one hexaploid species (H. tuberosus) were selected.
To achieve our task, we sequenced genomic DNA from one individual for each species, treating it as a “type” representative of the species. Concerning H. annuus, previous studies documented high variability of the repetitive component between wild and cultivated genotypes [31]. Since the present analysis has focused on wild species of the genus Helianthus, a wild accession from Illinois was chosen to represent H. annuus; this particular accession exhibits average features among wild H. annuus genotypes [31].
Since genome size was not available for some species (H. porteri, H. californicus, H. hirsutus and H. laevigatus), we measured genome size of all species to obtain comparable values. Genome size was evaluated cytophotometrically, measuring the absorption of prophase nuclei (which have a 4C-DNA content) of root apices after Feulgen staining. Moreover, for one species, chromosome number was not precisely ascertained. Since rare diploid forms were reported for tetraploid H. hirsutus [32], we decided to check the chromosome number of our materials. All chromosome numbers were in agreement with previous works (reviewed in [32]). The H. hirsutus accession used in our experiments resulted tetraploid, as expected. Table 2 reports the chromosome number and the relative genome size of the analysed species.
Interestingly, diploid H. agrestis showed a 4C-DNA absorption almost two times higher than the largest value measured for a diploid species (H. giganteus), similarly to what has been previously reported [33]. Note that the tetraploid species H. californicus and H. laevigatus had larger genome sizes than expected, based on genome sizes of diploid species (ranging from 23.2 to 34.2 arbitrary units). On the contrary, the hexaploid H. tuberosus had a 4C-value smaller than expected, in agreement with other data in the literature [34, 35].
The repetitive component of each species was then characterized by applying the RepeatExplorer pipeline [36], using a number of reads proportional to the ploidy level of each species.
In Table 3, we report the number of clustered sequence reads, i.e. the repetitive DNA, for each species and their corresponding percentages within genomes (ranging from 73.6 to 84.2%). Helianthus agrestis and H. porteri showed the highest percentage of repeated sequences, H. petiolaris spp. fallax and H. californicus the lowest.
Composition of Helianthus repetitive fraction
The “metagenome” structure of the analysed pool of species is reported in Fig. 1 and Table 4, based on the genomic proportion of the 338 hybrid clusters, each representing >0.01% of the analysed reads. The LTR-RE-related clusters composed the bulk of highly and moderately repeated sequences in the Helianthus genomes, as previously reported for H. annuus [27, 29]. The DNA transposons and non-LTR-REs were under-represented; unannotated repeats accounted for 3.0% of the genome. It is also presumable that other repeat remnants could be found among low-repeated/single sequences.

The repeat class distribution of the 338 top (most frequent) clusters obtained performing a hybrid clustering with a random set of reads of sunflowers (proportionally to species ploidy level) using RepeatExplorer. The percentage of reads included in repeat class is reported inside brackets
Table 4 reports the classification of 248 clusters annotated as LTR-REs. We found 56 clusters for the Copia superfamily (11.48% of analysed reads) and 164 clusters for the Gypsy superfamily (45.65% of analysed reads).
Focusing on LTR-REs, these elements were characterized at the lineage level (Table 4): five lineages were identified among Copia retrotransposons (AleII, Angela, Bianca, Maximus/SIRE, and TAR/Tork) and three lineages were identified among Gypsy elements (Chromovirus, Ogre/Tat, and Athila).
The genome proportions of the different RE lineages in the selected species are reported in Table 5. Cluster-related repeats annotated as LTR-REs ranged from 58.36% of the genome in H. petiolaris ssp. fallax to 74.99% in H. agrestis. Gypsy LTR-REs ranged from 33.05% in H. porteri to 57.03% in H. agrestis, and they were overrepresented compared to Copia elements, whose percentages ranged between 8.31% in H. divaricatus and 22.79% in H. porteri.
The ratio between the genomic proportions of Gypsy and Copia elements differed among species, from 5.91 in H. divaricatus to 1.45 in H. porteri. Interestingly, species of the section Helianthus showed a ratio ranging from 3.23 to 3.66, i.e. lower than that of perennial species (from 3.84 in H. hirsutus to 5.91 in H. divaricatus); the monophyletic section H. agrestis and H. porteri showedthe extreme ratios (5.37 and 1.45, respectively). The H. porteri low ratio (1.45) is peculiar in the Helianthus genus, in which Gypsy elements are typically reported to be much more abundant than Copia ones.
The maximum percentage variation of genome proportion of each LTR-RE superfamily or lineage among the 10 species and one subspecies of sunflowers gave us an estimation of genome proportion variability of Gypsy and Copia elements within the genus Helianthus. Such variability was large for each superfamily, and it was especially larger for Copia (63.53%) than for Gypsy (42.04%) and unknown elements (27.59%).
Among Copia REs, only Maximus/SIRE elements showed an average genome proportion higher than 1%. In contrast, Bianca REs were barely represented. Each Gypsy lineage accounted on average for more than 3% of the genome, with Chromoviruses exceeding or being around 30% in each species, excluding H. porteri (25.25%); in H. agrestis, Chromoviruses accounted for about 50% of the genome.
The abundance of each cluster in the different Helianthus species as determined using RepeatExplorer was confirmed by mapping Illumina reads on the contigs belonging to each cluster and counting the number of mapped reads (Additional file 1: Figure S1).
LTR-retrotransposons and Helianthus phylogeny
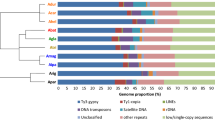
The results of hierarchical clustering of all genome proportions data concerning LTR-RE Copia and Gypsy related clusters were compared with a phylogeny obtained with rDNA ETS sequences [24] (Fig. 2). The dendrogram (Fig. 2a) highlights a division within the genus Helianthus, supporting separation among the three different sections analysed, with perennial species of the Divaricati section and species of the Helianthus section occupying close branches of the tree and H. agrestis and H. porteri being more distant.

a Dendrogram obtained by a hierarchical clustering analysis based on genome proportion data of Copia- and Gypsy-related clusters (as obtained by hybrid clustering using RepeatExplorer) of different Helianthus species. Asterisks indicate multiscale bootstrap resampling (only values >60% are given). The bar represents the genetic distance. b Maximum composite likelihood phylogram of the same Helianthus species based on ETS sequences isolated by Timme et al. (2007). Asterisks indicate multiscale bootstrap resampling (only values >60% are given). The bar represents the genetic distance. Colours indicate the different analysed sections: pink for Divaricati, light blue for Helianthus and green for Agrestis section. Acronyms as defined in Table 1
The two trees showed similar topologies, and the few differences observed concerned the Divaricati species. In fact, separation into distinct clades corresponding to the Divaricati subsections, as previously established [24], is not supported using LTR-RE genome proportion values (Fig. 2a). Furthermore, the occurrence of three distinct clades was more evident using LTR-RE genome proportion values than ETS sequences, suggesting that changes of the repetitive component have accentuated the differences among species.
Interestingly, both trees indicate a clear separation between annual and perennial species, with the annual H. porteri being separated from the other Divaricati species (which are perennial) and closer to the annual H. agrestis.
Furthermore, principal component analysis of genome proportion of the most abundant Copia and Gypsy lineages, Maximus/SIRE and Chromovirus, showed a significant (p < 0.05) separation between annual and perennial species (Fig. 3).

PCA plots of genome proportion values for Gypsy and Copia lineages with a percentage of reads >1% in annual (black dots) and perennial (grey dots) species. The percentage of variation accounted is reported on each axis. Asterisks mark permutational MANOVA significance with the following significance codes: 0.001 ‘***’ 0.05 ‘*’
LTR-retrotransposon dynamics during Helianthus evolution
To gain insight into the evolution of LTR-REs within the genus Helianthus, amino acid sequences of the RT domain were isolated from individual clustering analysis and aligned to produce distance trees that allowed us to evaluate the relationship among LTR-RE lineages for Gypsy and Copia superfamilies. Both trees showed a clear-cut separation of RT-encoding sequences according to their lineage (see Additional file 2: Figure S2).
The comparative timing of LTR-RE proliferation was inferred analysing sequence conservation, by mapping Illumina reads to the DNA sequences encoding the RT domains of LTR-RE-related clusters at different stringency conditions (Fig. 4). The more a sequence is conserved, the more recent should be the proliferation of the related element. Overall, results showed that Copia RT domains of H. agrestis and H. californicus are the most divergent and those of H. porteri are the most conserved ones. In contrast, Gypsy elements showed similar sequence conservation among species, with the exception of H. porteri, whose RT domains were highly divergent.

The previously reported comparative analysis (Table 5) also allowed us to infer both the evolutionary trend of each LTR-RE lineage and relationships among the species of the genus Helianthus. Separated clusters belonging to the same lineage presumably represent different sublineages according to their sequence similarity. Through hierarchical clustering analysis of LTR-RE clusters, based on the genomic proportion of each cluster, we identified and quantified groups of homologous clusters sharing similar abundance levels between the species [37].
The genome proportion of homologous clusters belonging to Gypsy superfamily, in the 10 species and one subspecies analysed, is reported in Fig. 5 (for the Copia superfamily see Additional file 3: Figure S3). Clusters were in turn grouped according to their abundance among the 11 genotypes: each group represents clusters showing a similar pattern of abundance.

Sequence composition of the LTR-Gypsy-RE-related clusters. The size of the rectangle is proportional to the genome proportion of a cluster for each species (acronyms as in Table 1). Bar plot in the top row shows the size of the clusters as number of reads in the comparative analysis. Upper lines label groups of clusters as assessed by a hierarchical clustering of the results. The percentage of reads included in the group is shown in parentheses. The colour of the rectangles corresponds to the lineage of the Gypsy LTR-RE
Gypsy-related clusters can be subdivided into 20 groups, representing from 0.02 to 6.80% of the genome (Fig. 5). All but one sublineage of Gypsy LTR-REs were represented in all species; this sublineage, which belongs to group 12, was abundant in the genome of all species and absent in H. porteri and in H. agrestis. It is evident from Fig. 5 that Gypsy sublineages showed different patterns among the 10 species and one subspecies, indicating that each group has experienced a different evolutionary dynamics. For instance, group 2 clusters (belonging to Chromovirus lineage), the most abundant in terms of genome proportions counting 6.80% of analysed reads, were especially represented in H. agrestis. Similarly, group 3, which had only one Chromovirus-related cluster, was highly specific for this species, being barely represented in the others. On the other hand, group 10, a group made of Chromovirus sublineages, which was the second most prominent group with regard to genome proportion, was overrepresented in perennial species compared to annuals.
In the absence of whole genome sequences of each species, we compared the tendency to produce solo-LTRs among the selected species by measuring the ratio between the average coverage of the LTR and the respective RE-coding portion of 41 full-length LTR-REs isolated from available genome scaffolds of H. annuus (Additional file 4: Table S1).
If all elements of a RE sublineage were intact, the average coverage of the 5′-LTR should be two-fold that of the respective inter-LTR DNA sequence. Ratios >2 should indicate the occurrence of solo-LTRs of that RE sublineage. On average, these ratios ranged from 0.0003 to 4.10, with only 3 out of 41 REs showing a ratio > 2, i.e. unequal homologous recombination should not be very common in sunflowers.
In Fig. 6, the distribution of ratios between average coverage of LTR and inter-LTR region of 41 REs in the species and subspecies is reported, keeping separated diploid, tetraploid and hexaploid species. Considering diploid species, significant differences occurred, indicating that some species should have accumulated more solo-LTRs than other. Interestingly, the highest values were measured in the species with the smallest genomes, suggesting that unequal homologous recombination occurred, leading to a reduction of genome size (Fig. 6). The negative correlation between LTR/inter-LTR average coverage ratio and genome size was significant for Gypsy elements (see Additional file 5: Figure S4).

Distribution (on a logarithmic scale) of the ratio between the average coverage of 5′-LTR and respective coding (inter-LTR) DNA sequence related to the 41 isolated REs grouped per species. Species are distributed by increasing genome size keeping separated ploidy levels. Diploid species are in red, tetraploid in blue and hexaploid in green. The boxes represent the 25–75%, whiskers the whole range of values and dots the outliers. The lines in the boxes represent the medians of the distributions. Within diploid or tetraploid species, those indicated by different letters are significantly different (p < 0.05) according to Tukey’s test
Genome doubling without polyploidization: H. agrestis
Relative 4C DNA absorption analysis (Table 2) indicated that H. agrestis genome size was almost two-fold larger than expected in a diploid species. The expansion of its genome is supported, at least in part, by the huge genome proportion of Gypsy-Chromovirus-related clusters (Table 5) compared to the other analysed species.
The involvement of Chromoviruses in the genome expansion of H. agrestis was confirmed by dot-blot hybridization experiments using a Chromovirus sequence isolated from H. annuus DNA as probe. The results of hybridization are reported in Fig. 7. It can be observed that the hybridization intensity in H. agrestis is more than two-fold that of H. annuus and H. divaricatus. The copy number of the analysed sequence in H. annuus amounted to 1600 per haploid genome, while in H. agrestis, it amounted to 4700. Considering that the probe used in this experiment was heterologous to H. agrestis, it can be hypothesized that the copy number in this species is even underestimated.

Histograms obtained by the densitometric scanning of slot blots of genomic DNAs of H. agrestis (AGR), H. annuus (ANN) and H. divaricatus (DIV), hybridized with a H. annuus probe, consisting in a DNA fragment belonging to a Gypsy-Chromovirus element. Each value is the mean (± SE) of three replicates
A neighbour-joining phylogenetic tree based on multiple alignment of sequences of Chromovirus-RT amino acid sequences of H. agrestis and RT amino acid sequences of Chromoviruses of several species (i.e. Beta vulgaris, Zea mays, Vitis vinifera) was performed to define the specific Chromovirus clade(s) to which the largely abundant Chromoviruses of H. agrestis belonged. The tree indicated that most sequences of H. agrestis belonged to Tekay-related elements, while Galadriel and other clades were barely represented (data not shown).
Finally, Illumina reads matching a Chromovirus RT-encoding domain of H. agrestis, H. annuus and H. divaricatus were pairwise compared and their distances [38] were converted into insertion dates according to SanMiguel et al. [39] and Piegu et al. [8], but using a mutation rate of 2 × 1 0^-8, i.e. 2-fold that calculated for sunflower gene sequences, to keep into account that mutation rate of retrotransposons is higher than that of genes, as it depends on error-prone action of reverse transcriptase during element retrotransposition besides on mutations occurring across generations. This analysis enabled the identification of one retrotranspositional wave, mostly overlapping in terms of time in the three species (Fig. 8). Although timing data should be taken cautiously, the proliferation burst should have started 10 MYA and reached its apex around 6-6.5 MYA. Chromovirus amplification has nearly ceased in H. agrestis and H. divaricatus, while an additional recent and smaller amplification peak was observed in H. annuus (Fig. 8). Because of the much larger proportion of Chromoviruses observed in H. agrestis than in the other two species (Table 5), it can be deduced that amplification of these elements occurred concurrently in the three analysed species, with large differences in the amplification rate, which was much higher in H. agrestis than in H. annuus and H. divaricatus.

Timing of the Chromovirus retrotranspositional activity in H. agrestis (AGR), H. annuus (ANN) and H. divaricatus (DIV). The y axis shows the percentage of the total pairwise comparisons of reads matching the Chromovirus RT domain in the three species
Discussion
Interspecific variation of LTR-RE abundance
The first goal of our research was to establish the extent of interspecific repetitive DNA variation related to LTR-RE abundance. Analysis of the genome size showed considerable variations among species, even in those with the same chromosome number. Conversely, comparative clustering of Illumina reads among species provided information about an “average” composition of this genus. Repetitive DNA represented 77.5% of this “metagenome”, similar to that already reported for Helianthus annuus [40]. Among species, the repetitive DNA ranged from 73.6% of the genome in H. petiolaris ssp. fallax to 84.2% in H. agrestis, i.e. it showed relative uniformity. Genome structure was also similar among the analysed species, with LTR-REs representing the vast majority of repetitive sequences.
Genome size variations in species with the same chromosome number are usually attributed to variations in the abundance of repetitive DNA [41]. Concerning LTR-REs, differences in abundance may be derived from massive amplification through retrotransposition or from DNA loss by unequal homologous recombination, which produced solo-LTRs [2].
As a result of the amplification burst(s) that may have occurred, our data on RT sequence conservation evidence that, for some elements, RE amplification have occurred in different times for different species. For instance, in H. porteri, the great abundance and the low sequence variability of Copia elements should imply that amplification of these REs has occurred in a more recent past than for the other species. On the contrary, the low number and large sequence variability of Gypsy elements indicates that, in H. porteri, proliferation of such elements ceased earlier than in the other species.
On the other hand, although indicating that solo LTRs (generally produced by unequal homologous recombination) are not common in sunflowers, differences in the extent of DNA removal can be inferred by our data. In fact, the ratio between the average coverage of the LTR and the respective RE-coding portion of 41 full-length LTR-REs suggest that diploid species of the Helianthus section show a higher frequency of putative solo-LTRs and hence, presumably, a stronger tendency to unequal homologous recombination compared to diploid Divaricati species, which contributed to the reduced genome size of the former species than of the latter.
We observed striking differences analysing the relative abundance of LTR-REs, from the superfamily to the sublineage level. These results suggest that the common ancestor of Helianthus contained different LTR-RE sublineages and that, after species separation, such sublineages were subjected to different rates of amplification/loss, while no new LTR-RE sublineage originated (by mutations or by horizontal transfer) in the genome.
One common feature of Helianthus genomes was the abundance of Gypsy elements, which was always higher than that of Copia ones, as observed in the cultivated sunflower (see [40]). However, it is to be noted that the ratio between Gypsy and Copia abundance is highly variable. Such variability is very large when compared to the observed interspecific variation of this ratio in all angiosperm species, ranging from 5:1 in the genome of papaya to 1:2 in that of grapevine (see [11, 42]).
Our data evidence the impact of RE dynamics on speciation within the genus Helianthus. In fact, it can be speculated that the huge variations observed have negatively affected chromosomal colinearity, favouring the reproductive isolation of the species.
LTR-RE abundance and evolution in Helianthus
The second point of our study was to investigate the relationship between changes of LTR-RE abundance and the evolution of the genus.
The different abundance of LTR-RE lineages was used to estimate the evolutionary relationship among the different species. Compared to the last reported Helianthus phylogeny [24], the LTR-RE genome-proportion-based dendrogram parallels Helianthus evolution in keeping the Helianthus and Agrestis sections separated. Although REs could be considered as autonomous entities, these data suggest that LTR-REs and their hosts in the genus Helianthus have generally evolved together.
However, results also indicate a firm separation between H. porteri and the other species of the Divaricati section, while clades within the branch corresponding to perennial species of the Divaricati section are poorly supported, confirming [28] that LTR-REs have continued to evolve during speciation. If one compares the two trees, although their topologies are very similar, genetic distances are larger using LTR-RE genome proportion values than rDNA-ETS sequences, suggesting the involvement of LTR-RE amplification/loss in increasing genetic differentiation between species.
LTR-RE abundance and annual or perennial habits of a species
The third point was to investigate whether there is a connection between annual or perennial habits of species and LTR-RE abundance. An interesting aspect of our analyses is that, considering only diploid species, the genome size of annual sunflowers is smaller than that of perennials, with the exception of H. agrestis (belonging to the monophyletic section Agrestis). This result is in line with cytophotometric determinations of genome size of Helianthus species previously provided [33]. Also, Southern blot hybridization analyses of Gypsy and Copia elements [25, 26, 43] evidenced a clear-cut separation between annual and perennial species.
The present data reinforce the hypothesis that LTR-RE abundance is more affected by species habit (i.e. annual vs. perennial) than by taxonomic relationships among species in determining the accumulation (or the reduction) of specific LTR-RE superfamilies or lineages. Keeping separated Gypsy and Copia lineages, PCA showed a significant separation between annuals and perennials for the most abundant lineages of both the Gypsy (Chromovirus) and the Copia superfamily (Maximus/SIRE). Chromovirus LTR-REs were more abundant in perennial species; Maximus/SIRE elements were more abundant in annuals. Such separation was not significant analysing the other LTR-RE lineages, suggesting that habit affected (with different outcomes) the abundance of specific types of LTR-REs.
The reasons for such differences between annual and perennial species can be only speculated. They might have established casually during Helianthus evolution. Another possibility is that the influence of the habits on RE abundance might be related to the different life cycle length between perennial and annual species. In fact, in case of RE proliferation or unequal homologous recombination burst, at each generation, the number of LTR-RE can be fixed and in the subsequent generation, new insertion/deletion events can occur, adding or losing other elements. Moreover, changes in LTR-RE abundance can be related to recombination events by which homologous chromosomes with high or low numbers of elements co-segregate in the subsequent generations.
The additive accumulation or loss of LTR-REs at each generation and the effect of co-segregation of chromosomes with increased or decreased numbers of LTR-REs change the number of these elements in the genome. The occurrence of both processes may be related to the number of generations in which accumulation or loss occur, that is larger in annuals than in perennials. Further studies, in other plant genera, are necessary to establish the occurrence of a relationship between habit and transposon accumulation and the reasons for which such a relationship occurs.
Coevolution of LTR-RE lineages and sublineages between and within species
The fourth aim was to determine the degree of coevolution of different LTR-RE lineages or sublineages between and within species.
At lineage level, Gypsy Chromovirus elements are by far the most abundant in all species, suggesting that probably the amplification of these LTR-REs largely predated species divergence in this genus. These variations indicate that high amplification rate was maintained in some species (as H. agrestis) even after speciation or that chromosome rearrangements, such as large duplications or deletions (often driven by retrotransposons, [2]) might have occurred. However, the large conservation of RT-domain sequences among species indicate that if LTR-RE lineages may have experienced amplification or loss during or after Helianthus speciation, these events have occurred in relatively recent times.
At sublineage level, the occurrence of different groups of Gypsy and Copia REs, with different patterns of abundance among species, reinforces the view of a very large variability in the abundance of LTR-RE sublineages originated after species separation.
Our data also point out the necessity of referring to single sublineages when studying retrotransposons, in fact, evaluating differences at superfamily or even at lineage level does not account for the existence of large differences among sublineages within a single lineage. Coevolved groups of LTR-RE in sunflowers are made of elements belonging to different lineages and, on the other hand, sublineages of the same lineage belong to groups with different evolutionary trends.
The impact of LTR-RE proliferation on genome size: the case of H. agrestis
We studied variations in the dynamics (amplification, loss, proliferation dating) of a specific LTR-RE lineage, Chromovirus, among species. We observed that H. agrestis, a diploid species, has a genome size more than two-fold that of the largest genome sized diploid species. This huge genome expansion is, at least in part, related to amplification of LTR-REs of the Chromovirus-Tekay lineage, as indicated by the average coverage of Chromovirus-related clusters and by slot-blot hybridization experiments. Such huge expansion is similar to others reported in different plant species, as Vicia pannonica [7] and Oryza australiensis [8]. As discussed before, it is possible that in H. agrestis, beside proliferation of Chromovirus LTR-REs, duplications of chromosomal regions and co-segregation of homologous chromosomes with high number of LTR-REs have also contributed to hugely increase the genome size.
Analysis of conservation of Gypsy RT-encoding sequences showed similar levels of conservation in all species, excluding H. porteri. This suggests that Gypsy proliferation bursts have occurred in the same period in all analysed species except H. porteri. Dating proliferation burst of Chromovirus LTR-REs also showed that it occurred in the same period in the three analysed species (H. agrestis, H. divaricatus and H. annuus). It can be inferred that, in that time span, proliferation of Chromoviruses in H. agrestis was by far more active than in the other species. Interestingly, proliferation seems to be exhausted in H. agrestis and H. divaricatus while it is continuing in H. annuus, again indicating the peculiarity of LTR-RE dynamics among species.
Conclusions
Our study exploits the potentiality of massively parallel sequencing technologies applied to the analysis of genome structure and evolution. It shows a great variability of LTR-RE abundance at superfamily, lineage and sublineage levels and suggests that the evolution of the LTR-RE component of the genome in Helianthus species is partly independent of the evolution of such species. This is not surprising, because LTR-REs are (at least potentially) autonomous in their reproduction [9]. Indeed, cases of species-specific huge amplification of LTR-RE lineages were already known, even in sunflowers [30, 44], and LTR-REs were still active in retrotransposition in H. annuus [45].
The availability of the forthcoming reference genome for H. annuus [46] in conjunction with new sequencing technologies, allowing for the production of very long DNA sequences, will be useful for further elucidating many aspects of genome evolution in this genus.
Methods
Data availability
Whole-genome shotgun sequences described are available on NCBI Sequence Read Archive under the accession numbers SRR5804988 (H. agrestis), SRR2919251 (H. annuus), SRR5713981 (H. petiolaris ssp. fallax), SRR5713980 (H. petiolaris ssp. petiolaris), SRR5804989 (H. porteri), SRR5713976 (H. divaricatus), SRR5713977 (H. giganteus), SRR5713975 (H. californicus), SRR5713978 (H. hirsutus), SRR5713979 (H. laevigatus), SRR5713974 (H. tuberosus). Clusters and contigs assembled by RepeatExplorer are available at the Sequence Repository Page of the Department of Agriculture, Food, and Environment of the University of Pisa (http://pgagl.agr.unipi.it/sequence-repository/).
The Chromovirus-related sequence used as probe in molecular analyses is available on the NCBI website (https://www.ncbi.nlm.nih.gov/) under the accession number MF448448.
Plant materials and DNA isolation
The Helianthus species used are listed in Table 1. Seeds were obtained from USDA, ARS, National Genetic Resources Program, USA (ARS-GRIN) (https://npgsweb.ars-grin.gov/gringlobal/search.aspx). Seeds were germinated in moistened paper in Petri dishes and 2-3 cm long plantlets were grown in pots in the greenhouse. Leaf tissue was sampled from single individuals of each genotype and total genomic DNA was extracted using the CTAB procedure [47].
Cytological analyses
Primary and secondary root apices were collected from three plantlets per species, treated with 2 mM 8-hydroxyquinoline for 90 min and fixed in ethanol: acetic acid (3:1, v/v). The roots were washed in an aqueous solution of 6 mM sodium citrate plus 4 mM citric acid, treated with a mixture of 3% pectinase (Sigma), 4.5% cellulase (Calbiochem) and 0.5% cellulase Onozuka (Serva) in citrate buffer pH 4.6 for 15 min at 37 °C and subsequently squashed under a coverslip in a drop of 60% acetic acid. The coverslips were removed after freezing at −80 °C. The preparations were air-dried and Feulgen-stained after hydrolysis in 1 N HCl at 60 °C for 8 min. After staining, the slides were subjected to three 10-min washes in SO2 water prior to dehydration and mounting in distyrene-dibutylphthalatexylene (DPX; BDH Chemicals). Feulgen DNA absorptions in individual prophase nuclei were measured in images captured by a charge-coupled-device camera on a Leica DMRB microscope, using a Leica Q500MC image analyser. On the same slides, metaphase plates were scored to determine chromosome number.
Illumina sequencing
The DNA samples were sheared into fragments for sequencing. Paired-end (insert size) libraries were prepared as recommended by Illumina (Illumina Inc., San Diego, CA), with minor modifications. Paired reads were first tested for quality and trimmed at 90 nt in length, using Trimmomatic [48] with the following parameters: ILLUMINACLIP:2:30:10 CROP:90 MINLEN:90, to remove adapters and low-quality regions. All reads containing organellar DNA sequences were removed using CLC-BIO Genomic Workbench 7.0.4 (CLC-BIO, Aarhus, Denmark), against chloroplast and mitochondrial sequences of H. annuus (NCBI reference sequence NC_007977 and KF815390, respectively).
Identification of LTR-RE sequences
In order to perform a comparative analysis of the repetitive component of 10 species and one subspecies of the genus Helianthus, a hybrid graph-based clustering method (RepeatExplorer, [36]) was applied allowing de novo identification of repeats and their proportion in each genome. Accordingly, a random set of sequences composed of reads of each species was used, choosing a number of reads proportional to the ploidy level of the species to ensure that the clusters obtained were comparable.
RepeatExplorer output was filtered to collect the clusters identified as REs. To increase the number of such clusters, similarity searches on the remaining unknown clusters were performed by BLASTN and tBLASTX against a library of sunflower repetitive sequences, SUNREP [27]. All annotated clusters were collected to prepare an in-house reference library of Helianthus LTR-REs.
Additionally, de novo identification of full-length LTR-REs was performed on 40 genome scaffolds of Helianthus annuus, downloaded from the NCBI website (www.ncbi.nlm.nih.gov/; Additional file 4: Table S2), by searching structural features with LTR-FINDER [49] and DOTTER [50]. All putative LTR-REs were annotated using BLASTX and BLASTN against the nr database of NCBI and transferring the annotation from the best hit.
Mapping procedure for abundance estimation
Abundance values of sequences were estimated for each species by counting the total number of reads (per million) mapping to cluster sequences. This method has already been used for many plant species [42, 51,52,53], including sunflower [27, 31]. CLC-BIO Genomic Workbench was used to perform mapping at high stringency with the following parameters: mismatch cost = 1, deletion cost = 1, insertion cost = 1, similarity = 0.9 and length fraction = 0.9.
In another analysis, to estimate the occurrence of solo-LTRs in all the species, every read set was mapped onto each isolated full-length RE, keeping separated the 5′-LTR region and the inter-LTR one.
Phylogenetic trees
All species were analysed one-by-one using RepeatExplorer to perform graph-based clustering on a random set of genomic sequences. Subsequently, the protein domain tool was used to identify and extract conserved regions of RT protein domains for Gypsy and Copia RE superfamilies.
Afterwards, the multiple protein alignment was calculated using Clustal Omega [54] and the phylogenetic trees were built using a neighbour joining clustering method (NJ) (1000 bootstrap replications).
In another analysis, the external transcribed spacer of ribosomal DNA (rDNA-ETS) sequences reported in Timme et al. [24] were used to draw a dendrogram of the species used in this work. An aligned data set was prepared for phylogenetic analyses concatenating the 5′ and 3′ single copy regions of the each ETS into one partition after removing subrepeats (as described in [24]). The alignment was performed using CLUSTAL X [55] and the phylogenetic trees were built using NJ (1000 bootstrap replications).
Finally, a dendrogram based on the genome proportions data of each LTR-RE analysed was built by using R package pvclust version 1.3-2 [56], which allowed the assignment of the uncertainty in hierarchical cluster analysis (10,000 bootstrap replications).
Retrotransposon insertion time analysis
For a comparative estimation of the age of each RE lineage the sequence conservation of the RT domain was analysed in all species. Illumina reads were mapped onto RT domain encoding sequences and counted using CLC-BIO Genomic Workbench at different stringencies (high, medium or low). We kept fixed mismatch cost, deletion cost and insertion cost at 1, changing similarity and length fraction at 0.9, 0.7 or 0.5, respectively. The ratio between the number of reads mapping onto a given lineage at medium and high stringencies reflects the sequence conservation level of the elements that belong to that lineage: assuming similar evolutionary rates in each lineage, the lower the ratio, the higher the sequence conservation.
Timing of Chromovirus LTR-REs proliferation bursts in H. agrestis, H. annuus and H. giganteus were also estimated according to Piegu et al. [8] and Ammiraju et al. [57] through analysis of the distribution of divergence values between pairwise comparisons of sequences belonging to the same lineage.
After collecting Chromovirus RT domain-related sequences (90 nt-long) from RepeatExplorer, cluster mapping was performed using CLC-BIO Genomic Workbench for isolating reads homologous to RT in each species or subspecies. Then, paralogous reads were pairwise compared (using MEGA version 7; [58]) within each species or subspecies and Kimura distances [38] were calculated. Kimura distances were converted to millions of years ago (MYA) using a substitution rate of 2*10-8, i.e. two-fold the value determined for gene sequences of sunflower as already used for sunflowers [30, 59]. In fact it is to be noted that, beside accumulating mutations as time passes, REs accumulate further mutation during retrotranscription, being the RT error-prone [10]. Hence, an increased substitution rate is to be used for calculating retrotransposition time periods.
Dot blot hybridization and calculation of sequence copy number
A 678 bp-long Gypsy fragment was amplified by PCR from 50 ng H. annuus genomic DNA. Primers were designed onto an integrase encoding sequence (forward primer: 5′-AAACGGATGGACAAACTGAACG-3′) and a chromodomain (reverse primer: 5′-CCTTGACTATGCGAATCTTGCT-3′) of a Chromovirus-related cluster from the hybrid graph-based clustering. The PCR conditions were as follows: at 94 °C for 4 min, then 30 cycles of 94 °C for 30 s, 58 °C for 30 s, 72 °C for 40 s. Final extension was performed at 72 °C for 7 min. The PCR products were purified with Wizard SV gel and PCR clean-up system (Promega) and cloned into the pGEM-T Easy plasmid vector (Promega). The cloned fragments were sequenced and one clone was selected (EMBL accession number MF448448).
Dot blot was prepared by applying dilution series (three replicates) of DNA to positively charged nylon membranes (Roche) using a Bio-Dot apparatus (Biorad). Based on 4C Feulgen absorptions and using a H. annuus C-value estimation of 3.3 pg [20], H. agrestis, H. annuus and H. divaricatus denatured genomic DNAs were spotted in a dilution series from 20 × 10^3 to 5 × 10^3 genomes. Similarly, dilutions of denatured PCR product of the Chromovirus fragment of 678 bp, were applied to filters in a dilution series representing 5 × 10^7 to 0.625 × 10^7copies.
The probe used for hybridization was digoxigenin-labelled by PCR using 1× PCR buffer, 0.5 μl Taq DNA polymerase (Promega), dNTP labelling mix (final concentrations 200 μM dATP, 200 μM dCTP, 200 μM dGTP, 190 μM dTTP, 10 μM digoxigenin-11-dUTP, alkaline labile; Roche), 2.5 mM MgCl2, 0.8 μM each forward and reverse primers, 1 ng plasmid DNA derived from selected clone as template (total volume 50 μl). Samples were heated at 94 °C for 4 min and the PCR reaction was performed as described above. The digoxigenin-labelled PCR product was purified with Wizard SV gel and PCR clean-up system (Promega).
Hybridization was performed using 15 ng/ml probe at 65 °C for 12 h in deionized water, 5 × SSC, 2% blocking reagent (Roche), 0.02% SDS and 0.2% SLS. The filter was washed twice in 2 × SSC, 0.1% SDS for 15 min at room temperature, once in 1 × SSC 0.1% SDS for 30 min at 68 °C and once in 0.5 × SSC, 0.1% SDS for 30 min at 68 °C. The temperature of the final wash was calculated in order to ensure hybridization of DNA sequences sharing at least 85% similarity with the probe. Detection was performed using the DIG-Nucleic Acid Detection Kit (Roche) according to the manufacturer’s instructions.
Finally, the membrane was scanned densitometrically using a UVP System 5000 equipped with GelBase-GelBlot software. Estimation of the copy number of the sequence probed in the genomic DNA was carried out as described previously [25].
Statistical analyses
Genomic proportions of the most abundant Gypsy and Copia RE lineages were subjected to principal component analysis (PCA) using the implementation of the R package FactoMineR version 1.26 [60] and to permutational MANOVA [61] with R package vegan version 2.0-10 [62]. Differences among average coverage of LTR over coding region ratios were tested by using the non-parametrical method of Tukey. A separate test was performed for each group of species with the same ploidy level.
Abbreviations
- CTAB:
-
Cetyl trimethyl-ammonium bromide
- ETS:
-
External transcribed spacer
- LTR:
-
Long-terminal-repeat
- MANOVA:
-
Molecular analysis of variance
- MYA:
-
Millions of years ago
- NJ:
-
Neighbor joining
- ORF:
-
Open reading frame
- PCA:
-
Principal component analysis
- PCR:
-
Polymerase chain reaction
- RE:
-
Retrotransposon
- RT:
-
Reverse transcriptase
- SDS:
-
Sodium dodecyl sulfate
- SSC:
-
Saline-sodium citrate
- TE:
-
Transposable element
- USDA:
-
United States Department of Agriculture
References
Britten RJ. Transposable element insertions have strongly affected human evolution. Proc Natl Acad Sci U S A. 2010;107:19945–8.
Lisch D. How important are transposons for plant evolution? Nat Rev Genet. 2013;14:49–61.
Morgante M, De Paoli E, Radovic S. Transposable elements and the plant pan-genomes. Curr Opin Plant Biol. 2007;10:149–55.
Morgante M, Brunner S, Pea G, et al. Gene duplication and exon shuffling by helitron-like transposons generate intraspecies diversity in maize. Nat Genet. 2005;37:997–1002.
Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nature Rev Genet. 2007;8:272–85.
Hawkins JS, Kim H, Nason JD, Wing RA, Wendel J. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16:1252–61.
Neumann P, Koblizkova A, Navratilova A, Macas J. Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics. 2006;173:1047–56.
Piegu B, Guyot R, Picault N, et al. Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006;16:1262–9.
Wicker T, Sabot F, Hua-Van A, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–82.
Kumar A, Bennetzen JL. Plant retrotransposons. Ann Rev Genet. 1999;33:479–532.
Vitte C, Fustier MA, Alix K, Tenaillon MI. The bright side of transposons in crop evolution. Brief Funct Genom. 2014;13:276–95.
Wicker T, Keller B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007;17:1072–81.
Llorens C, Futami R, Covelli L, et al. The Gypsy database (GyDB) of mobile genetic elements: release 2.0. Nucl Acids Res. 2011;39:D70–4.
Neumann P, Požárková D, Macas J. Highly abundant pea LTR-retrotransposon Ogre is constitutively transcribed and partially spliced. Plant Mol Biol. 2003;53:399–410.
Wright DA, Voytas DF. Athila4 of Arabidopsis and Calypso of soybean define a lineage of endogenous plant retroviruses. Genome Res. 2002;12:122–31.
Gorinsek B, Gubensek F, Kordis D. Evolutionary genomics of chromoviruses in eukaryotes. Mol Biol Evol. 2004;21:781–98.
Weber B, Heitkam T, Holtgräwe D, et al. Highly diverse chromoviruses of Beta vulgaris are classified by chromodomains and chromosomal integration. Mob DNA. 2013;4:8.
Schnable PS, Ware D, Fulton RS, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5.
International Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012;491:711–6.
Cavallini A, Zolfino C, Cionini G, et al. Nuclear DNA changes within Helianthus annuus L.: cytophotometric, karyological and biochemical analyses. Theor Appl Genet. 1986;73:20–6.
Heiser CB, Smith DM, Clevenger SB, Martin WC. North American sunflowers (Helianthus). Mem Torrey Bot Club. 1969;22:1–218.
Rosenthal DM, Schwarzbach AE, Donovan LA, Raymond O, Rieseberg LH. Phenotypic differentiation between three ancient hybrid taxa and their parental species. Int J Plant Sci. 2002;163:387–98.
Rosenthal DM, Rieseberg LH, Donovan LA. Re-creating ancient hybrid species complex phenotypes from early-generation synthetic hybrids: three examples using wild sunflowers. Am Naturalist. 2005;166:26–41.
Timme RE, Simpson BB, Linder CR. High-resolution phylogeny for Helianthus (Asteraceae) using the 18S-26S ribosomal DNA external transcribed spacer. Am J Bot. 2007;94:1837–52.
Santini S, Cavallini A, Natali L, et al. Ty1 /copia- and Ty3 /gypsy-like DNA sequences in Helianthus species. Chromosoma. 2002;111:192–200.
Natali L, Santini S, Giordani T, Minelli S, Maestrini P. Distribution of Ty3-gypsy- and Ty1-copia-like DNA sequences in the genus Helianthus and other Asteraceae. Genome. 2006;49:64–72.
Natali L, Cossu RM, Barghini E, et al. The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads. BMC Genomics. 2013;14:686.
Cavallini A, Natali L, Zuccolo A, et al. Analysis of transposons and repeat composition of the sunflower (Helianthus annuus L.) genome. Theor Appl Genet. 2010;120:491–508.
Staton SE, Bakken BH, Blackman BK, et al. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012;72:142–53.
Ungerer MC, Strakosh SC, Stimpson KM. Proliferation of Ty3/gypsy-like retrotransposons in hybrid sunflower taxa inferred from phylogenetic data. BMC Biol. 2009;7:40.
Mascagni F, Barghini E, Giordani T, et al. Repetitive DNA and plant domestication: variation in copy number and proximity to genes of LTR-retrotransposons among wild and cultivated sunflower (Helianthus annuus) genotypes. Genome Biol Evol. 2015;7:3368–82.
Rogers CE, Thompson TE, Seiler GJ. Sunflower species of the United States. Fargo: National Sunflower Association; 1982.
Sims LE, Price HJ. Nuclear DNA content variation in Helianthus (Asteraceae). Am J Bot. 1985;72:1213–9.
Ingle J, Sinclair J. Ribosomal RNA genes and plant development. Nature. 1972;235:30–2.
Natali L, Giordani T, Polizzi E, et al. Genomic alterations in the interspecific hybrid Helianthus annuus × Helianthus tuberosus. Theor Appl Genet. 1998;97:1240–7.
Novak P, Neumann P, Pech J, Steinhaisl J, Macas J. RepeatExplorer: a galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next generation sequence reads. Bioinformatics. 2013;296:792–3.
Novák P, Hřibová E, Neumann P, et al. Genome-wide analysis of repeat diversity across the family Musaceae. PLoS One. 2014;9:e98918.
Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
SanMiguel P, Tikhonov A, Jin YK, et al. Nested retrotransposons in the intergenic regions of the maize genome. Science. 1996;274:765–8.
Giordani T, Cavallini A, Natali L. The repetitive component of the sunflower genome. Curr Plant Biol. 2014;1:45–54.
Flavell RB. Repetitive DNA and chromosome evolution in plants. Phil Trans R Soc London B: Biol Sci. 1986;312:227–42.
Barghini E, Natali L, Giordani T, et al. LTR retrotransposon dynamics in the evolution of the olive (Olea europaea) genome. DNA Res. 2015;22:91–100.
Vukich M, Schulman AH, Giordani T, et al. Genetic variability in sunflower (Helianthus annuus L.) and in the Helianthus genus as assessed by retrotransposon-based molecular markers. Theor Appl Genet. 2009;119:1027–38.
Ungerer MC, Strakosh SC, Zhen Y. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr Biol. 2006;16:R872–3.
Vukich M, Giordani T, Natali L, Cavallini A. Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol. 2009;9:150.
Langlade J. Result of the de novo sequencing of the complex sunflower genome using PacBio technology. XXIVth Plant & Animal Genome Conference, San Diego (CA), Abstract Book, p. W211, 2016.
Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus. 1989;12:13–5.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114–20.
Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucl Acids Res. 2007;35:W265–8.
Sonnhammer EL, Durbin R. A dot-matriprogram with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:GC1–GC10.
Swaminathan K, Varala K, Hudson ME. Global repeat discovery and estimation of genomic copy number in a large, complex genome using a high-throughput 454 sequence survey. BMC Genomics. 2007;8:132.
Tenaillon MI, Hufford MB, Gaut BS, Ross-Ibarra J. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol Evol. 2011;3:219–29.
Barghini E, Natali L, Cossu RM, et al. The peculiar landscape of repetitive sequences in the olive (Olea europaea L.) genome. Genome Biol Evol. 2014;6:776–91.
McWilliam H, Li W, Uludag M, Squizzato S, et al. Analysis tool web services from the EMBL-EBI. Nucl Acids Res. 2013;41:W597–600.
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res. 1997;25:4876–82.
Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540–2.
Ammiraju JS, Zuccolo A, Yu Y, et al. Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus Oryza. Plant J. 2007;52:342–51.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Buti M, Giordani T, Cattonaro F, et al. Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor Appl Genet. 2011;123:779–91.
Lê S, Josse J, Husson F. FactoMineR: an R package for multivariate analysis. J Stat Softw. 2008;25:1–18.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46.
Oksanen J, Blanchet FG, Kindt R, et al. vegan: community ecology package. R package, version 2.0-10. http://CRAN.R-project.org/package=vegan, 2013.
Acknowledgements
The authors thank to Prof. Brandon Gaut for his critical reading of the manuscript.
Funding
This research work was supported by PRIN-MIUR, Italy, Project “SUNREP: caratterizzazione molecolare della componente ripetitiva del genoma di girasole” and by DiSAAA-a, University of Pisa, Project “Plantomics”. Funding bodies approved the design of the study, collection, analysis, and interpretation of data, and the manuscript.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in http://pgagl.agr.unipi.it/sequence-repository/. All data generated or analysed during this study are included in this published article as Additional files.
Author information
Authors and Affiliations
Contributions
FM, AC, LN conceived and the study; FM, TG designed the study; FM, TG performed bioinformatics analyses; TG performed molecular analyses; MC performed cytological analyses; LN principal investigator for the “SUNREP” PRIN-MIUR project and coordinator of the study; FM, TG, MC, AC, LN wrote the paper; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All plant species were identified by USDA, ARS, National Genetic Resources Program, USA (ARS-GRIN). Seeds were obtained from the same source and are available at ARS-GRIN website (https://npgsweb.ars-grin.gov/gringlobal/search.aspx). Identification codes are reported in Table 1. No field studies were conducted and materials were discarded after DNA isolation.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figure S1.
Correlation plot between genome proportion and total read count per million reads related to the 248 clusters annotated as LTR-REs (acronyms as in Table 1). (PDF 222 kb)
Additional file 2: Figure S2.
Distance tree of LTR-Copia RT domains of 10 species and one subspecies of Helianthus subjected to NJ analysis. Bootstrap values higher than 0.6 are shown with asterisk. Bar represents the nucleotide distance. Outgroups are RT domains of other species. Distance tree of LTR-Gypsy RT domains of 10 species and one subspecies of Helianthus subjected to NJ analysis. Bootstrap values higher than 0.6 are shown with asterisk. Bar represents the nucleotide distance. Outgroups are RT domains of other species. (PDF 487 kb)
Additional file 3: Figure S3.
Sequence composition of LTR-Copia-RE-related clusters. The size of the rectangle is proportional to the genome proportion of a cluster for each species (acronyms as in Table 1). Bar plot in the top row shows the size of the clusters as number of reads in the comparative analysis. Upper lines label groups of clusters as asses by a hierarchical clustering of the results. The percentage of reads included in the group is shown in parentheses. The colour of the rectangles corresponds to the lineage of the Copia LTR-RE. All Copia-related repeats were shared among all the 10 species and one subspecies, with some peculiarities. In fact, differences were found even within lineages, producing 14 groups of RE sublineages with different abundance patterns, which accounted for 0.03 to 3.53% of the genome. For example, group 10, although being the most abundant (3.53% of the genome on the whole), was made up by nine clusters (eight annotated as Maximus/SIRE and one as Copia-unknown), that were represented in all species, but showed the largest abundance in H. porteri. On the contrary, the eight sublineages of group 12, annotated as Maximus as well, showed the highest genome proportions especially in H. agrestis and, to a lesser extent, in the Helianthus section. (PDF 454 kb)
Additional file 4: Table S1.
List of 40 genomic scaffolds of Heliantus annuus downloaded from NCBI. Table S2. LTR RE-coding portion of 41 full-length LTR-REs isolated from available genome scaffolds of H. annuus. (XLS 373 kb)
Additional file 5: Figure S4.
Distribution (on a logarithmic scale) of the ratio between the average coverage of 5′-LTR and respective coding (inter-LTR) DNA sequence related to 25 Copia (left) and 16 Gypsy (right) isolated REs, grouped per species. Species are distributed by increasing genome size keeping separated ploidy levels. Diploid species are in red, tetraploid in blue and hexaploid in green. The boxes represent the 25–75%, whiskers the whole range of values and dots the outliers. The lines in the boxes represent the medians of the distributions. Within diploid or tetraploid species, those indicated by different letters are significantly different (p < 0.05) according to Tukey’s test. (PDF 426 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mascagni, F., Giordani, T., Ceccarelli, M. et al. Genome-wide analysis of LTR-retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genomics 18, 634 (2017). https://doi.org/10.1186/s12864-017-4050-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4050-6