
Abstract
Background
With the release of genomic data for B.rapa, B.oleracea, and B.napus, research on the genetic and molecular functions of Brassica spp. has entered a new stage. PEBP genes in plants play an important role in the transition to flowering as well as seed development and germination. Molecular evolutionary and functional analyses of the PEBP gene family in B.napus based on molecular biology methods can provide a theoretical basis for subsequent investigations of related regulators.
Results
In this paper, we identified a total of 29 PEBP genes from B.napus that were located on 14 chromosomes and 3 random locations. Most members contained 4 exons and 3 introns; motif 1 and motif 2 were the characteristic motifs of PEBP members. On the basis of intraspecific and interspecific collinearity analyses, it is speculated that fragment replication and genomic replication are the main drivers of for the amplification and evolution of the PEBP gene in the B.napus genome. The results of promoter cis-elements prediction suggest that BnPEBP family genes are inducible promoters, which may directly or indirectly participate in multiple regulatory pathways of plant growth cycle. Furthermore, the tissue-specific expression results show that the expression levels of BnPEBP family genes in different tissues were quite different, but the gene expression organization and patterns of the same subgroup were basically the same. qRT‒PCR revealed certain spatiotemporal patterns in the expression of the PEBP subgroups in roots, stems, leaves, buds, and siliques, was tissue-specific, and related to function.
Conclusions
A systematic comparative analysis of the B.napus PEBP gene family was carried out at here. The results of gene identification, phylogenetic tree construction, structural analysis, gene duplication analysis, prediction of promoter cis-elements and interacting proteins, and expression analysis provide a reference for exploring the molecular mechanisms of BnPEBP family genes in future research.
Similar content being viewed by others


Background
As proteins with conserved functions throughout evolution, the phosphatidylethanolamine-binding protein (PEBP) family are widespread among archaeans, prokaryotes, and eukaryotes [1, 2]. In animals, as RAF kinase inhibitors, PEBP family members regulate the cell cycle and growth [3, 4]. In plants, the PEBP family has been shown to play a crucial role in regulating growth and morphogenesis. Phosphatidylethanolamine is a major component of plant biofilms and is responsible for protein recognition and signal transduction in signal transmission [5]. Studies have shown that the PEBP gene family has been conserved during evolution. The proteins encoded by family members contain a single and highly conserved PEBP/RKIP domain, which constitutes 80% of the gene coding sequence [6, 7]. PEBP genes in plants were originally cloned from inflorescence mutants, including the snapdragon CENTRORADIALIS (CEN) gene [8], the TERMINAL FLOWER 1 (TFL1) gene in A.thaliana [5], and the SELF-PRUNING (SP) gene in tomato [9]. It is believed that PEBP family genes are generally key regulators of the transition from vegetative growth to reproductive growth and determine the morphological structure of plants [8, 10, 11].
Currently, the PEBP gene family in plants is divided into three subfamilies, namely, FLOWERING LOCUS T (FT)-like proteins, MOTHER OF FT AND TFL1 (MFT)-like proteins, and TERMINAL FLOWER 1 (TFL1)-like proteins. Among them, the FT-like subfamily has two members, FLOWERING LOCUS T (FT) and TWIN SISTER FT (TSF); the MFT-like subfamily has only one member, the MFT gene; and the TFL1-like subfamily has three members: BROTHER OF FT and TFL1 (BFT), TERMINAL FLOWER 1 (TFL1), and A.thaliana CENTRORADIALIS (ATC) [12, 13].
The FT gene and its homologues have been identified and cloned in a variety of plant species. In the photoperiod pathway of flowering, long days can induce the expression of FT, and in the vernalization pathway, long-term light induction can increase the instantaneous accumulation of FT mRNA and promote the flowering of vernalized material [14,15,16]. TSF, as a member of the FT-like subgroup, performs the same function as FT. TSF has been shown to be a flowering integration factor and to promote flowering under short-day conditions by inducing cytokines [17, 18]. The main functions of TFL are to maintain vegetative growth and to promote indeterminate inflorescence growth. There are two TFL1-like homologous genes in pea: DETERMINATE (DET) and LATE FLOWERING (LF). DET maintains infinite inflorescence growth and is expressed in the apical meristem after the completion of the floral transition. When DET is mutated, the apex changes from infinite growth to limited growth, but the flowering time does not change, which is similar to the effects of the CEN gene in snapdragon, where LF induces flower formation. Both shoot apical meristem sites and shoot tips in the vegetative growth stage express CEN. When LF is mutated, flowering is advanced, but the state of the apical meristem does not change [19]. In A.thaliana, TFL1 controls plant morphological structure by regulating the LEAFY (LFY) and APETALA 1 (AP1) genes in the apical meristem. In wild-type A.thaliana, the LFY and AP1 genes are upregulated at the top of the floral meristem, thus producing a flower at the top of the stem and transforming the indeterminate inflorescence into a determinate inflorescence, which delays the flowering of the tfl1 mutant [5, 20]. The ATC gene in A.thaliana generates flower buds in a non-cell-autonomous manner. The ATC gene is specifically expressed in vascular tissue to produce ATC proteins. ATC proteins are then transported from vascular tissue to the shoot apex and bind to the FD protein to inhibit the expression of AP1, thus delaying flowering [21, 22]. RAS-RELATED GTP-BINDING NUCLEAR PROTEIN 2 (RAN2) is the homologous gene of ATC in rice. Overexpression of RAN2 in A.thaliana delays flowering and increases the number of branches. Overexpression of RAN2 in rice prolongs vegetative growth and increases branching [23]. Studies have shown that overexpression of BFT in RAN2 A.thaliana delays flowering and produces inflorescence structures similar to those produced in response to overexpression of the TFL1 gene. Thus, it is speculated that the BFT and TFL1 genes have similar functions [24]. In addition, under high-salt stress, BFT protein and FT protein compete for binding of the transcription factor FD in the nucleus, and interference with the binding of FT-FD also leads to delayed flowering [25, 26]. A previous study found that after the MFT gene was overexpressed in A.thaliana, the flowering time decreased slightly, but the response was not as significant as that to overexpression of the FT gene [27]. A study of A.thaliana MFT showed that MFT was specifically expressed in seeds in response to abscisic acid (ABA) and gibberellic acid (GA) signaling pathways and participated in the regulation of seed germination [28]. The wheat MFT homologue plays a key role in regulating seed germination during wheat seed development and regulating the effect of temperature on seed dormancy [29].
B.napus (Brassica napus, AACC, 2n = 38), the largest oil crop species in China, belongs to the cruciferous family, to the same as A.thaliana. The growth cycle and flowering time of plants directly determine their ecological adaptability, and studies have confirmed that the flowering time and maturity of B.napus are significantly positively correlated [30]. In recent years, in some areas of southern China with the "rape-rice-rice" farming method of three crops a year, the contradiction between B.napus and rice stubble has continued to intensify, resulting in a large amount of farmland being idle in the winter and the area of rapeseed planting continuing to decrease. In northwestern China, B.napus does not mature properly due to the high altitude and low temperature. The key factor to solve these problems is using heterosis to cultivate early-maturing B.napus varieties [31, 32]. The molecular mechanisms regulating the growth and development of A.thaliana have been studied. By means of map-based cloning, some quantitative trait loci (QTLs) and genes related to growth and development were successively obtained in B.napus. It has been demonstrated that members of the PEBP gene family play an important role in the growth and development of soybean, cotton, wheat, and other crop species [33]. However, studies on the identification, evolution, and expression of the PEBP gene family in B.napus are limited. In this study, the members of the A.thaliana PEBP gene family were used as reference sequences, and the BLASTP method was used to determine the PEBP gene family members in B.napus obtain information on this family in the ancestor species B.rapa and B.oleracea, construct a phylogenetic tree, and analyse gene structure. Analysis of protein physicochemical properties, selection pressure, predicted promoter cis-elements and interacting proteins, and tissue-spiecific expression can provide a theoretical reference for the study of candidate genes for flowering traits in B.napus.
Results
Identification of PEBP family members
To screen and identify members of the PEBP gene family in B.napus, B.rapa, and B.oleracea, six PEBP protein sequences from A.thaliana were used as query sequences for BLASTP analysis via the BRAD website. Ultimately, 14, 14, and 29 PEBP gene family members were identified in B.rapa, B.oleracea, and B.napus, respectively. Among the 29 BnPEBP genes identified, BnaC03G0275900ZS was the longest (over 3724 bp), and BnaA04G0179000ZS was the shortest (only 445 bp). The three subgroups FT-like, MFT-like, and TFL-like of the PEBP gene family in B.napus contain 8, 4, and 17 members, respectively. FT has six homologous genes, TSF has two homologous genes, MFT has four homologous genes, TFL1 has six homologous genes, ATC has nine homologous genes, and BFT has two homologous genes. The data in Table 1 suggest that the A.thaliana AT1G65480 and AT2G27550 genes have no homologues in B.rapa and B.oleracea. However, there was 1 BnaFT-C04 homologue and 3 genes (Bnascaffold0139G0000200ZS, Bnascaffold0105G0000300ZS, and Bnascaffold0027G0054400ZS) homologues in B.napus, and it is preliminarily speculated that the genes formed in B.napus through the distant hybridization of B.rapa and B.oleracea. Genome doubling leads to gene expansion, which can persist after tens of thousands of years of evolution in B.napus, indicating that these genes have an important impact on the growth and development of this species. In addition, BolTFL-C04, BraATC-A04, and BraATC-Ann have no homologues in B.napus. There are two reasons for this speculation. First, these three genes may have been eliminated during the genome evolution of B.rapa and B.oleracea; second, they may have been pressnt in B.rapa and B.oleracea during hybridization but doubled in B.napus. It is possible that some functionally similar genes replaced the redundant PEBP genes. In general, although the PEBP gene family has been lost during the evolution of B.napus, the FT-like and ATC-like genes in the B.napus genome have been amplified to a certain extent, which means that gene replication plays a very important role in the evolution of B.napus.
Phylogenetic analysis and classification of PEBP genes
To explore the phylogenetic relationships of the PEBP gene family in cruciferous species, we investigated a total of 63 PEBP members from B.napus (29 genes), B.rapa (14 genes), B.oleracea (14 genes), and A.thaliana (6 genes). Sequence alignment was performed using Jalview software, and a phylogenetic tree was constructed by the neighbour-joining method using MEGA software (Fig. 1). The phylogenetic tree divided these 63 proteins into five subgroups: the ATC-like subgroup, the TFL1-like subgroup, the TSF/FT-like subgroup, the BFT-like subgroup, and the MFT-like subgroup. Among them, the ATC-like subgroup and TSF/FT-like subgroup had the most members at 18 each, while the BFT-like subgroup had the fewest members at 5 (Fig. 1). According to the clustering in Fig. 1, the PEBP family members of the A sub-genome in B.napus may be derived from the A genome of B.rapa, and the corresponding members of the C sub-genome may be derived from the C genome of B.oleracea. These results further suggest that genome doubling is responsible for the increase in the gene copy number of PEBP family members in B.napus.

Phylogeny of the PEBP family in B.napus , B.rapa, B.oleracea, and A.thaliana. Note: The proteins are clustered into six subgroups. Yellow, light blue, orchid, light green, orange and pink sections indicate the subgroups of PEBP proteins. BnaPEBPs are marked with green dots, BraPEBPs are marked with purple dots, BolPEBPs are marked with blue stars, AtPEBPs are marked with red triangles, and 9 BnPEBPs core genes are marked with red stars
Gene structure and motif compositions
It is known that structure determines function. To analyses the function of the PEBP gene family, the structure of 29 BnPEBP gene family members was analysed, and the results showed that the gene structure of BnPEBP family members was highly conserved (Fig. 2). The motif prediction results (Fig. 2a) show that motif 3, motif 4, motif 5, and motif 6 are characteristic motifs of the BnPEBP gene family, except for BnTSF-C06, BnATC-A04, and BnBFT-C04, which contain only motif one and motif two. These results suggest that BnPEBP members from different subgroups have a simple structure and that specific motifs may lead to different roles for these genes based on the function of the protein produced. Figure 2b shows that the BnPEBP gene family has the same conserved domain. Gene structure analysis (Fig. 2c) showed that, except for BnTSF-C06, BnATC-A04, and BnBFT-C04, which contain 2 exons and 1 intron, most members contain 4 exons and 3 introns. Combined with phylogenetic tree analysis results, these results revealed that there were also differences in terms of the unified score, such as for BnTSF-C06 in BnTSF and for BnATC-C04 and BnATC-A04 in BnATC. The structures of these three genes are relatively similar, while the length of their introns is different; however, since the intron is removed during transcription and translation, the function of the gene is not affected. This suggests that transposable elements may have been inserted into these genes during evolution, resulting in the fragmentation of the coding sequence into multiple segments. In addition, the different combinations of introns and exons may be due to the different splicing methods used by genes during evolution, which can lead to proteins with specific functions being produced to meet certain biochemical requirements.

Construction of a phylogenetic tree of 29 PEBP genes in B.napus with TBtools. Note: The visual display of the BnPEBP evolutionary tree, gene motifs (a), domains (b) and structure (c)
Chromosomal distribution and homology analysis
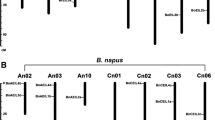
Analysis of the chromosomal location of 29 members of the PEBP gene family in B.napus revealed that these genes were not distributed evenly on each chromosome. Among them, 3 genes were on unassembled scaffold segments, and the remaining 26 BnPEBP genes were distributed on 14 chromosomes. There were 12 BnPEBP genes in the A genome and 14 BnPEBP genes in the C genome. There were no BnPEBP genes on chromosomes A01, A05, and C01, and most genes were distributed on chromosomes A07, C02, C03, and C04, all of which contained 3 genes (Fig. 3).

Distribution of PEBP family genes on the chromosomes of B.napus
There are many duplicated genes in the B.napus PEBP gene family. Because the B.napus genome has undergone large-scale chromosomal doubling and duplication, these duplicated genes exist not only in the form of adjacent genes but also as genes on different chromosomes. In this study, to understand the potential duplication events between BnPEBP genes, TBtools software was used to analyse the gene duplication types of the BnPEBP family. A Circos plot was constructed (Fig. 4). The Intraspecific collinearity analysis of the PEBP genes in B.napus was carried out, and a total of 35 collinear genes were detected. Among them, there were 3, 9, and 23 pairs of homologous genes (Supplementary Table S1) between the A-A, C–C, and A-C genomes, respectively. These genes were distributed on 14 chromosomes in sub-genomes A and C of B.napus, and each chromosome contained one ~ six genes, and the mode of gene replication was fragment replication. In addition, some genes did not show intraspecific collinearity. Finally, we speculate that gene fragment replication may be the main mode of PEBP gene replication mode in the B.napus genome.

Duplication of BnPEBP genes on the chromosomes of B.napus L.
Based on the genome annotation information in the Ensembl Plants database, in this study, the PEBP genes of A.thaliana, B.oleracea, B.rapa, and B.napus were subjected to collinearity analysis and visualized by TBtools (Fig. 5). The results of the collinearity analysis showed that there were 43, 49, and 18 pairs of collinearity relationships between BnPEBP gene family members with BoPEBP, BrPEBP, and AtPEBP, respectively (Supplementary Table S2). The BnPEBP gene family experienced a whole-genome duplication (WGD) event during its evolution. There are 29 B.napus PEBPs, among which there may be at least 10 BnPEBPs from the B.rapa genome and at least 9 BnPEBPs from the B.oleracea genome. That is, genome duplication is the main reason for the rapid expansion of the BnPEBP gene family in B.napus. On the other hand, it is speculated that some members of the PEBP gene family formed after the duplication of the genome.

Synteny analysis of PEBP genes between B.napus and its progenitors. Note: The grey lines in the background indicate the collinear blocks within B.napus and its progenitor species. The purple-red lines highlight the syntenic PEBP gene pairs. (a) indicates the genome collinearity of B.napus (AACC) and B.rapa (AA), and (b) indicates the genome collinearity of B.napus (AACC) and B.oleracea (CC). (c) indicates B.napus (AACC) and A. thaliana genome collinearity. To show the completeness of the results, genes located in random regions are not shown in the figure
Physicochemical properties and subcellular localization
Physicochemical analysis of BnPEBP family protein members showed that the number of amino acids encoded by the 29 BnPEBP genes varies from 76 (BnaATC-C04) to 202 (BnaTSF-C02). The molecular weight is between 8.41 and 22.96 kDa; the gene with the highest molecular weight is BnaTSF-C02, and the gene with the lowest is BnaATC-C04. Members of the BnPEBP protein family have isoelectric points (pIs) ranging from 6.38 to 9.56. The pIs of BnPEBP protein members ranged from 6.38 to 9.56. Val is the most abundant amino acid in the proteins of this family. The instability coefficient of BnPEBP protein members ranges between 26.9 and 51.58. An instability coefficient greater than 40 was used as the threshold; thus, there are 16 stable proteins in B.napus, while the remaining 13 proteins are unstable. The fatty acid coefficients of the 29 BnPEBP proteins range from 74.86 to 100.70, and the hydrophilicity (GRAVY) values (J) of the proteins are all less than 0, indicating that they are all hydrophilic. The subcellular localization results of 29 BnPEBP genes showed that 23 genes are expressed in the cytoplasm, 5 are expressed in the periplasm, and 1 gene is expressed in the extracellular space (Table S3).
Selection pressure analysis
There may be selection pressure due to environmental stress and changes in survival during the process of species differentiation. Some genetic changes are related to the environment; that is, environmental variables can directly modify a gene, and the development of particular mutations promotes this phenomenon. However, some mutations are neutral, while others have deleterious effects, and such mutations are easily eliminated during the evolutionary process. Therefore, a mutation that is beneficial to the biological adaptation to the environment is considered to be under positive selection; however, neutral selection and purifying selection do not conducive to biological adaptations to the environment. To explore the evolutionary pressure on the BnPEBP genes, we calculated the nonsynonymous substitution rate (Ka), the synonymous substitution rate (Ks), and their ratio (Ka/Ks) between homologous gene pairs (Table S4). The Ka/Ks ratios of the PEBP gene family in B.napus were all less than 1, indicating that the evolution of the PEBP gene family in B.napus was relatively conserved and subject to strict purifying selection. The Ka/Ks ratios of the TSF and FTL1 subgroups were larger than 0.2, while the Ka/Ks ratios of the ATC, MFT, FT, and BFT subgroups were smaller, indicating that the TSF and TFL1 subfamilies are more conserved.
Promoter cis-acting element Analysis
Cis-regulatory sequences can regulate plant growth, development, and physiological metabolism by regulating gene expression. To better understand the transcriptional regulation and potential function of the BnPEBP genes, in this study, the promoter sequence for investigation was the 2000 bp sequence upstream of the BnPEBP translation start site (ATG), and the cis-elements in the promoter sequences were examined using the PlantCARE database. The results are shown in Fig. 6. The promoter sequences of all BnPEBP genes contained multiple cis-acting elements, indicating that BnPEBP genes are involved in a complex regulatory network. The analysis revealed 39 cis-acting elements with known functions in the BnPEBP gene and several cis-acting elements with unknown functions. Cis-acting elements with known functions can be categorized into five groups: the first group consists of core promoter elements, which include the TATA-box and CAAT-box; the second group consists of light-responsive elements, including the GT1 motif, the AAAC motif, SP1, the TCC motif, the GTGC motif, the TCT motif, and the AE-box; the third group consists of hormone-responsive elements, including auxin response elements (TGA elements and AUXRR-CORE elements), ABA response elements (ABREs) and gibberellin response elements (GARs); the fourth group consists of several abiotic stress response elements, including anaerobic induction elements (AREs) and defence response elements; and the fifth group consists of transcription factor-binding sites and others, including AT-rich elements and CCAAT-boxes. In addition, some elements of unknown function were also predicted. Furthermore, studies have found that some gene promoter regions contain other cis-regulatory elements related to plant development, including meristem expression (CAT-boxes), endosperm expression (GCN4 motifs), circadian rhythm regulation-related elements, and palisade mesophyll cell differentiation (HD-Zic1)-related cis-regulatory elements. Except for the core promoter element, which is the most abundant, the remaining elements are shown in Fig. 6. Several types of cis-elements are related to plant growth and development, which further indicates that the PEBP gene family regulates plant growth and development and plays an important role in this process.

Analysis of cis-acting elements in the BnPEBP gene promoter. Notes: The image does not contain any unannotated cis-acting elements
Interaction protein prediction
A protein interaction network comprising the members of the BnPEBP gene family identified in B.napus was constructed; the prediction results are shown in Fig. 7. Ten BnPEBP proteins interacted with each other and with ATBZIP, GI, SPY, ABI5, and HDG7. Five proteins were found to interact with a PPI enrichment p value of 1.0e-16, which means that the interactions between the proteins were stronger than those expected for a random set of proteins of the same size and degree distribution extracted from the larger set. Using the betweenness centrality index (BC) index to screen core proteins in Cytoscape software, we found that FT, TSF, MFT, and TFL1 in the BnPEBP protein family were regulated by ATBZIP and GI proteins. Various proteins in the entire protein interaction network are involved in the regulation of flowering. Among them, ATBZIP, a transcription factor, is involved in the regulation of plant light responses, while GI is a circadian rhythm gene that responds to photoperiod signals together with CO and acts on downstream FT genes to regulate flowering. As shown in Fig. 6, in addition to gene family members, SPY regulates flowering in response to the gibberellin pathway, and ABI5 is involved in ABA signaling during seed maturation and germination, including sensitivity and regulation to ABA inhibition of seed germination. The expression of some ABA genes is involved in the regulation of the plant growth process. HDG7 is a transcription factor that is widely involved in plant development and participates in the development of plant vascular tissue, the formation of organs, and the response to stress. Therefore, the network diagram of protein interactions indicates that BnPEBP family members directly or indirectly participate in multiple regulatory pathways during plant growth and development.

Predicted BnPEBP gene family protein interaction network
Tissue-specific expression analysis
To understand the role of PEBP family genes during plant development, we used published data corresponding to different tissues of the B.napus cultivar "Zhongshuang 11" (cotyledon, root, vegetative rosette, stem, leaf, sepal, petal, filament, pollen, bud, silique, silique wall, and seed tissue) and performed a transcriptomic data analysis of the expression of BnPEBP family genes (Fig. 8). A gene expression heatmap was constructed and showed that the BnaPEBP family genes show tissue-specific expression in B.napus. The BnaFT gene in the BnaFT-like subgroup was highly expressed in the stem, leaf, and silique wall tissues, which is consistent with the production and transport of FT from leaves to stem apical meristems to promote plant flowering. BnaTSF-C02, which is typically found in the roots and during the vegetative stage, was expressed at higher levels in the rosette leaves, and BnaTSF-C02 was expressed in the stems and leaves. The BnaMFT-like subgroup genes showed higher expression in the seeds than in the filaments and pollen. BnaACT in the BnaTFL-like subgroup was highly expressed in the cotyledons, but the level was low or not detected in the petal, filament, pollen, and bud tissues; BnaTFL1 gene expression was relatively low in the early vegetative growth stage but high in the late siliques, silique walls, and seeds. The BnaBTF gene showed low expression across the different tissues. The above results show that the BnPEBP family genes in B.napus have different expression levels in different tissues, but the expression patterns of genes in the same subgroup are essentially the same, which may be related to their evolution from the same gene.

Tissue-specific expression analysis of PEBP gene members in B.napus
Expression pattern analysis of BnPEBP genes by qRT‒PCR
The B.napus cultivar "Zhongshuang 9" was chosen as the experimental object based on the tissue-specific expression analysis of BnPEBP family genes to verify and compare the expression patterns of the same group of homologous genes in roots, stems, leaves, buds, and siliques of three plants with consistent growth at 10 days after flowering. First of all, it can be seen from Figs. 8 and 9 show that in these five tissues, the expression of genes belonging to the same subgroup was quite different. The BnTFL1-C02 gene in the BnTFL1-like subgroup has a high expression level in roots and siliques, which is significantly different from the pattern in other genes, and the gene BnTFL1-A03 has a high expression level in buds, which is significantly different from the expression pattern of other genes. Three genes in the BnFT/TSF-like subgroup are highly expressed in roots and stems, and most of them are expressed in leaves, buds, and siliques, but the expression level is low with little variation. The expression of the BnATC-like subgroup gene varies greatly among tissues, especially that of BnATC-027, which is high in leaves, while other genes are expressed at low or notat all. The expression level of the BnMFT-like subgroup gene differed significantly among the five tissues. In addition to that of BnMFT-C08, the expression levels of three other genes in leaves, buds, and siliques were very low, which was significantly different from that of BnMFT-C08; the expression level of the BnBFT-like subgroup was low, and there was a significant difference between the two genes in buds and siliques. Different from that in the reference genome "Zhongshuang 11,"the expression level showed an overall increase in expression in "Zhongshuang 9" It is speculated that the growth period is approximately 25 days earlier in "Zhongshuang 9" than in "Zhongshuang 11," as the BnPEBP family is related to plant growth and development. Therefore, there was a trend towards higher expression in the early-maturing materials. In addition, different sampling periods and different plant growth conditions are also reasons for large differences in expression patterns. Second, BnPEBP family genes were expressed in different tissues of B.napus, and the BnaTFL1 subgroup was more highly expressed in buds than other tissues, which can be explained by the function of this gene in flowering and inflorescence growth. The BnaMFT subgroup and the BnaATC subgroup were highly expressed in the roots, which is unique. The expression of BnaFT/TSF in these five tissues was similar to that in the reference genome, with higher values in the aboveground tissues. The overall expression of the BnaBFT subgroup was low, but stable expression was observed in roots, stems, leaves, buds, and siliques.

Expression levels of 5 subgroups of PEBP genes in roots, stems, leaves, buds, siliques of B.napus by qRT-PCR
Discussion
B.napus, a naturally doubled allotetraploid crop species, has a complex genome. With the release of genomic data from B.rapa, B.oleracea, and B.napus, research on the genetic and molecular functions of B.napus has entered a new stage [34,35,36]. PEBP gene belong to a relatively small gene family in plants and play an important role in the flowering transition, seed development, and germination of plants [37]. To date, 19 PEBP genes have been reported in rice [38], 25 in maize [39], and 23 in soybean [40]. In this study, 14, 14, and 29 PEBP gene family members were identified in B.rapa, B.oleracea, and B.napus, respectively, by genome-wide searches. This shows that the number of PEBP genesdiffers among apecies, possibly related to species specificity. According to Table 1, the number of PEBP genes in B.napus is not the sum of the numbers in B.rapa and B.oleracea, indicating that members of the PEBP gene family were also lost or expanded with the genome duplication of B.napus. Phylogenetic analysis (Fig. 1) revealed that the PEBP genes of B.napus and its progenitor species, unlike those of other plant species, were divided into five subgroups, the ATC-like subgroup, the TFL1-like subgroup, the TSF/FT-like subgroup, the BFT-like subgroup, and the MFT-like subgroup, which were different from the three main subfamilies identified in previous research [41]. The amino acid sequences of FT and TFL1 are similar, but their functions are opposite. Hanzawa performed a detailed study on this conjecture, proving that the histidine residue (His88) at the 88th position of the TFL1 protein and the tyrosine residue (Tyr85) at the 85th position of the FT protein play a decisive role in the opposite functions [42]. The number of members of the ATC-like subgroup is significantly higher than that of other subgroups, indicating that this subgroup has undergone gene expansion as part of the evolutionary process and may play a more important role in the growth and development of B.napus than other subgroups.
Collinearity analysis can be used to predict inheritance and potential functions. The analysis of intraspecific and interspecific collinearity of the PEBP gene in B.napus shows that the mode of PEBP gene replication in this species is fragment replication, and the mode of gene expansion is genome doubling. There are some genes without a collinear relationship, and it is possible that chromosome recombination and homologous replacement caused by genome doubling and subsequent doubling events in B.napus have caused complex changes in gene expansion and loss to varying degrees. In a study of the wheat PEBP gene family [43], it was found that other genes also display this phenomenon.
In addition, we also predicted the cis-elements in the promoter; except for the core components, a large number of which are related to environmental response, such as light-responsive elements, hormone-responsive elements, and abiotic stress response elements. Combined with the function of the PEBP gene family in plant growth and development, it demonstrates that the promoter of PEBP gene family plays an important role in regulating plant growth and development. It can be inferred that the type of PEBP gene promoter may be an inducible promoter. In previous articles, there was little information about this; this finding can be used as a reference in the future study of gene function in this family. Furthermore, a protein interaction network comprising the members of the BnPEBP gene family identified in B.napus was constructed; the prediction results are shown in Fig. 7. We found that various proteins in the entire protein interaction network that are involved in the regulation of plant flowering. Among them, CO and SPY regulate plant flowering by participating in the photoperiodic and gibberellin pathways, respectively. ABI5 is involved in ABA signal transduction during seed maturation and germination, That is, the interacting proteins also prove that the PEBP protein plays an important role in the development of plant vascular tissue and the formation of organs. From this point of view, there is a certain relationship between the cis-element of the promoter and protein interaction, which can be used as an analytical direction in future research.
Tissue expression data and qRT‒PCR analysis results collected throughout the growth period revealed that BnPEBP genes were expressed at varying levels in distinct tissue regions. In this study, it was found that the MFT-like subgroup was specifically expressed in fruits and highly expressed in mature fruits. This result is consistent with the expression characteristics of GmMFT of Glycine max in fruit clips [44], suggesting that the MFT-like subgroup plays an important role in seed maturation. In addition, the ATC-like subgroupwas also highly expressed in a specific tissue: cotyledons. The FT/TFL1-like subgroup was expressed in multiple tissues, showing pan-tissue expression, Extensive tissue expression characteristics also indicate that FT/TFL1-like subgroup plays an important role in the growth and development of B.napus. Furthermore, we used qRT‒PCR to measure the expression levels in the roots, stems, leaves, buds, and siliques of the B.napus cultivar "Zhongshuang 9." The expression patterns of most BnPEBP homologous genes were similar and tissue specific. However, the pattern was not consistent with the tissues showing high expression in the reference cultivar "Zhongshuang 11," which may be due to the different material. In addition, different sampling periods and different plant growth conditions are also reasons for the large differences in expression patterns.
Conclusions
In conclusion, 29 PEBP genes identified in B.napus were systematically studied in terms of gene evolution, gene structure, collinearity, cis-acting elements, physical and chemical properties of proteins, and gene expression patterns by bioinformatics methods. The evolution of the PEBP genes in B.napus was relatively conserved and it was proved that genome doubling and fragment duplication are the ways by which PEBP genes expand and replicate in B.napus. Prediction of cis-elements and analysis of interacting proteins provide new research evidence for the role of PEBP gene in the growth and development of B.napus, and the tissue expression patterns of the PEBP gene are closely related to their function. To date, only a few genes representing the PEBP gene family have been characterized in detail in B.napus. This study provides nearly complete information on the BnPEBP gene family, and our findings point to a new direction for further research into the evolution and function of PEBP family genes in B.napus and its progenitors.
Methods
Identification of the PEBP gene family members
The Stockholm model of PEBP (IPR008914) was obtained from the InterPro website (https://www.ebi.ac.uk/interpro/), and then all A.thaliana PEBP protein data were retrieved from the HMMER website (https://www.ebi.ac.uk/Tools/hmmer/) [45]. The CDSs of members of the A.thaliana PEBP gene family () were downloaded from the Arabidopsis Information Resource (TAIR) website (https://www.arabidopsis.org/). To identify PEBP gene candidates, the CDSs and protein sequences of the A.thaliana PEBP gene family members were used to search the protein database of the B.napus genome and its ancestral species by BLASTP (e value ≤ 1e−5) with the BRAD database (http://brassicadb.cn). Among them, Zhongshuang 11 was selected as the reference genome (Brassica napus, AACC, 2n = 38) for B.napus, Brara_Chiifu_V3.0 was used as the reference genome (Brassica rapa, AA, 2n = 20) for B.rapa, and Braol_JZS_V2.0 was used for the B.oleracea reference genome (Brassica oleracea, CC, 2n = 18).
Multiple sequence alignment and evolutionary analysis of PEBP gene family
The 29 PEBP gene family sequences obtained in B.napus were completely aligned using MEGA 11.0 software [46]. First, MEGA 11.0 software was used to perform multiple sequence alignment of selected gene coding region sequences and translate them into amino acid sequences to construct an evolutionary tree through ClusterW. The Allgnment parameter was selected from the ClusterW options. Pairwise alignment was performed with gap opening penalty and gap extension penalty values of 15.00 and 6.66, respectively. Multiple Alignment was performed with a gap opening penalty and gap extension penalty of 15.00 and 6.66, respectively. Multiple sequences were aligned,and and the results were saved in MEGA format. A molecular evolutionary phylogenetic tree was constructed by the neighbor-joining (NJ) method, and the bootstrap parameter was 1000. The substitution type in the "substitution model" was "Nuckeotide," and the "Model/Method" as the "Tamura Nei model." Other parameters were defined as follows: Rates among sites in "Rates and patterns" as " Uniform rates, " " the Gap/Sending data treatment in Data subset to use" as "use all sites, " the ML heuristic method in "Tree input options" as "Nearest Highborn Interchange (NNI) ", and "the Initial tree for ML" as "Make initial tree automatically (Default NJ/BioNJ), " to build an evolutionary tree, and "Tree Style" to "Circle.". After preliminary construction of the evolutionary tree, the EvolView website (www.evolgenius.infi/evolview/#/terrview) was used for further beautification.
Gene structure and motif prediction of BnPEBP genes
The annotation files of PEBP gene family members were downloaded from the BnTIR website (http://yanglab.hzau.edu.cn/). Gene structure was subsequently analysed, and a gene structure map was drawn by online Gene Structure Display Server (GSDS) analysis (http://gsds.gao-lab.org/) [47]. MEME software online (http://meme-suite.org/tools/meme) was used to identify the conserved motifs of the PEBP protein sequence in B.napus [48]. The number of motifs was set to 10, and the remaining parameters were set to default conditions. Finally, TBtools was used to visualize the gene evolutionary tree, gene structure, domains, and conserved motifs [49].
Chromosomal distribution and homology analysis of BnPEBP genes
According to the information within the BnTIR database (http://yanglab.hzau.edu.cn/), the specific positions of the PEBP gene family members on the chromosomes of B.napus were analysed. Then, TBtools software was used to map these genes to individual chromosomes.
Gene duplication is considered one of the main drivers of the evolution of genomes and genetic systems [50]. It includes six different mechanisms (WGD, tandem, proximate, DNA-based transmission, retrotransmission, and separation). Among these patterns, fragments and tandem repeats are considered to be two main methods for the expansion of plant gene families [51]. Segmental duplication replicates multiple genes by genome doubling, usually on different chromosomes, and tandem duplications are characterized by regions of chromosomal recombination where multiple gene members are arranged in the same intergenic region or adjacent intergenic regions on the same chromosome [52, 53]. B.napus is an allotetraploid plant species formed by natural genome duplication. Many gene chromosomal segmental duplication events have occurred during the long-term evolutionary process. We are interested in segmented and tandem repeat events. FASTA genomic data files and GFF3 gene annotation files of B.napus, B.rapa, B.oleracea, and A.thaliana were downloaded from the Ensembl Plants website (http://plants.ensembl.org/index.html), TBtools was used to analyse the collinearity genes of the PEBP gene within B.napus species, between B.napus and B.rapa, B.oleracea, and A.thaliana, and visualize the results.
Physicochemical properties and subcellular localization of BnPEBP genes
The physicochemical properties, including the number of amino acids, most abundant amino acids, molecular weight (MW), isoelectric point (pI), instability coefficient, hydrophobicity coefficient, grand average of hydropathy (GRAVY) value, and subcellular localization, of all BnPEBP proteins were predicted using the ExPASy website (http://web.expasy.org/protparam/) [54].
Selection pressure analysis
In the process of evolutionary analysis, the Ka/Ks value is used to infer natural selection on genes and to determine the degree of environmental influence on homologous genes during the evolutionary process, as well as the direction of gene mutation. If Ka/Ks is greater than one, there is a positive selection effect; if Ka/Ks is less than one, there is neutral selection; and if Ka/Ks is one, there is purifying selectivition. The ratio of non-synonymous mutations (Ka) to synonymous mutations (Ks) was calculated by the Tbtools built-in Ka/Ks Calculator estimate the selection pressure on the 29 PEBP gene family members in B.napus over the course of evolution, with A.thaliana PEBP family genes as a reference.
Analysis of cis-acting elements in the BnPEBP promoter
The 2000 bp sequence upstream of the BnPEBP genes was extracted using TBtools to analyses cis-acting elements, sequences were organized in one text file, and then PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) [55] was used to identify cis-acting elements. The files were upload, searches were performed in batches, and the final result was fed back as a TXT file. TBtools was then used to visualize the results.
Interacting protein predictions
The STRING database contains a collection of protein interaction data from many species [56]. Sequences of the 29 BnPEBP genes orthologous to those in A.thaliana were input into the STRING database to identify the homologous BnPEBP genes in B.napus. The source genes were predicted via protein interaction networks, the prediction results were saved as a text file, and Cytoscape 3.7.2 was ultimately used to visualize the results [57].
Tissue-specific expression analysis of BnPEBP family genes
To further analyse the expression of BnPEBP family genes in B.napus tissue, the expression data of 29 BnPEBP family genes in different tissues and at different times of B.napus cultivar "Zhongshuang 11 were downloaded from the BnTIR website (http://yanglab.hzau.edu.cn/). TBtools was used to draw a heatmap based on the log2-transformed expression data from each family member.
qRT‒PCR analysis of BnPEBP family genes
Based on the expression analysis results of PEBP gene in different tissues of B.napus, we selected B.napus "Zhongshuang 9" as the experimental material and collected five tissues (root, stem, leaf, bud, and silique) of three plants with consistent growth at 10 days after flowering for a real-time fluorescence quantitative PCR assay. The qRT‒PCR quantitative experiment was completed with a kit from Shanghai Yeasen Biological Company. The first round of reverse transcription was performed with Hifair® III 1st Strand cDNA Synthesis SuperMix for qPCR® and QPCR SYBR Green Master Mix (Low Rox Plus). Primer 5.0 software was used to design specific primers for quantitative analysis (Supplementary Table S5), and Actin7 (GeneBank ID:GBEQ01027912.1) was used as an internal reference gene. Real-time PCR was performed with an AB-7500 fluorescence quantitative PCR apparatus, with three biological replicates and three technical replicates for each tissue, Relative expression of genes was quantified using the 2−△△CT method to identify the expression patterns of the BnPEBP genes in different tissues of B.napus.
Availability of data and materials
All data generated or analysed during this study are contained in the following persistent WEB links or access dataset.
InterPro website: (https://www.ebi.ac.uk/interpro/) the PEBP family is included in the InterPro database under number ‘IPR008914’.
HMMER website: https://www.ebi.ac.uk/Tools/hmmer/.
Arabidopsis Information Resource website: https://www.arabidopsis.org/. The PEBP family member is included in the Arabidopsis database under number ‘AT1G65480, AT4G20370, AT1G18100, AT5G03840, AT2G27550, AT5G62040’.
BRAD database (B.napus Zhongshuang11 genome, B.rapa Chiifu_V3.0 genome and Brassica oleracea JZS_V2.0 genome information): http://brassicadb.cn.
Gene expression data were downloaded through the BnIR website: http://yanglab.hzau.edu.cn/, which integrates transcriptome data for all genes of Zhongshuang 11. Open the website and enter the gene name to download gene expression data without accession numbers.
ExPASy online website: https://web.expasy.org/protparam/.
Ensembl Plants website: http://plants.ensembl.org/index.html.
PlantCARE website: http://bioinformatics.psb.ugent.be/webtools/plantcare/html/.
References
Chautard H, Jacquet M, Schoentgen F, Bureaud N, Bénédetti H. Tfs1p, a member of the PEBP family, inhibits the Ira2p but not the Ira1p Ras GTPase-activating protein in Saccharomyces cerevisiae. Eukaryot Cell. 2004;3:459–70.
Banfield MJ, Barker JJ, Perry ACF, Brady RL. Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction. Structure. 1998;6:1245–54.
Schoentgen F, Jollès P. From structure to function: possible biological roles of a new widespread protein family binding hydrophobic ligands and displaying a nucleotide binding site. FEBS Lett. 1995;369:22–6.
Bernier I, Jollés P. Purification and characterization of a basic 23 kDa cytosolic protein from bovine brain. Biochim Biophys Acta. 1984;790:174–81.
Bradley D, Ratcliffe O, Vincent C, Carpenter R, Coen E. Inflorescence commitment and architecture in A.thaliana. Science. 1997;275:80–3.
Chardon F, Damerval C. Phylogenomic analysis of the PEBP gene family in cereals. J Mol Evol. 2005;61:579–90.
Hanzawa Y, Money T, Bradley D. A single amino acid converts a repressor to an activator of flowering. Proc Natl Acad Sci U S A. 2005;102:7748–53.
Bradley D, Carpenter R, Copsey L, Vincent C, Rothstein S, Coen E. Control of inflorescence architecture in Antirrhinum. Nature. 1996;379:791–7.
Pnueli L, Carmel-Goren L, Hareven D, Gutfinger T, Alvarez J, Ganal M, et al. The SELF-PRUNING gene of tomato regulates vegetative to reproductive switching of sympodial meristems and is the ortholog of CEN and TFL1. Development. 1998;125:1979–89.
Kobayashi Y, Kaya H, Goto K, Iwabuchi M, Araki T. A pair of related genes with antagonistic roles in mediating flowering signals. Science. 1999;286:1960–2.
Kardailsky I, Shukla VK, Ahn JH, Dagenais N, Christensen SK, Nguyen JT, et al. Activation tagging of the floral inducer FT. Science. 1999;286:1962–5.
Danilevskaya ON, Meng X, Hou Z, Ananiev EV, Simmons CR. A genomic and expression compendium of the expanded PEBP gene family from maize. Plant Physiol. 2008;146:250–64.
Karlgren A, Gyllenstrand N, Källman T, Sundström JF, Moore D, Lascoux M, et al. Evolution of the PEBP gene family in plants: functional diversification in seed plant evolution. Plant Physiol. 2011;156:1967–77.
Corbesier L, Vincent C, Jang S, Fornara F, Fan Q, Searle I, et al. FT protein movement contributes to long-distance signaling in floral induction of A.thaliana. Science. 2007;316:1030–3.
Mouradov A, Cremer F, Coupland G. Control of flowering time: interacting pathways as a basis for diversity. Plant Cell. 2002;14(Suppl):S111–30.
Valverde F, Mouradov A, Soppe W, Ravenscroft D, Samach A, Coupland G. Photoreceptor regulation of CONSTANS protein in photoperiodic flowering. Science. 2004;303:1003–6.
Yamaguchi A, Kobayashi Y, Goto K, Abe M, Araki T. TWIN SISTER OF FT (TSF) acts as a floral pathway integrator redundantly with FT. Plant Cell Physiol. 2005;46:1175–89.
D’Aloia M, Bonhomme D, Bouché F, Tamseddak K, Ormenese S, Torti S, et al. Cytokinin promotes flowering of A.thaliana via transcriptional activation of the FT paralogue TSF. Plant J. 2011;65:972–9.
Foucher F, Morin J, Courtiade J, Cadioux S, Ellis N, Banfield MJ, et al. DETERMINATE and LATE FLOWERING are two TERMINAL FLOWER1/CENTRORADIALIS homologs that control two distinct phases of flowering initiation and development in pea. Plant Cell. 2003;15:2742–54.
Liljegren SJ, Gustafson-Brown C, Pinyopich A, Ditta GS, Yanofsky MF. Interactions among APETALA1, LEAFY, and TERMINAL FLOWER1 specify meristem fate. Plant Cell. 1999;11:1007–18.
Mimida N, Goto K, Kobayashi Y, Araki T, Ahn JH, Weigel D, et al. Functional divergence of the TFL1-like gene family in A.thaliana revealed by characterization of a novel homologue. Genes Cells. 2001;6:327–36.
Huang N-C, Jane W-N, Chen J, Yu T-S. A.thaliana thaliana CENTRORADIALIS homologue (ATC) acts systemically to inhibit floral initiation in A.thaliana. Plant J. 2012;72:175–84.
Nakagawa M, Shimamoto K, Kyozuka J. Overexpression of RCN1 and RCN2, rice TERMINAL FLOWER 1/CENTRORADIALIS homologs, confers delay of phase transition and altered panicle morphology in rice. Plant J. 2002;29:743–50.
Yoo SJ, Chung KS, Jung SH, Yoo SY, Lee JS, Ahn JH. BROTHER OF FT AND TFL1 (BFT) has TFL1-like activity and functions redundantly with TFL1 in inflorescence meristem development in A.thaliana. Plant J. 2010;63:241–53.
Ryu JY, Park C-M, Seo PJ. The floral repressor BROTHER OF FT AND TFL1 (BFT) modulates flowering initiation under high salinity in A.thaliana. Mol Cells. 2011;32:295–303.
Ryu JY, Lee HJ, Seo PJ, Jung JH, Ahn JH, Park CM. The A.thaliana floral repressor BFT delays flowering by competing with FT for FD binding under high salinity. Mol Plant. 2014;7:377–87.
Yoo SY, Kardailsky I, Lee JS, Weigel D, Ahn JH. Acceleration of flowering by overexpression of MFT (MOTHER OF FT AND TFL1). Mol Cells. 2004;17:95–101.
Xi W, Liu C, Hou X, Yu H. MOTHER OF FT AND TFL1 regulates seed germination through a negative feedback loop modulating ABA signaling in A.thaliana. Plant Cell. 2010;22:1733–48.
Nakamura S, Abe F, Kawahigashi H, Nakazono K, Tagiri A, Matsumoto T, et al. A wheat homolog of MOTHER OF FT AND TFL1 acts in the regulation of germination. Plant Cell. 2011;23:3215–29.
Campbell DC, Kondra ZP. A genetic study of growth characters and yield characters of oilseed rape. Euphytica. 1978;27:177–83.
Cai Z. Fine mapping of QTL qFT c2–1 for a major flowering period of Brassica napus. Wuhan: Huazhong Agricultural University; 2016.
Du D, Nie P, Xu L, et al. Heterosis performance of different ecological types of Brassica napus under ecological conditions in Qinghai. Chin J Oil Crops. 2012;34:180–6.
Shah S, Weinholdt C, Jedrusik N, Molina C, Zou J, Große I, et al. Whole-transcriptome analysis reveals genetic factors underlying flowering time regulation in rapeseed (Brassica napus L.). Plant Cell Environ. 2018;41:1935–47.
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43:1035–9.
Liu S, Liu Y, Yang X, et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Communication. 2014;5:3930.
Sun F, Fan G, Hu Q, et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017;92(3):452–68.
Pnueli L, Carmel-Goren L, Hareven D, Gutfinger T, Alvarez J, Ganal M, Lifschitz E. The SELF-PRUNING gene of tomato regulates vegetative to reproductiveswitching of sympodial meristems and is the ortholog of CEN and TFL1. Development. 1998;125(11):1979–89.
Chardon F, Damerval C. Phylogenomic analysis of the PEBP gene family in cereals. J Mol Evol. 2005;61(5):579–90.
Danilevskaya ON, Meng X, Hou Z, et al. A genomic and expression compendium of the expanded PEBP gene family from maize. Plant Physiol. 2008;146(1):250–64.
Wang Z, Zhou Z, Liu Y, et al. Functional evolution of phosphatidylethanolamine binding proteins in soybean and A.thaliana. Plant Cell. 2015;27(2):323–36.
Jin H, Yang Bo, Li Y, et al. Identification and expression analysis of PEBP gene family in oilseed rape. J Crops. 2019;45(3):354–64.
Hanzawa Y, Money T, Bradley D. A single amino acid converts a repressor to an activator of flowering. Proc Natl Acad Sci U S A. 2005;102(21):7748–53.
Dong L, Lu Y, Liu S. Genome-wide member identification, phylogeny and expression analysis of PEBP gene family in wheat and its progenitors. PeerJ. 2020;8:e10483.
Li Q, Fan C, Zhang X, et al. Identification of a soybean MOTHER OF FT AND TFL1 homolog involved in regulation of seed germination. PLoS ONE. 2014;9(6):e99642.
Zhang G, Jin X, Li X, Zhang N, Li S, Si H, et al. Genome-wide identification of PEBP gene family members in potato, their phylogenetic relationships, and expression patterns under heat stress. Mol Biol Rep. 2022;49:4683–97.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31:1296–7.
Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–73.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202.
Shi T, Rahmani RS, Gugger PF, et al. Distinct expression and methylation patterns for genes with different fates following a single whole-genome duplication in flowering plants. Mol Biol Evol. 2020;37(8):2394–413.
Wang Y, Wang X, Tang H, et al. Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS ONE. 2011;6(12):e28150.
Kashkush K, Feldman M, Levy AA. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat Genet. 2003;33(1):102–6.
O’Neill RJ, O’Neill MJ, Graves JA. Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature. 1998;393(6680):68–72.
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–8.
Lescot M, Déhais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30:325–7.
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–13.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Acknowledgements
Not applicable.
Funding
This study was supported by the Qinghai Province "Handsome Scientist Responsibility System" Project (Grant No. 2022-NK-170); China Agriculture Research System (Grant No. CRAS-12); Qinghai Natural Science Foundation Program-innovation team (Grant No. 2022-ZJ-902) and Qinghai innovation platform construction project (Grant No. 2022-ZJ-Y01 and 2022-ZJ-Y13). The funding bodies played no role in the design of the study and collection, analysis, interpretation of data, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YLL conceived the structure of the article and wrote the first draft. LX and ZZ made detailed revisions to the article. HPZ participated in part of the data analysis, and DZD provided detailed reviews and guidance for the overall article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The gene pairs and duplicated type of PEBP genes in B. napus.
Additional file 2: Table S2.
The list of orthologous PEBP gene pairs between B. napus, B. oleracea, B. rapa, and A. thaliana.
Additional file 3: Table S3.
Physicochemical properties and subcellular localization of PEBP gene family members in B.napus.
Additional file 4: Table S4.
Ka/Ks values of BnPEBP genes between B.napus and A.thaliana.
Additional file 5: Table S5.
Specific primers of BnPEBP genes for qRT-PCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Y., Xiao, L., Zhao, Z. et al. Identification, evolution and expression analyses of the whole genome-wide PEBP gene family in Brassica napus L.. BMC Genom Data 24, 27 (2023). https://doi.org/10.1186/s12863-023-01127-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-023-01127-4