
Abstract
Background
Vertebrate mitochondrial genomes are optimized for fast replication and low cost of RNA expression. Accordingly, they are devoid of introns, are transcribed as polycistrons and contain very little intergenic sequences. Usually, vertebrate mitochondrial genomes measure between 16.5 and 17 kilobases (kb).
Results
During genome sequencing projects for two novel vertebrate models, the invasive round goby and the sand goby, we found that the sand goby genome is exceptionally small (16.4 kb), while the mitochondrial genome of the round goby is much larger than expected for a vertebrate. It is 19 kb in size and is thus one of the largest fish and even vertebrate mitochondrial genomes known to date. The expansion is attributable to a sequence insertion downstream of the putative transcriptional start site. This insertion carries traces of repeats from the control region, but is mostly novel. To get more information about this phenomenon, we gathered all available mitochondrial genomes of Gobiidae and of nine gobioid species, performed phylogenetic analyses, analysed gene arrangements, and compared gobiid mitochondrial genome sizes, ecological information and other species characteristics with respect to the mitochondrial phylogeny. This allowed us amongst others to identify a unique arrangement of tRNAs among Ponto-Caspian gobies.
Conclusions
Our results indicate that the round goby mitochondrial genome may contain novel features. Since mitochondrial genome organisation is tightly linked to energy metabolism, these features may be linked to its invasion success. Also, the unique tRNA arrangement among Ponto-Caspian gobies may be helpful in studying the evolution of this highly adaptive and invasive species group. Finally, we find that the phylogeny of gobiids can be further refined by the use of longer stretches of linked DNA sequence.
Similar content being viewed by others

Background
Gobiids (Teleostei, Gobiidae, sensu Gill and Mooi 2011 [1]) are a diverse and fascinating group of small, predominately bottom-dwelling fish species with a world-wide distribution. The Gobiidae family contains more than 1700 species in more than 200 genera and is therefore one of the largest vertebrate families [2, 3]. Gobiids display a wide range of very special adaptations. Several species are able to breathe air and display an amphibious lifestyle. Other species spend early and late life stages at different salinities, or are euryhaline and can cope with sudden salinity shifts [2]. Some species display alternative reproductive tactics [4, 5]. Others are tremendously successful bioinvaders [6]. In recent years, two Eurasian gobiid species received particular attention. The sand goby Pomatoschistus minutus became an evolutionary model species for behavioural studies [4, 7–11], while the invasive round goby Neogobius melanostomus became a model species for invasion ecology and invasion genetics [6, 12–14].
Most West Eurasian gobiid species, including the sand goby and the round goby, lack molecular resources. Therefore, West Eurasian gobiids are under-represented in some phylogenetic studies. Sequence-based phylogenies based on up to five nuclear and mitochondrial markers have outlined two major clades within Gobiidae, the gobiine-like and the gobionelline-like gobiids (sensu Agorreta et al. [15]) [3, 16, 17]. West Eurasian gobiid species cluster with both clades. While the round goby and its Ponto-Caspian relatives belong to the gobiine-like gobiids [16], the sand goby group [18] belongs to the gobionelline-like gobiids [17].
Complete mitochondrial genomes can provide both sequence- and non-sequence based phylogenetic information of high resolution. A high rate of sequence evolution, a lack of recombination, its inheritance as one locus, and a short coalescence time compared to bi-parentally inherited nuclear loci [19] render the mitochondrial genome a valuable sequence-based phylogenetic marker. Also, size variations and non-coding sequence insertions and deletions [20, 21] as well as gene order variations between species and taxonomic groups [22, 23] are common and provide additional non-sequence based phylogenetic information. To confirm and to better understand phylogenetic relationships, molecular trees can then be interpreted with regard to physiological and ecological traits to identify branch-specific and ancestral adaptations [24].
The aim of this paper is to enhance our molecular understanding of the round and the sand goby by reporting the annotated mitochondrial genomes of these two West Eurasian gobiid research model species, and to place these genomes within the mitochondrial phylogeny of Gobiidae using forty Gobiidae species, nine species from other gobioid families, and of Siganus guttatus and Lactoria diphana. We extend current phylogenies using the additional power of gene arrangement analyses and whole mitochondrial genome tree building, and make sense of these phylogenies by linking them to the ecological properties of the analysed species. We also aim to identify novel molecular markers and to explore the origin and the potential implications of a novel, large non-coding sequence insertion in the round goby mitochondrial genome.
In this paper, we use the term “non-coding region” for all sequence of unknown function which is not coding for either protein or tRNAs. We use the term “control region” to refer to non-coding sequences which contain functional elements that control mitochondrial genome replication.
Results
Mitochondrial genome size
During genome sequencing projects for the round goby and the sand goby, we found that both the sand goby and the round goby mitochondrial genome were unusual with regard to their lengths. The sand goby mitochondrial genome (16,396 bp) was the second smallest mitochondrial genome of all gobiid species sequenced to date, while the round goby mitochondrial genome (18,999 bp) was the longest of all gobiid mitochondrial genomes known (Fig. 1a). The round goby mitochondrial genome also was larger than most mitochondrial genomes of ray-finned fish (Fig. 1b; Additional file 1: Table S2) and other vertebrates (Fig. 1c; Additional file 1: Table S3). Only 1.83% of sequenced vertebrate species (n (total) = 3652) and 3.18% of sequenced animal species (n (total) = 5567) had larger mitochondrial genomes. We found that the large size was caused by two instances of sequence insertions. First, the noncoding region between tRNA Proline and tRNA Phenylalanine that contains the control region was larger than in other species (1920 bp in the round goby, compared to 720 bp in sand goby). Second, the round goby genome featured a 1250 bp insert between tRNA Phenylalanine and the 12S rRNA gene (Fig. 2).

The round goby mitochondrial genome is one of the largest vertebrate mitochondrial genomes. a Barplot of gobioid mitochondrial genome sizes available on NCBI. Species are ordered according to phylogenetic relatedness as determined in this study. Round goby and sand goby genomes are highlighted in orange. b Histogram of mitochondrial genome sizes of 1708 ray-finned fish for which complete mitochondrial genomes were available. Orange arrowheads indicate the sizes of sand goby and round goby mitochondrial genomes. c Boxplots illustrating the mitochondrial genome size distributions for major vertebrate classes. For 2427 species, mitochondrial genomes as well as class annotations were available from NCBI and IUCN respectively. “Reptiles” includes all members of the reptilia clade (turtles, lizards, snakes, crocodiles etc.) except birds. Orange arrowheads and dotted lines indicate the sizes of sand goby and round goby mitochondrial genomes

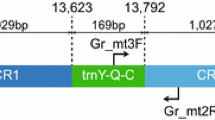
Sand and round goby mitochondrial genome gene arrangements. Left panel, sand goby mitochondrial genome annotation. Right panel, round goby mitochondrial genome annotation. The round goby genome features an enlarged D-loop region, a non-coding sequence insertion downstream of tRNA Phe, and an inverted arrangement of tRNA Ile and tRNA Gln
Characterisation of non-coding sequence inserts
To better understand the source of the inserted sequences, we annotated functional regions and repeats. We identified the control region, which directs mitochondrial genome replication and transcription, by searching for Transcription Associated Site (TAS)- and Conserved Sequence Block (CSB)-like elements in conserved regions between tRNA Proline and tRNA Phenylalanine. Three canonical TAS sites are conserved in gobiids (Additional file 2: Figure S1 and Additional file 3: Figure S3). In addition, the round goby mitochondrial genome contains many canonical TAS sites in the context of tandem repeats. CSB sites were more difficult to identify. Only when relaxing search criteria (larger spacer sequence or relieved identity constraints on the third base), we were able to identify CSB-I like sequences in conserved regions downstream from the TAS motif. We also identified CSB II and CSB III-like sequences in conserved regions (Additional file 2: Figure S1).
The round goby non-coding sequence insert between tRNA Phenylalanine and the 12S rRNA gene did not contain TAS or CSB motifs, and was not similar to any published sequence, but bore similarity to sequence parts upstream of tRNA Phenylalanine. By mapping repeated sequence motifs between sand goby, round goby, and its closest sequenced relative, the bighead goby Ponticola kessleri, we identified five repeated motifs, which we called NM_1 (106 bp), NM_2 (100 bp), NM_3 (120 bp), NM_4 (202 bp), and PK_1 (56 bp; Fig. 3; Additional file 4: Figure S2). NM_3 and PK_1 are both located between CSB III and tRNA Phenylalanine and share a 14 bp core motif (TAATAATCATTTTA in bighead goby, TAATAATACATTTTTA in round goby). NM_4, NM_3, and NM_2 occur both upstream and downstream of tRNA Phenylalanine. All repeat motifs are predicted to form secondary structures (Additional file 4: Figure S2).

Architecture of non-coding sequences. Non-coding regions between tRNA Pro and the 12S gene of sand goby, bighead goby (the closest sequenced relative of round goby), and round goby are drawn to scale below each other. Conserved and/or putatively functional motifs of the control region such as TAS and CSB elements, tandem repeats, repeated sequences, and the location of tRNA Phe are indicated by coloured bars
Gene arrangement analysis
To compare gene arrangements between the analysed species, we performed automatic annotations. They revealed two instances of non-canonical gene arrangements involving tRNA genes, one in Ponto-Caspian gobies and one in Odontobutis platycephala (Additional file 3: Figure S3). In most gobioids analysed in this study, the tRNAs for Isoleucine, Glutamine and Methionine come in the order Ile/Gln/Met. In the two Ponto-Caspian species, the round goby and the bighead goby, these tRNAs are arranged as Gln/Ile/Met (Fig. 2). Strandedness differed between the two species, with Gln (-)/Ile (+)/Met (+) in the bighead goby and Gln (+)/Ile (-)/Met (+) in the round goby. In Odontobutis platycephala, we found that the tRNAs for Serine, Leucine and Histidine came as Ser/Leu/His instead of the canonical His/Ser/Leu arrangement (see also [25]).
Mitochondrial phylogeny
To complement previous gene-centered studies, we buildt whole mitochondrial genome phylogenies using Bayesian and Maximum Likelihood approaches. The resulting phylogenies were consistent with phylogenies from previous studies (Fig. 4), with two exceptions (the placement of Micropercops swinhonis and Oxyurichthys formosanus, see below). As expected, we identified two major clades within Gobiidae, gobionelline-like gobiids and gobiine-like gobiids (Fig. 4; sensu Agorreta et al. [15]). Members from Butidae, Eleotridae, Rhyacichththydae, and Odontobutidae clustered as sister groups to the Gobiidae (Table 1).

Phylogenetic analysis matched with biological and ecological species characteristics. a Phylogenetic tree of twelve concatenated mitochondrial genes obtained from MrBayes. Black dots indicate branches with a posterior probability value from MrBayes > 0.95 and a bootstrap value from maximum likelihood analysis in RAxML >75. b Mitochondrial genome features of the respective species. c Ecological characteristics of the respective species. Features and characteristics in b and c are indicated by the colour and/or size of the dot and/or symbol. Body size corresponds to maximal total length (TL) observed
Gobionelline-like gobiids split into two major groups, with members from the Acanthogobius- and Mugilogobius-lineages (sensu Agorreta et al. [15]) in one, and members from the Stenogobius- and Periophthalmus-lineages (sensu Agorreta et al. [15]) in the other group. The sand goby clustered as sister taxon to all other gobionelline-like gobiids. Since statistical support was too low to determine the exact branching order among all three major groups within gobionelline-like gobiids, the sand goby may cluster as sister taxon to either one of the two other large gobionelline-like gobiid groups.
Gobiine-like gobiids clustered into two major groups. Unexpectedly, one of those contained Micropercops swinhonis, a member of Odontobutidae [26], and Oxyurichthys formosanus, which was expected to cluster with the gobionelline-like lineage Stenogobius [15], with high statistical support. The round goby clustered with the bighead goby, the only other West Eurasian gobiine-like gobiid species included into this study. These two species grouped with two Glossogobius species, albeit with low statistical support.
Linking ecology, biology, and mitochondrial phylogeny
To complement the molecular phylogeny, we mapped genetic, biological, and ecological traits such as mitochondrial genome size, gene rearrangements, geographical occurrence, body length, preferred salinity, and specialized adaptations to the mitochondrial phylogenetic tree. We found that mitochondrial genome size, body length, and specialized adaptations were linked to particular branches with high statistical branch support, while the other features were independent from the mitochondrial phylogeny.
Extremely large mitochondrial genomes were distributed randomly throughout the gobiid mitochondrial phylogeny, but smaller-than-average and larger-than-average genomes clustered together with high statistical branch support. The four species with exceptionally large mitochondrial genomes exceeding 17 kb, round goby, Chaeturichthys stigmatias, Amblychaeturichthys hexanema, and Odontobutis platycephala, clustered with the gobiine-like gobiids, the gobionelline-like gobiids, and Gobiidae outgroups, respectively. However, when considering only mitochondrial genomes smaller than 17 kb, smaller genomes (up to 16,600 bp) were particularly prevalent in an Acanthogobius/Mugilogobius-lineage (e.g. Gillichthys mirabilis, Pseudogobius javanicus) while larger genomes were more common in a clade formed exclusively by species related to members of the Acanthogobius-lineage (e.g. Acanthogobius hasta) and in a clade consisting of members of the Periophthalmus-lineage (e.g. Odontamblyopus rubicundus) (lineage designations sensu Agorreta et al. [15]; Fig. 4).
For the ecological parameters compiled, we found that salinity preference and geographical occurrence were independent from the mitochondrial phylogeny, while body size and specialized adaptations occurred within certain clades or groups for the species included in this study that received high statistical branch support. Specifically, many of the analyzed members of the Rhinogobius group (gobionelline-like gobiids) were small. Burrowing life-style appeared to be a feature of gobionelline-like gobiid species only. Nonetheless, the trait appeared to have evolved twice independently based on the mitochondrial phylogeny. Similarly, air-breathing and amphibious life style was restricted to the gobionelline-like clade, but appeared to have several independent origins.
Discussion
Functional implications of the round goby sequence insertions
While sequence variations and repetitive elements flanking the control regions are common in a wide range of species [27–29], the non-coding sequence inserted downstream of tRNA Phenylalanine in the round goby mitochondrial genome is located at an unusual position and may therefore either come at a cost and/or have a novel function. Repeated sequence motifs and a degenerate copy of tRNA Phenylalanine at the end of the insertion suggest that the insertion arose from a duplication event involving the 3’ end of the control region. Since the insert does not contain TAS or CSB-like sites, it is quite unlikely that the insert represents a functional duplicated control region as observed in parrots [21], mites and ticks [30], silk moths [31], or millipedes [32]. However, the insert may be transcribed into RNA, since the origins of heavy strand transcription lie upstream of tRNA Phenylalanine [33–35].
Transcription and replication are rate limiting steps in tissues with high energy demands [35]. Therefore, animal mitochondrial genomes are under selection for small size and are depleted of non-coding sequences, they lack introns and intergenic sequences (discussed in [20, 21]). The retention of a non-coding sequence in the round goby thus suggests functional relevance. Mitochondrial variants can have great impact on the fitness of an organism (reviewed in [19, 35]). Also, size selection on mitochondrial genomes is stronger in endotherms than in ectotherms, which indicates that metabolic rates and mitochondrial genotypes may be closely linked [20]. In this context, it is of particular interest that the highly invasive round goby has lower metabolic rates, and controls metabolic rates better at high temperatures than other less invasive Ponto-Caspian goby species [36].
Phylogenetic origins of mitochondrial genome size
Our results indicate that the tendency to harbour a smaller- or larger-than-average mitochondrial genome may be a feature of entire mitochondrial lineages. Mitochondrial genome size has been linked to body temperature and metabolism [20]. Thus, the observed size patterns may be linked to ecological parameters such as water temperature that were not covered in this study. Alternatively, the propensity of certain lineages to generate and tolerate sequence insertions on the one hand or to select for small mitochondrial genome size on the other hand may possibly depend on evolutionary differences in the DNA replication or repair machineries of those lineages.
Potentials and limitations of gobiid mitochondrial genome phylogenies
Our phylogenetic reconstructions recovered the two previously described clades within Gobiidae, gobiine-like gobiids and gobionelline-like gobiids [15], and confirmed the placing of round goby in the former and sand goby in the latter group [3, 15–17, 37]. For gobionelline-like gobiids, our results agree with previous studies, except for members of the Mugilogobius-lineage. We find Mugilogobius species nested within an Acanthogobius-lineage, while they were considered sister groups, albeit with low statistical support, by two recent studies [3, 15]. The gobiine-like gobiid clade also contains two unexpected members: Oxyurichthys formosanus and Micropercops swinhonis. Previously, other members of the Oxyurichthys genus grouped with gobionelline-like gobiids (e.g. Oxyurichthys stigmalophius; [15], Oxyurichthys lonchotus and Oxyurichthys ophthalmonema [3]). Micropercops swinhonis was previously placed with Odontobutidae [26], which form a sister lineage to Gobiidae [38]. However, Micropercops swinhonis does not always group with Odontobutis [39], and its developmental process is different from Odontobutis but resembles that of Gobiidae [39]. Also, it contains the same His-Ser-Leu tRNA arrangement as all Gobiidae, while Odontobutis platycephala shows a unique Ser-Leu-His arrangement. Thus, Micropercops swinhonis may be a true member of Gobiidae.
For sand goby, our results reflect previous uncertainties about the exact phylogenetic placement of this species. The position as sister taxon to either an Acanthogobius/Mugilogobius lineage, to a Stenogobius/Periophthalmus lineage, or to all four gobionelline-like gobiid lineages is consistent with [37] (sister taxon to Acanthogobius species), [3] (sister taxon to Mugilogobius/Acanthogobius), [17] (sister taxon to Mugilogobius), but conflicting with [40] (sister taxon to all gobiine-like gobiids), and [16] (clustering with other members of the Pomatoschistus-lineage among gobiine-like gobiids).
As expected, the round goby groups together with the Ponticola kessleri, the bighead goby, which is at present the only other representative of Benthophilinae with an available complete mitochondrial genome. The Ponto-Caspian species group and their immediate relatives have been previously suggested to have experienced an evolutionary burst that led to the formation of three Benthophilinae tribes, Ponticolini, Neogobiini, and Benthophilini [16]. Both round goby (Neogobiini) and bighead goby (Ponticolini) feature a particular tRNA arrangement (Gln-Ile-Met instead of the common Ile-Gln-Met arrangement). If this arrangement is also present in Benthophilini, it may be a specific signature of the entire group, and may be related to the suggested historic radiative event promoting the diversification of Benthophilinae. The differential orientation of these tRNAs on the heavy and light strand in the round goby and the bighead goby may in turn help to shed light on the radiation within Benthophilinae.
Many previous phylogenetic studies of gobiids were based on a combination of individual nuclear and mitochondrial markers or on nuclear markers alone. Differences among topologies obtained from different regions of the genome (“gene trees”) are expected and can be explained by different evolutionary processes acting on those regions, such as incomplete lineage sorting, gene duplications and hybridization [41–43]. All in all, our results strongly speak for further examination of Gobiidae, in particular West Eurasian gobiids, using denser taxon sampling for the mitochondrial genome and additional nuclear markers.
Ion-transport capacities may explain the tremendous success of Gobiidae
One striking feature of gobiids is their capacity to colonize very different habitats and even the land. We support the idea that this capacity, albeit it manifests itself in restricted lineages only, may be linked to a more ancient ability of this family to deal with fluctuating ion concentrations. Euryhalinity and amphidromous life cycle are present in many gobiid clades, and species with a preference of high or low salt conditions are evenly distributed across the mitochondrial phylogeny including outgroup species from other Gobioidei lineages, indicating that adaptation to novel salinity levels may be a capacity of all gobioids. Similarly, several gobioid mitochondrial lineages contain amphidromous species, indicating that the ability to breed in fresh water, develop in sea water, and then return to freshwater may be an ancient adaptation in the gobioid group. In the gobionellinae-like gobiids, amphidromous species belong to the Stenogobius- and Acanthogobius-lineages (sensu Agorreta et al. [15]), represented by Sycopterus, Stiphodon and Rhinogobius in the present study, and in gobiine-like gobiids to Glossogobius [44]. The amphidromous life cycle is additionally present in the gobioid families Eleotridae and Ryacichthyidae, which are sister taxa to Gobiidae in the gobioid phylogeny.
In contrast, the ability to breathe air is mostly restricted to the Periophthalmus-lineage (sensu Agorreta et al. 2013; recovered with high statistical branch support in this study). Scartelaos, Boleophthalmus and Periophthalmus, the so-called mudskippers, spend a large time of their time above water [45]. In agreement with [15] and [46], mudskippers are a paraphyletic group in the mitochondrial phylogeny and appear to have evolved twice independently. Alternatively, the mud-burrowing ‘eel gobies’ represented by Odontamblyopus rubicundus and Trypauchen vagina in the present study, may have evolved from mudskippers. Also, the mud flat living non-mudskipper Oxyderces dentatus may actually have a partly amphibious lifestyle [45]. Thus, all members of this lineage follow land- and air-oriented lifestyles.
While euryhalinity and air breathing may seem like very different features, euryhalinity, amphidromous life cycle, and amphibious life style all expose organisms to salinity oscillations, and require the ability to tolerate and excrete ammonia independently of ambient levels. Thus, they all demand excellent and adaptable active ion transport capacities [45, 47]. These observations suggest that the entire gobiid family may have evolved the ability to deal with fluctuating ion gradients, which in turn may be the key to their world-wide success.
Conclusion
In conclusion, we find that the sand goby mitochondrial genome closely resembles other gobiid mitochondrial genomes. The round goby on the other hand has an unusually large mitochondrial genome featuring tandem repeat expansions in the control region and a non-coding and potentially transcribed sequence inserted downstream of the putative origin of transcription. We may thus speculate that the tremendous colonization abilities of this species may be linked to special features of its mitochondrial metabolism. While we were not able to further resolve the phylogenetic placement of the sand goby and the round goby, we provided additional information on the placement of Oxyurichthys formosanus and Micropercops swinhonis in the mitochondrial phylogeny. Also, we identified a novel molecular marker for Ponto-Caspian species in the gene arrangement of the tRNAs Gln-Ile-Met. From analysing the ecological traits of air-breathing and amphibious lifestyle we speculate that the entire species group may be adapted to deal with challenging ion transport conditions, which may be the critical factor in their world-wide success.
Methods
Sequencing of the round goby mitochondrial genome
The round goby mitochondrial genome was obtained by PacBio single molecule sequencing. Genomic DNA was extracted from the liver of one male individual of round goby caught in Basel, Switzerland (47° 35′ 18″ N, 7° 35′ 26″ E) in spring 2015. At the Genome Center Dresden, Dresden, Germany, 300 mg of liver tissue were ground by mortar and pestle in liquid nitrogen and lysed in Qiagen G2 lysis buffer with Proteinase K. RNA was digested by RNase A treatment. Proteins and fat were removed with two cycles of phenol-chloroform extraction and two cycles of chloroform extraction. Then, the DNA was precipitated in 100% ice cold Ethanol, spooled onto a glass hook, and eluted in 1x TE buffer and stored at 4 °C. 10 μg of DNA were cleaned up on AMPure beads. From this DNA, five long insert libraries were prepared for PacBio sequencing according to the manufacturer’s protocols. Genomic DNA was sheared to 35 kb using the Megaruptor device. The PacBio libraries were size selected for fragments larger than 15 kb using the BluePippin device. PacBio SMRT sequencing was performed with the P6/C4 chemistry using 240 min sequencing runs.
Sequencing of the sand goby mitochondrial genome
The sand goby mitochondrial genome was obtained by Illumina sequencing. Genomic DNA was extracted from the tail fin of one individual male Pomatoschistus minutus caught in summer 2010 in Bökevik Bay (58° 14′ 55″ N, 11° 26′ 51″ E) close to the Sven Lovén Centre For Marine Sciences, Kristineberg, Sweden. The tissue was digested in CTAB-buffer, mercaptoethanol and Proteinase K [48]. RNA was digested by RNase A treatment. Thereafter, DNA was purified by chloroform-isoamylate extraction and precipitated with isopropanol. The DNA was washed with 70% ethanol and stored in 10% TE-buffer at -70 °C. One 300 bp paired-end library was sequenced on an Illumina HiSeq2000 (2x101 bp) at the national sequencing facility, Science for Life Laboratory, in Stockholm, Sweden.
Assembly of the round goby mitochondrial genome
The round goby mitochondrial genome was assembled at the Heidelberg Institute for Theoretical Studies HITS gGmbH. First, consensus reads covering mitochondrial sequences were pulled out via the Pacbio long read aligner (blasr [49]) using the bighead goby mitochondrial genome [50] as query. Local multiple alignments of the mitochondrial reads were computed with daligner [51]. From the resulting overlaps, five high quality reads were selected as template genomes for the error correction step. Then, these were extended on both sites by the overhang sequence of overlapping highest quality reads to ensure that each read contained at least one copy of the mitochondrial genome. Errors were corrected with PacBio’s quiver program [52]. Thereafter, all mitochondrial reads were mapped back onto the five mitochondrial template genomes with blasr. Coverage was on average 170x. The resulting consensus sequences were aligned in CLC Main Workbench 6.8.1 (https://www.qiagenbioinformatics.com/). The resulting consensus was further corrected by majority vote. Open reading frame shifts after homopolymers were attributed to sequencing errors and were manually corrected by adapting homopolymer length.
Assembly of the sand goby mitochondrial genome
The sand goby mitochondrial genome was assembled at 1573x coverage in CLC Main Workbench 4.06beta.67189 (https://www.qiagenbioinformatics.com/). A second assembly was performed using SOAPdenovo v1.3 (http://soap.genomics.org.cn/).
Verification of the round goby mitochondrial genome
We verified the round goby mitochondrial genome by two methods. First, we aligned the round goby and the bighead goby mitochondrial genomes in ClustalOmega [53] and assumed that conserved regions were correctly sequenced. Second, we identified non-conserved regions and confirmed them by PCR and Sanger sequencing on five additional round goby individuals caught in Basel, Switzerland, in 2015. The first such region spanned the tRNAs Glutamine, Isoleucine, and Methionine. The second such region spanned the non-coding region between tRNA Proline and the 12S gene. Overlapping amplicons were designed to amplify and sequence those regions. Primer sequences were, 5′–3′: fw1, CCCGATTCCGATATGACCAAC; rev1, GGCTGGATTTTAACCGGCATG; fw2, AGAGCGCCGGCCTTGTAAG; rev2, CAGGTCTTAACTTGGTGTGAG; fw3, ACCCAACTCGAGATTTTCCTG; rev3, CATCAACAATCATTCAAGAATGC; fw4, ATATCATGAGCATAAGTAATTGAC; rev4, GATTGGGTGCAGATCACAGTG; fw5, TACAAAATTGCCCATAATTATGAC; rev5, GGGGTGAGGAGACTTGCATG. We purified DNA from lateral muscle with the Qiagen Blood and Tissue kit. PCR was performed on 50 ng of DNA using FastStart Taq DNA Polymerase from Roche. PCR conditions for amplicon 1 (fw1 and rev1) were: 4′ at 94 °C; 35 cycles of 30″ denaturing at 94 °C, 30″ annealing at 56 °C, 40″ or 1 min/kb extension at 72 °C; 5′ at 72 °C. PCR conditions for amplicons 2, 3, 4, and 5 were adapted for amplification of AT-rich sequences [54] so that extension temperature was 65 °C for amplicons 2, 4, and 5, and 72 °C for amplicon 3, and so that extension time was 2 min for all four amplicons. Amplified fragments were TA-cloned and sequenced at Microsynth, Switzerland. All resulting sequences were identical to those from the PacBio based assembly.
Verification of the sand goby mitochondrial genome
Since repetitive regions which are often present around the mitochondrial control region are difficult to assemble from short reads, the sand goby mitochondrial genome was assembled a second time from 300 bp paired-end reads from a 500 bp insert library, with identical results. We did not identify regions of higher than average coverage in the control region, which would indicate collapsed repeats.
Comparing mitochondrial genome lengths
To understand whether the size of the round goby and sand goby mitochondrial genomes was in the normal range for fish and vertebrates, we compared them to other available mitochondrial genomes. We collected complete mitochondrial genomes of animals, vertebrates, and bony fish from the NCBI database, using the search terms “vertebrate mitochondrion complete genome”, “animalia mitochondrion complete genome”, and “actinopterygii mitochondrion complete genome”. Partial mitochondrial sequences were removed from the dataset in R [55]. Some species, such as human or mouse, were overrepresented in the dataset. Also, for some species with more than one mitochondrial genome available, entries differed in length. To avoid overrepresentation bias and to obtain a single length value for each species, a median length was calculated for each species in R [55]. Those median values were plotted by taxonomic class as obtained from the IUCN database [56].
Gene arrangement analysis
To analyse gene arrangement conservation in an unbiased manner, we annotated or re-annotated all complete mitochondrial goby genomes available on NCBI using MitoFish v2.95 [57]. Species names and accession numbers are listed in Additional file 1: Table S1. Annotations were then visually screened for deviations from the common gene arrangement.
Annotation of round and sand goby mitochondrial genomes
We annotated the sand and round goby mitochondrial genomes using MitoFish v2.95 [57]. We corrected the automated annotation manually in Geneious 5.6.7 [58] using an alignment with complete mitochondrial genomes from Ponticola kessleri, Amoya chusanensis, Glossogobius circumspectus, Glossogobius olivaceus, Oxyeleotris marmorata, Siganus guttatus, Mogurnda adspersa, Bostrychus sinensis, Scartelaos histophorus, Boleophthalmus boddarti, Odontamblyopus rubicundus, Oxuderces dentatus, and Lactoria diaphana, including their annotations (see Additional file 1: Table S1 for accession numbers).
Analysis of non-coding sequence arrangements
To understand the evolution of the novel non-coding sequences in the round goby mitochondrial genome, we compared the non-coding regions of sand goby, round goby, and its closest sequenced relative, the bighead goby. Repetitive regions were identified with the dot plot tool of CLC Main Workbench 6.8.1 with window lengths of 10 nucleotides (nt) and 15 nt. Sequence repeats were identified through manual searches using consecutive 10 nt-blocks of the non-coding sequences as query and all three non-coding regions as target. The identified repeats were extracted and aligned in CLC biobench to generate a consensus for each repeat. Since mitochondrial non-coding sequence repeats have been shown to form secondary structures with functions in protein binding, replication or transcription [59–61], we tested the ability of the consensus sequences to form secondary structures with a minimum free energy approach based on the Zuker algorithm [62] implemented in CLC Main Workbench 6.8.1.
To identify functional regions in the non-coding sequence blocks, we searched for the conserved sequence blocks, CSB I to III, and the termination associated site, TAS [27, 63–65]. To this end, we extracted the sequences between tRNA Proline and tRNA Phenylalanine from the analyzed species, aligned them with ClustalOmega [53] and identified regions of high conservation with the Percentage Identity coloration of Jalview 2.9.0b2. Then, we searched for TAS motifs using the query ATGN(8–9)CAT [65]. We searched for CSB-like motifs [27] using the following queries: CSB I, AT or ATG followed in some distance by GACA; CSB II, AAACCCCCCNNNCCC; CSB III, AAACCCC within an otherwise A/C-homopolymer rich region.
Phylogenetic analyses
We aligned the annotated mitochondrial genomes of forty Gobiidae species, nine species from other gobioid families, and of Siganus guttatus and Lactoria diphana with the round and the sand goby mitochondrial genomes using Geneious 5.6.7 [58]. We kept only protein-coding regions in the alignment, excluded the gene ND6, which is evolving under different constraints than other mitochondrial genes [66], and manually removed all gaps, double checking for correct reading frames using the translated amino acid sequences. We then used MEGA7.0.14 [67] to determine the best-fitting model of sequence evolution. Thereafter, two independent phylogenetic trees were calculated, one based on Bayesian inferences and one based on maximum likelihood analyses. For Bayesian inferences in MrBayes 3.2 [68], we used one cold and three heated chains and ran the analyses for 10,000,000 Markov chain Monte Carlo generations sampling every 2′000th generation, with a burnin of 25%. Convergence was confirmed in Tracer v1.5 (effective sampling size > 200). For maximum likelihood analyses in raxmlGUI 1.3.1 [69], we used the rapid hill-climbing algorithm [70] with 1000 bootstrap replicates [71].
Mapping ecological properties to the phylogenetic tree
We retrieved information on body length, preferred salinity, geographical distribution, and specialized adaptations for all analysed gobioid species from FishBase [72]. We then verified the information using published scientific literature (see Additional file 1: Table S1 for references) and mapped it to the phylogenetic tree.
Abbreviations
- CSB:
-
Conserved Sequence Block
- TAS:
-
Transcription Associated Site
References
Gill AC, Mooi RD. Thalasseleotrididae, new family of marine gobioid fishes from New Zealand and temperate Australia, with a revised definition of its sister taxon, the Gobiidae (Teleostei: Acanthomorpha). Zootaxa. 2011;3266:41–52.
Patzner RA. The biology of gobies. 1st ed. Enfield, Boca Raton: Science Publishers; 2011.
Tornabene L, Chen Y, Pezold F. Gobies are deeply divided: phylogenetic evidence from nuclear DNA (Teleostei: Gobioidei: Gobiidae). Syst Biodivers. 2013;11(3):345–61.
Svensson O, Kvarnemo C. Parasitic spawning in sand gobies: an experimental assessment of nest-opening size, sneaker male cues, paternity, and filial cannibalism. Behav Ecol. 2007;18(2):410–9.
Mazzoldi C, Patzner RA, Rasotto MB. Morphological organization and variability of the reproductive apparatus in gobies. In: Patzner RA, VanTassell JL, Kovacic M, Kapoor BG, editors. The biology of gobies. Enfield: Science Publishers; 2012. p. 367–402.
Roche KF, Janac M, Jurajda P. A review of Gobiid expansion along the Danube-Rhine corridor - geopolitical change as a driver for invasion. Knowl Manag Aquat Ecosyst. 2013;411:01.
Forsgren E, Kvarnemo C, Lindstrom K. Mode of sexual selection determined by resource abundance in two sand goby populations. Evolution. 1996;50(2):646–54.
Pampoulie C, Gysels ES, Maes GE, et al. Evidence for fine-scale genetic structure and estuarine colonisation in a potential high gene flow marine goby (Pomatoschistus minutus). Heredity. 2004;92(5):434–45.
Larmuseau MHD, Van Houdt JKJ, Guelinckx J, et al. Distributional and demographic consequences of Pleistocene climate fluctuations for a marine demersal fish in the north-eastern Atlantic. J Biogeogr. 2009;36(6):1138–51.
Larmuseau MH, Raeymaekers JA, Ruddick KG, et al. To see in different seas: spatial variation in the rhodopsin gene of the sand goby (Pomatoschistus minutus). Mol Ecol. 2009;18(20):4227–39.
Saaristo M, Craft JA, Lehtonen KK, et al. Disruption of sexual selection in sand gobies (Pomatoschistus minutus) by 17alpha-ethinyl estradiol, an endocrine disruptor. Horm Behav. 2009;55(4):530–7.
Stepien CA, Tumeo MA. Invasion genetics of Ponto-Caspian gobies in the Great Lakes: a ‘cryptic’ species, absence of founder effects, and comparative risk analysis. Biol Invasions. 2006;8(1):61–78.
Brown JE, Stepien CA. Invasion genetics of the Eurasian round goby in North America: tracing sources and spread patterns. Mol Ecol. 2009;18(1):64–79.
Kornis MS, Mercado-Silva N, Vander Zanden MJ. Twenty years of invasion: a review of round goby Neogobius melanostomus biology, spread and ecological implications. J Fish Biol. 2012;80(2):235–85.
Agorreta A, San Mauro D, Schliewen U, et al. Molecular phylogenetics of Gobioidei and phylogenetic placement of European gobies. Mol Phylogenet Evol. 2013;69(3):619–33.
Neilson ME, Stepien CA. Escape from the Ponto-Caspian: Evolution and biogeography of an endemic goby species flock (Benthophilinae: Gobiidae: Teleostei). Mol Phylogenet Evol. 2009;52(1):84–102.
Thacker CE. Phylogenetic placement of the European sand gobies in Gobionellidae and characterization of gobionellid lineages (Gobiiformes: Gobioidei). Zootaxa. 2013;3619(3):369–82.
Huyse T, van Houdt J, Volckaert FA. Paleoclimatic history and vicariant speciation in the “sand goby” group (Gobiidae, Teleostei). Mol Phylogenet Evol. 2004;32(1):324–36.
Ballard J, William O, Whitlock MC. The incomplete natural history of mitochondria. Mol Ecol. 2004;13(4):729–44.
Rand DM. Endotherms, ectotherms, and mitochondrial genome-size variation. J Mol Evol. 1993;37(3):281–95.
Schirtzinger EE, Tavares ES, Gonzales LA, et al. Multiple independent origins of mitochondrial control region duplications in the order Psittaciformes. Mol Phylogenet Evol. 2012;64(2):342–56.
Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27(8):1767–80.
Xu W, Jameson D, Tang B, Higgs PG. The relationship between the rate of molecular evolution and the rate of genome rearrangement in animal mitochondrial genomes. J Mol Evol. 2006;63(3):375–92.
Block BA, Finnerty JR, Stewart AF, Kidd J. Evolution of endothermy in fish: mapping physiological traits on a molecular phylogeny. Science. 1993;260(5105):210–4.
Ma Z, Yang X, Bercsenyi M, et al. Comparative mitogenomics of the genus Odontobutis (Perciformes: Gobioidei: Odontobutidae) revealed conserved gene rearrangement and high sequence variations. Int J Mol Sci. 2015;16(10):25031–49.
Birdsong RS, Murdy EO, Pezold FL. A study of the vertebral column and median fin osteology in gobioid fishes with comments on gobioid relationships. Bull Mar Sci. 1988;42(2):174–214.
Sbisa E, Tanzariello F, Reyes A, et al. Mammalian mitochondrial D-loop region structural analysis: identification of new conserved sequences and their functional and evolutionary implications. Gene. 1997;205(1-2):125–40.
Wilkinson GS, Mayer F, Kerth G, Petri B. Evolution of repeated sequence arrays in the D-loop region of bat mitochondrial DNA. Genetics. 1997;146(3):1035–48.
Nesbø CL, Arab MO, Jakobsen KS. Heteroplasmy, length and sequence variation in the mtDNA control regions of three percid fish species (Perca fluviatilis, Acerina cernua, Stizostedion lucioperca). Genetics. 1998;148(4):1907–19.
Shao R, Barker SC, Mitani H, et al. Evolution of duplicate control regions in the mitochondrial genomes of metazoa: a case study with Australasian Ixodes ticks. Mol Biol Evol. 2005;22(3):620–9.
Yukuhiro K, Sezutsu H, Itoh M, et al. Significant levels of sequence divergence and gene rearrangements have occurred between the mitochondrial genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori. Mol Biol Evol. 2002;19(8):1385–9.
Lavrov DV, Boore JL, Brown WM. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Mol Biol Evol. 2002;19(2):163–9.
Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410(2):103–23.
Falkenberg M, Larsson N, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu Rev Biochem. 2007;76:679–99.
Chinnery PF, Hudson G. Mitochondrial genetics. Br Med Bull. 2013;106(1):135–59.
O’Neil JA. Determination of standard and field metabolic rates in two great lakes invading fish species: round goby (Neogobius melanostomus) and tubenose goby (Proterorhinus semilunaris). PhD Thesis. University of Windsor; 2013. http://scholar.uwindsor.ca/cgi/viewcontent.cgi?article=5988&context=etd. Accessed 19 Dec 2016.
Thacker CE. Biogeography of goby lineages (Gobiiformes: Gobioidei): origin, invasions and extinction throughout the Cenozoic. J Biogeogr. 2015;42(9):1615–25.
Thacker CE, Hardman MA. Molecular phylogeny of basal gobioid fishes: Rhyacichthyidae, Odontobutidae, Xenisthmidae, Eleotridae (Teleostei: Perciformes: Gobioidei). Mol Phylogenet Evol. 2005;37(3):858–71.
Iwata A, Sakai H, Shibukawa K, Jeon SR. Developmental characteristics of a freshwater goby, Micropercops swinhonis, from Korea. Zool Sci. 2001;18(1):91–7.
Thacker CE, Roje DM. Phylogeny of Gobiidae and identification of gobiid lineages. Syst Biodivers. 2011;9(4):329–47.
Nichols R. Gene trees and species trees are not the same. Trends Ecol Evol. 2001;16(7):358–64.
Nakhleh L. Computational approaches to species phylogeny inference and gene tree reconciliation. Trends Ecol Evol. 2013;28(12):719–28.
Kutschera VE, Bidon T, Hailer F, Rodi JL, Fain SR, Janke A. Bears in a forest of gene trees: phylogenetic inference is complicated by incomplete lineage sorting and gene flow. Mol Biol Evol. 2014;31(8):2004–17.
Keith P, Lord C. Systematics of sicydiinae. In: Patzner RA, VanTassell JL, Kovacic M, Kapoor BG, editors. The biology of gobies. Enfield: Science Publishers; 2011. p. 119–28.
Ishimatsu A, Gonzales TT. Mudskippers: front runners in the modern invasion of land. In: Patzner RA, VanTassell JL, Kovacic M, Kapoor BG, editors. The biology of gobies. Enfield: Science Publishers; 2011. p. 609–38.
Thacker CE. Molecular phylogeny of the gobioid fishes (Teleostei: Perciformes: Gobioidei). Mol Phylogenet Evol. 2003;3:354–68.
Wilson JM, Randall DJ, Donowitz M, et al. Immunolocalization of ion-transport proteins to branchial epithelium mitochondria-rich cells in the mudskipper (Periophthalmodon schlosseri). J Exp Biol. 2000;203:2297–310.
Panova M, Aronsson H, Cameron AR, Dahl P, Godhe A, Lind U, et al. DNA extraction protocols for whole genome sequencing in marine organisms. In: Bourlat S, editor. Marine genomics: Methods and Protocols. New York: Springer; 2016. p. 13–44.
Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 2012;13:238.
Kalchhauser I, Kutschera VE, Burkhardt-Holm P. The complete mitochondrial genome of the invasive Ponto-Caspian goby Ponticola kessleri obtained from high-throughput sequencing using the Ion Torrent Personal Genome Machine. Mitochondrial DNA. 2014;27(3):1887–1889.
Myers G. Efficient local alignment discovery amongst noisy long reads. In: Brown D, Morgenstern B, editors. Algorithms in Bioinformatics. WABI 2014. Lecture Notes in Computer Science, vol. 8701. Berlin: Springer; 2014. p. 52–67.
Chin C, Alexander DH, Marks P, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563–9.
Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539.
Su XZ, Wu Y, Sifri CD, Wellems TE. Reduced extension temperatures required for PCR amplification of extremely A + T-rich DNA. Nucleic Acids Res. 1996;24(8):1574–5.
R Development Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2008. http://www.r-project.org/. Accessed 19 Dec 2016.
IUCN. The IUCN Red List of Threatened Species: Version 2015-4. 2015. http://www.iucnredlist.org/. Accessed 19 Dec 2016.
Iwasaki W, Fukunaga T, Isagozawa R, et al. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol. 2013;30(11):2531–40.
Drummond AJ, Ashton B, Buxton S, et al. Geneious. v5.6. 2012. http://www.geneious.com/. Accessed 19 Dec 2016.
La Roche J, Snyder M, Cook DI, et al. Molecular characterization of a repeat element causing large-scale size variation in the mitochondrial DNA of the sea scallop Placopecten magellanicus. Mol Biol Evol. 1990;7(1):45–64.
Arnason E, Rand DM. Heteroplasmy of short tandem repeats in mitochondrial DNA of Atlantic cod, Gadus morhua. Genetics. 1992;132(1):211–20.
Ghivizzani SC, Mackay SL, Madsen CS, et al. Transcribed heteroplasmic repeated sequences in the porcine mitochondrial DNA D-loop region. J Mol Evol. 1993;37(1):36–7.
Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9(1):133–48.
Xu B, Clayton DA. RNA-DNA hybrid formation at the human mitochondrial heavy-strand origin ceases at replication start sites: an implication for RNA-DNA hybrids serving as primers. EMBO J. 1996;15(12):3135–43.
Nicholls TJ, Minczuk M. In D-loop: 40 years of mitochondrial 7S DNA. Exp Gerontol. 2014;56:175–81.
Jemt E, Persson O, Shi Y, et al. Regulation of DNA replication at the end of the mitochondrial D-loop involves the helicase TWINKLE and a conserved sequence element. Nucleic Acids Res. 2015;43(19):9262–75.
Asakawa S, Kumazawa Y, Araki T, et al. Strand-specific nucleotide composition bias in echinoderm and vertebrate mitochondrial genomes. J Mol Evol. 1991;32(6):511–20.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016. doi:10.1093/molbev/msw054.
Ronquist F, Teslenko M, van der Mark P, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42.
Silvestro D, Michalak I. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol. 2012;12(4):335–7.
Stamatakis A, Blagojevic F, Nikolopoulos DS, Antonopoulos CD. Exploring new search algorithms and hardware for phylogenetics: RAxML meets the IBM cell. J VLSI Sig Proc Syst. 2007;48(3):271–86.
Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol. 2008;57(5):758–71.
Froese R, Pauly D. FishBase. World Wide Web electronic publication. 2016. www.fishbase.org. Accessed 19 Dec 2016.
Thacker CE. Phylogeny of Gobioidei and placement within Acanthomorpha, with a new classification and investigation of diversification and character evolution. Copeia. 2009;1:93–104.
Thacker CE. Species and shape diversification are inversely correlated among gobies and cardinalfishes (Teleostei: Gobiiformes). Org Divers Evol. 2014;14(4):419–39.
Acknowledgements
We are grateful to Ulrika Lind, Marina Panova, Ricardo Pereyra and Vincent Somerville for help in the lab, to Sandrine Straub for support in automated mitochondrial genome annotations, to Céline Mäder for reference proofreading, to Gene Myers for supporting a collaboration on sequencing and assembling the round goby genome, to Tomas Larsson and Mats Töpel for help in genome assembly, and to Filip Volckaert for support and discussions.
Funding
Sequencing of the round goby mitochondrial genome was performed at the Genome Center Dresden c/o Deep Sequencing Group of the TU Dresden, Germany, and was supported by a grant from the Freiwillige Akademische Gesellschaft Basel to Irene Adrian-Kalchhauser.
Sequencing of the sand goby mitochondrial genome was performed within the Centre for Marine Evolutionary Biology at the University of Gothenburg (www.cemeb.science.gu.se), and was supported by a Linnaeus-grant from the Swedish Research Councils VR and Formas.
The funding sources had no role in the design of the study, in collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
The sequenced round goby specimen is currently being deposited at the Basel Natural History Museum, Switzerland. The catalogue number will be available from the first author IAK on request. The Genbank Accession number of the annotated sequence is KU755530. The head of the sequenced sand goby specimen is deposited in the Ichthyology Collection of the Swedish Museum of Natural History (NRM) with catalogue number NRM 69326. Submission to Genbank is ongoing. The accession number will be available from the second author OS.
Authors’ contributions
IAK conceived and planned the study, verified the round goby mitochondrial genome sequences, performed mitogenome length analyses, analysed and compared the control region sequences, analysed gene arrangements, wrote the manuscript and prepared the figures. OS participated in the planning of the study, coordinated the sand goby genome sequencing effort together with AB, analysed ecological traits and was a major contributor in writing the manuscript. VEK participated in planning the study, annotated the round and sand goby mitochondrial genomes, generated alignments, performed phylogenetic analyses, and was a major contributor in writing the manuscript. MAR, SW, MP and SS assembled and verified the round and sand goby mitochondrial genome sequences from next generation sequencing data. MAR and MP contributed to the writing of the manuscript. AB and PBH provided critical intellectual contributions to data analysis and to drafting and revising the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Fish used in this study were caught in the wild with permission GS 18-07-01 from the environmental departement Basel-Stadt and permission 1022H from the animal welfare committee Basel-Stadt, and with ethical permit 135-2010 from the Animal Ethics Committee of Gothenburg, according to best practise regulations from local fishery authorities.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Contains information on the sequences used for phylogenetic analysis and the references used for each species to extract biological and ecological characteristics. Table S2. Contains median mitochondrial genome sizes for ray finned fish. Table S3. Contains median mitochondrial genome sizes and class annotations for vertebrates. (XLSX 126 kb)
Additional file 2: Figure S1.
This figure shows the aligned non-coding sequences of all gobioids included into this study, with the query sequences of TAS and CSB elements indicated above the alignments, and hits for this query coloured in the alignment. (PDF 25604 kb)
Additional file 3: Figure S3.
This figure shows graphical representations of the MitoFish annotation results for all gobioid mitochondrial genomes. (PDF 28930 kb)
Additional file 4: Figure S2.
This figure shows sequence alignments of the individual occurrences of the repeat motifs identified in the round and the bighead goby, the extracted consensus sequence and the predicted secondary structure of the consensus. (PDF 1954 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Adrian-Kalchhauser, I., Svensson, O., Kutschera, V.E. et al. The mitochondrial genome sequences of the round goby and the sand goby reveal patterns of recent evolution in gobiid fish. BMC Genomics 18, 177 (2017). https://doi.org/10.1186/s12864-017-3550-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-3550-8