
Abstract
Background
Quantitative traits are common in nature, but quantitative pathogenicity has received only little attention in phytopathology. In this study, we used 100 Fusarium culmorum isolates collected from natural field environments to assess their variation for two quantitative traits, aggressiveness and deoxynivalenol (DON) production on wheat plants grown in four different field environments (location-year combinations). Seventeen Fusarium graminearum pathogenicity candidate genes were assessed for their effect on the aggressiveness and DON production of F. culmorum under field conditions.
Results
For both traits, genotypic variance among isolates was high and significant while the isolate-by-environment interaction was also significant, amounting to approximately half of the genotypic variance. Among the studied candidate genes, the mitogen-activated protein kinase (MAPK) HOG1 was found to be significantly associated with aggressiveness and deoxynivalenol (DON) production, explaining 10.29 and 6.05% of the genotypic variance, respectively.
Conclusions
To the best of our knowledge, this is the first report of a protein kinase regulator explaining differences in field aggressiveness and mycotoxin production among individuals from natural populations of a plant pathogen.
Similar content being viewed by others
Background
Quantitative traits are a key feature in nature [1]. They are controlled by many genes, each contributing with a small effect to the overall phenotypic expression of a trait. Surprisingly, quantitative traits of pathogenicity have received only little attention in fungal biology [2, 3]. The expression of quantitative pathogenicity is not only controlled by the pathogen and the host, but also by the environment and their interaction [3]. Association mapping employing mixed models is a common method to dissect the genetic architecture of quantitative traits. Originally designed for the analysis of human diseases, association mapping is now extensively used in plant genetic research [4] either as genome-wide association study (GWAS) using anonymous molecular markers distributed across the whole genome or as candidate gene association by studying single nucleotide polymorphisms within candidate genes. The genetic basis of both methods is a linkage disequilibrium (LD) between molecular polymorphisms and phenotypic traits [4]. Therefore, a sound phenotypic trait evaluation is urgently required. Moreover, proper assessment of quantitative pathogenicity in natural field habitats, which accounts for host-by-environment and pathogen-by-environment-interaction, is indispensable for predicting disease risk and design effective breeding programs for durable resistance.
While qualitative plant-pathogen molecular interactions, especially gene-for-gene interactions, have been widely studied, quantitative realtionships have received far less attention [3, 5]. It is known that genes associated with qualitative pathogenicity frequently encode secreted effectors, which disassemble the host defense response [6, 7]. From the point of view of the host, some hypotheses for explaining the molecular mechanisms that control quantitative disease resistance have been suggested, although the authors remark that this is a poorly understood field [5]. From the point of view of the pathogen, the current understanding of the molecular mechanisms that control quantitative disease pathogenicity is even more scarce. Plant-pathogen qualitative interactions are frequent in biotrophic and obligate pathogens, while hemibiotrophic/necrotrophic pathogens more often show quantitative interactions. It has been proposed that host range, life style and speciation of pathogens are shaped by the type of molecular communication with the host, e.g. the ability to rapidly evolve pathogen effector repertoires [8]. In this context, attempts to answer the following questions are highly relevant: Which are the key molecular players in the quantitative disease pathogenicity? Are they located in the first line of molecular communication with the host, as effectors which are secreted, or are they masters of the molecular regulatory cascades, as transcription factors? Furthermore, are there different allelic variants of those “key players” and what is their role in aggressiveness? Fusarium culmorum is a hemibiotroph, with a wide host range encompassing most of the cereals [9], which makes it a perfect model for the study of quantitative interactions.
Fusarium head blight (FHB) is a devastating disease of bread wheat (Triticum aestivum L.) and other small-grain cereals worldwide [10]. It leads to significant losses not only in terms of yield but also quality, because of the contamination of kernels by mycotoxins, which pose a significant risk to human and animal health [11]. Isolates causing FHB can be classified by their production of type B trichothecenes, including nivalenol (NIV), deoxynivalenol (DON) and its acetylated forms 3-acetyl-deoxynivalenol (3-ADON) and 15-acetyl-deoxynivalenol (15-ADON), as well as estrogen analogues like zearalenone (ZEA) and other mycotoxins [12]. DON is the predominant and economically most important trichothecene detected in cereals in Europe [12]. Therefore, the European Union has set a limit for DON in unprocessed bread wheat for human consumption of 1.25 mg/kg [13]. Various Fusarium species have been reported as causal agents of FHB, among them the haploid ascomycetes Fusarium graminearum and Fusarium culmorum are two of the most important. F. culmorum has been traditionally related with FHB epidemics in the Mediterranean region [14,15,16] as well as northern, central and western Europe [17,18,19,20]. In contrast to F. graminearum, which has been extensively studied and whose full genome sequence has been published [21], a draft of F. culmorum genome was just recently released [22].
Aggressiveness, i.e. the quantitative pathogenicity as described by Van der Plank [23], and DON production are important determinants of parasitic fitness in Fusarium species [24, 25]. There is no generally recognized specific interaction between wheat genotypes and FHB causal agents [26, 27]. Resistant cultivars stay resistant even when challenged by highly aggressive isolates [28]. Aggressiveness is usually evaluated by directly measuring epidemic rates in this monocyclic disease. Higher aggressiveness is associated with faster symptom development and a larger amount of mycelium within the host tissue [29]. DON plays a key role in F. graminearum aggressiveness [30, 31] enabling faster pathogen spread from infected florets into the wheat rachis [32]. A distribution indicative of a quantitative inheritance was demonstrated in F. graminearum segregating progenies of three biparental crosses of genetically diverse isolates for both, aggressiveness and DON production [28, 33]. In contrast, the perfect stage (teleomorph) of F. culmorum is unknown, and therefore such studies could to date not be performed [34]. In order to understand the inheritance and the genetic control of aggressiveness in F. culmorum, the historically accumulated variation in natural populations can be exploited.
Thousands of genes have been characterized affecting host-fungus interaction in agricultural pathosystems, and many of them were found to have a pleiotropic effect [35, 36]. These experiments mainly worked with knock-out or deletion mutants, which are not adequate for explaining quantitative differences among isolates, because frequently the expression of aggressiveness is confounded with basic survival functions of the fungi in these studies. Candidate-gene association mapping appears as a promising and powerful tool to detect functional polymorphisms associated with differences in aggressiveness and DON production in F. culmorum populations.
To enhance our understanding of quantitative pathogenicity, we performed a candidate gene association mapping based on pathogenicity-related candidate genes that firstly, have been reported or predicted as related with quantitative variations in pathogenicity, but do not affect the survival of the fungus and secondly, play a role in the pathogenicity signaling cascade. Based on the sequenced F. graminearum genome, F. culmorum homologues for four transcription factors, eight signal transducers, two membrane receptors and three secreted proteins were sequenced. Using a natural population of 100 isolates of F. culmorum of diverse origin, we aimed to (i) determine the phenotypic variance for aggressiveness and DON production in replicated field experiments across two locations and 2 years with a moderately susceptible bread wheat cultivar as host, (ii) study sequences of the 17 candidate genes to identify nucleotide diversity within selected gene regions, (iii) test these quantitative trait nucleotides (QTNs) for their association with aggressiveness and DON production and estimate the proportion of genotypic variance explained by them.
Methods
Fungal materials and field trials
One hundred F. culmorum isolates from a previous diversity study [18] were phenotyped under field conditions for aggressiveness and deoxynivalenol (DON) production following a chessboard design described previously [37, 38]. In short, field plots were arranged such that each inoculated entry plot of bread wheat was surrounded by four border plots of a tall triticale cultivar (x Triticosecale) to minimize plot-by-plot interference. Fungal material consisted of four field populations (one from Russia and three from Germany), one transect population from Syria and one international collection (Additional file 1). The field populations were randomly collected isolates from symptom-bearing FHB infected heads from individual commercial wheat fields as described earlier [18]. All isolates were used as single-spore derived isolates.
Field experiments with these isolates were conducted during 2014 and 2015, and across two locations: Hohenheim (HOH, longitude 9° 11′ 23″ E, latitude 48° 42′ 54″ N, altitude 403 m) and Oberer Lindenhof (OLI, longitude 9° 18′ 17″, latitude 48° 28′ 25″ N, altitude 702 m) in Southwestern Germany. The experiments followed an incomplete block design (α design) with two replications. Experimental units were three-row plots (1.0 m long, 0.625 m wide), which were machine sown with 220 kernels per m2, a seeding rate that results in a homogeneous wheat stand. A moderately susceptible winter wheat cultivar was used as host (“Inspiration”, KWS LOCHOW GMBH, Bergen, Germany) with a FHB rating of 6 on the 1–9 scale, where 1 = without disease and 9 = fully infected.
Inoculum production was done in shaking cultures according to an existing procedure [39]. For inoculation, a dose of 100 ml suspension per square meter in a concentration of 2 × 105 conidiospores ml−1 was sprayed onto wheat heads during full flowering. Inoculum for each isolate was sprayed on its corresponding plot, according to the randomization of the experiment design, using a hand atomizer with constant air pressure of 3 bar from a tractor to ensure full coverage of all heads of the plot with the same dosage. All plots flowered simultaneously, because only one homogeneous wheat cultivar was used. This allowed inoculation and ratings for all plots at the same dates per location. Contamination with natural inoculum was negligible as verified by the border plots which had always a FHB rating < 5%.
Phenotyping aggressiveness and DON production
To assess aggressiveness of isolates, FHB symptoms were visually rated in each plot at least three times starting with the onset of symptom development, about 14 days after inoculation and was continued at 3- to 5-day intervals until the beginning of the yellow ripening stage. Rating was performed as percentage of infected spikelets per plot (0–100%). This reflects both the percentage of infected spikes per plot and the percentage of infected spikelets per spike in a single rating. The arithmetic mean of at least three ratings (i.e. the mean FHB ratings was used as aggressiveness trait).
To measure DON production, wheat plots were harvested by hand at full ripening, carefully threshed in a single-head thresher (Walter-Wintersteiger, Austria) and cleaned with reduced wind speed. The remaining fragments of glumes and rachis were manually picked out to retain highly shrunken kernels in the sample. Cleaned wheat grain was ground in a commercial laboratory mill with a sieve size of 1 mm. Later, the coarse meal was analyzed to quantify the amount of DON by a commercially available immunotest (R-Biopharm AG, Darmstadt, Germany).
Phenotypic data analyses
Phenotypic data of aggressiveness and DON production were obtained from one experiment performed during 2 years and across two locations, yielding four test environments. Aggressiveness data and DON production were arcsin transformed to meet the required normal distribution and subjected to outlier detection with the method BH-MADR (Bonferroni–Holm with re-scaled MAD standardized residuals) suggested by Bernal-Vasquez et al. [40]. For the description of the model, the syntax suggested by Patterson [41] was followed, where fixed effects (E, R, I) appear before the colon and random effects (I × E, B) after the colon. In order to obtain best linear unbiased estimators (BLUE) of each isolate genotype, the phenotypic data were analyzed based on the following linear mixed model:
where E, R, I, and B denote environment, replication, isolate, and block or their interaction, respectively. Variance components were determined by the restricted maximum likelihood (REML) method. Significance of variance components was tested by model comparisons with likelihood ratio tests [42]. Heritability (h 2) was estimated as heritability on an entry-mean basis following the approach suggested by Piepho and Möhring [43]. All statistical analyses were performed using ASReml 3.0 [44] and R [45] combined with the graphical user interface RStudio.
Gene selection and sequencing
Seventeen candidate genes with a confirmed or predicted role in pathogenesis of Fusarium spp., aggressiveness and/or trichothecene biosynthesis were used in our study (Table 1). Based on previous studies suggesting that genes associated with aggressiveness are involved in regulation or transport activities [37, 38], we focused mainly on those groups of genes, although few genes codifying for proteins located in membrane and secreted proteins were also included. The most variable regions of the selected genes were identified using BLAST analysis. Specific primers (Additional file 2) for amplification of those regions in each gene were designed using the software Primer3 - version 0.4.0 [46]. DNA extracted from each of the 100 isolates was used in a polymerase chain reaction (PCR) with the designed primers for each gene, following a standard protocol. Amplicons for each isolate and gene combination were sequenced once using the Sanger method. The sequences were aligned against the reference sequence of F. graminearum in the revised complete genome of the strain PH-1, RRes v4.0 [47], available within ENSEMBL fungi (http://fungi.ensembl.org) using the program MEGA6 [48] to identify single nucleotide polymorphisms (SNPs) among the 100 isolates. Polymorphisms that had more than 20% missing values or a minor allele frequency (MAF) of <5% were not considered for further analyses.
Association mapping
Modified Rogers genetic distances (RD; Rogers [49]) based on 10 SSR markers were computed among all isolates under study, using the software Plabsoft [50]. Principal coordinate analysis (PCoA) was conducted on the Modified Rogers’s distances. Additionally, pairwise kinship coefficients [51] among individuals in the collection were estimated as 1 minus the modified Rogers’ distance [52]. A mixed linear model incorporating the BLUES obtained from the phenotypic analysis (Additional file 3), SNP’s information (Additional file 4), the three main principal coordinates as fixed effect and a kinship matrix for the random genotypic isolate effect was used to identify marker-trait associations [53]. The p values obtained from the one-degree-of-freedom score test were corrected for possible inflation [53]. The proportion of genotypic variance (ρG) explained by each SNP was derived from the sums of squares of the SNP in a linear model using aggressiveness or DON production as dependent variables. As we found high multi-colinearity among SNPs within the candidate genes, only one SNP out of a group of highly linked SNPs (HOG1–380) was used in the linear model to assess the proportion of explained genotypic variance [54]. All calculations were done with the open-source statistical software R [45] including packages GenABEL [53] and APE [55]. The squared correlation coefficient (r 2) was used to estimate linkage disequilibrium (LD) between each pair of marker loci [56] using TASSEL [57].
Results
Phenotypic data
All F. culmorum isolates successfully produced symptoms on the inoculated wheat spikes and differences among isolates were observed at all environments. The overall mean of aggressiveness was 15.97%, varying from a minimum of 4.2% for isolate FC60 and a maximum of 19.8% for the isolate S109. The overall mean for DON production was 12.98 mg kg−1 ranging from 0.7 mg kg−1 to 22.95 mg kg−1 for FC50 and 9D22, respectively. Nivalenol (NIV) chemotypes displayed always the lowest DON production values as well as lowest aggressiveness values. Significant correlation between aggressiveness and DON production was observed (r = 0.67, p < 0.001, Fig. 1). The overall entry-mean heritability was 0.87 for aggressiveness and 0.90 for DON production, illustrating a high relevance of genotypic variance. Accordingly, genotypic variances across all environments were significantly (p < 0.001) different from zero for both traits. Isolate-by-environment interaction was also significantly different from zero and about half that of the genotypic isolate variance (Table 2). The frequency distribution of aggressiveness and DON production was continuous and followed a normal distribution for both traits (Additional file 5). Variability within and among field populations, for aggressiveness and DON production, were observed. While the population from Russia produced on average the highest values for aggressiveness and DON production, the population 7D from Entringen, Germany, as well as the international collection had the lowest values for both traits (Fig. 2). Transect population from Syria displayed a large range for both trait values, almost the same range as displayed by the international collection (Fig. 2).

Relationship between mean aggressiveness and deoxynivalenol (DON) production across 100 F. culmorum isolates

Violin boxplots of mean aggressiveness and deoxynivalenol (DON) production for populations under study after inoculation on bread wheat across four environments. Field populations consisted in Entringen [7D], Herrenberg [8D], Nufringen [9D] and Novgorod [R], one transect population from Syria [S], and the collection of 22 isolates of Fusarium culmorum [FC]. Horizontal line within boxes = median, • = outliers
Genotypic and molecular analysis
A principal coordinate analysis based on the modified Rogers’ distances among the 100 entries as revealed by SSR markers was performed [18]. The first three main coordinates explained 17, 10 and 10% of the molecular variance and were used to correct for population stratification in the subsequent association analysis (Additional file 6). In accordance with previous results, we observed two clusters setting Syrian samples apart from the German isolates, but not from the international collection.
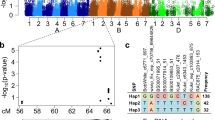
Analysis of the sequences of 17 genes for 100 F. culmorum isolates included in our study revealed 34 SNPs (Additional file 7) with a minor allele frequency higher than 5%, distributed in ten out of the 17 analyzed genes (Table 1). The gene displaying the highest number of SNPs was CUT with 17 SNPs, followed by FGL1 with four SNPs. Different numbers of SNPs were found in genes with different function and cellular location of the encoded proteins. Genes encoding secreted proteins displayed the majority of SNPs, while no SNPs were found in any of the genes encoding transcription factors included in this study (FGP1, SCH9, TOP1). High LD was detected among SNPs within individual genes (Fig. 3) and within SNPs from two genes located on the same chromosome (MSB2-FGL1). Additionally, some SNPs in genes on different chromosomes showed high LD, for instance, the genes TRI6/MSB2, TRI6/CUT, MSB2/CUT. HOG1 displayed the lowest level of LD with any of the other studied genes.

Pairwise linkage disequilibrium (LD) within and among 10 candidate genes based on 100 Fusarium culmorum isolates. LD measured as r 2 between all pairs of selected SNP loci (above diagonal) and significance (below diagonal). The horizontal and vertical lines separate the candidate genes and chromosomes
Association analysis for aggressiveness and DON production
Three SNPs closely linked to each other and located in HOG1 were found to be significantly associated (p < 0.05) with aggressiveness and DON production. Given high collinearity, all three SNPs were analyzed as only one haplotype, which explains 10.29 and 6.05% of the genotypic variance of aggressiveness and DON production, respectively. Any haplotype was restricted to a single population, so no bias by population structure can be expected. Additionally, we used principal coordinate and kinship matrix analysis to correct for population structure. Associated polymorphisms are located within the gene at positions 380, 382 (intron 3) and 724 (intron 5) relative to the start codon (Fig. 4) and correspond to changes in base pairs C/T, C/T and G/A, respectively. Five more polymorphisms in introns and exons were found in HOG1, but were excluded from the analysis due to the minor allele frequency limit of 5%. Isolates having the most frequent HOG1 haplotype expressed on average highest values for aggressiveness and DON production (Fig. 4). An association close to the significant threshold was also detected for one SNP in TRI6 (TRI147, p = 0.057) with DON production and one insertion in CUT (CUT536 + 3, p = 0.07) with aggressiveness (Additional file 7). Both were excluded from further analysis because the p values were slightly higher than the threshold 0.05.

HOG1 sequenced region, SNPs found and genetic effects of the two haplotypes. a Gene structure taken from ENSEMBL fungi database [116] modified to show HOG1 sequenced region on chromosome four and all SNPs found. The location of the gene in F. graminearum genome is expressed in base pairs. Minor allele frequency expressed in percentage. Boxplots diagrams depicting the genetic effects of two HOG1 haplotypes with significant associations to (b) aggressiveness and (c) DON production
Discussion
Given the economic and public health importance of Fusarium head blight [10, 35], the causal fungi have been under intensive investigation and became a model to study quantitative plant-microbe interactions. The release of the F. graminearum genome sequence in 2007 [21] motivated research on this pathogen using a wide variety of approaches to better understand the basis of pathogen biology. As a result, genes related to pathogenesis and other responses have been identified. Special attention has been given to genes encoding proteins of the secretome [58,59,60,61], kinome [62] and phosphatome [63]. However, little is known about which of those identified genes play an active role in explaining quantitative differences in aggressiveness among isolates occurring in natural populations, as the vast majority of studies have used only one Fusarium isolate. Furthermore, aggressiveness is a quantitative trait that is highly influenced by the environment. Thus, it is not surprising that candidate genes identified under controlled environments may not be relevant under field conditions [64,65,66]. In this study, we used 100 F. culmorum isolates collected from natural environments to evaluate the effect of previously identified candidate genes on aggressiveness and DON production across four field environments.
Large genetic variation of isolates from individual fields for aggressiveness and DON production
Quantitative variation within natural field populations has previously only been reported for a few plant pathogens like Rhynchosporium commune [67], Zymoseptoria tritici [68,69,70] and the close relative of Fusarium culmorum, F. graminearum [71]. To our knowledge, this is the first study reporting variability in aggressiveness and DON production within F. culmorum isolates from natural field populations.
Both traits under study followed a continuous distribution, as expected for quantitative traits (Additional file 5). We observed a high correlation between DON production and aggressiveness (r = 0.67, p < 0.001, Fig. 1), which is consistent with a previous study using 42 F. culmorum isolates [72] and F. graminearum [71]. Even if the correlation cannot conclude causality of DON in aggressiveness [72], this evidence confirms an important role of DON in aggressiveness. Moreover, high infestation on the field is represented by high levels of DON contamination on harvested grain, despite the fact that highly aggressive isolates do not necessarily produce more DON per mycelium unit than less aggressive ones [72].
Large isolate-by-environment interaction highlights the importance to study FHB using multi-environmental field trials
The genotypic variance is the component of phenotypic variability that could be exploited by selective forces [73, 74]. The observed genotypic variance, which corresponds to the isolate variance component in this study, was high and significant for both traits. In the light of evolution, our results suggest, that if aggressiveness increases the fitness of the population even during the saprophytic phases of the life cycle, the most aggressive genotypes will, with time and under a constant selection pressure, predominate in the population. However, the role of aggressiveness during the saprophytic phases of the life cycle are still to be studied.
Conducting experiments as multi-environment trials, using plots rather than single plants as experimental units and applying mixed models in the statistical analysis are standard practices in the study of quantitative traits of plants [75]. These approaches have only recently been applied to study quantitative plant-pathogen relationships [37, 38]. Here we report an isolate-by-environment interaction significantly different from zero and amounting to about half that of the isolate genotypic variance. These results are in accordance with previous studies [38, 76] and illustrate the relevance of methods which account for the isolate-by-environment interaction in the study of quantitative disease pathogenicity.
High nucleotide variation found in candidate genes encoding secreted proteins
The F. culmorum draft genome sequence is currently under study, but gene annotation has not been completed yet [22]. Therefore, a wide array of information has been inferred from the well annotated reference genome sequence of its close relative F. graminerum [17, 77]. We designed primers for known genes in F. graminearum and used them successfully for amplification of all homologous genes in F. culmorum isolates. This further supports a high synteny and sequence homology among F. culmorum and F. graminearum genomes [78, 79].
The fact that the majority of SNPs were found in genes encoding secreted or membrane proteins, could be explained by their role in the molecular communication with the host and signal perception from the environment, which might be subject to positive selection pressure as widely reported [80,81,82,83,84,85]. By contrast, no informative SNPs were found in any of three out of four transcription factors included in this study (FGP1, SCH9, TOP1). This result and previous information concerning the function of those genes, suggested that they might be involved in the pleiotropic control of several basic physiological functions, among them pathogenicity [86,87,88,89,90,91,92,93].
High and significant LD was found among genes from different chromosomes, as for example TRI6 and CUT (Fig. 3). Similar results were found in a study on F. graminearum reporting high levels of LD between genes of the TRI cluster and pathogenicity related candidate genes located on different chromosomes e.g. TRI5/MetAP1, TRI10/MetAP1 [37]. Several hypotheses could explain these results: (i) simultaneous selection on the linked genes as a result of a common role in aggressiveness or a coordinated involvement in the same response pathway or (ii) low physical distance with other genes, which are responsible for the LD between different chromosomes.
HOG1, a gene involved in osmotic and oxidative stress is associated with quantitative pathogenicity
HOG1 was significantly associated with aggressiveness and DON production in our study. The mitogen-activated protein kinase (MAPK) HOG1 is a core component of the high osmolarity glycerol (HOG) pathway and has been well characterized in S. cerevisiae [94]. MAPK pathways are three-tiered protein kinase modules that are present in all eukaryotic organisms and function in succession to transmit a variety of cellular signals [95]. Most fungal pathogens contain three MAPKs that are orthologues of the S. cerevisiae Fus3/Kss1, Slt2, and Hog1 MAPKs, and function in separate signaling cascades to regulate infection-related morphogenesis, cell wall remodeling, and high osmolarity stress response, respectively [96,97,98,99]. The first functional characterization of the kinome in a plant pathogenic fungi was developed using Fusarium graminearum as a model and generated deletion mutants for 96 protein kinase genes, out of them 42 kinase mutants were significantly reduced in virulence or non-pathogenic [62]. The FgHOG1 is a core component of the HOG pathway in F. graminearum, which has been involved in the response to various environmental stresses [92]. Based on our results, we hypothesize that changes in HOG1 regulation confer advantages in the response of F. culmorum to multiple stresses, especially to the osmotic and oxidative stresses resulting from the plant defense mechanisms. Our study further supports the observation of Talas et al. [38], suggesting that most of the genes associated with aggressiveness are involved in regulation or transport activities.
Quantitative traits are controlled by complex interactions of genes, and therefore single or few genes explaining a large percentage of genetic variation are not expected [1]. Consistently, the observed percentage of genetic variance explained by HOG1 was 10.29 and 6.05% for aggressiveness and DON respectively. Similar results have been reported for F. graminearum, where genome-wide association reveled quantitative trait nucleotides explaining from 9% up to a maximum of 24% of genotypic variance [38]. The positive results of our study validate the application of candidate gene association mapping strategies to validate factors associated with pathogen aggressiveness under field conditions.
Isolates having the most frequent HOG1 haplotype expressed on average the highest values for aggressiveness and DON production, which could be an effect of selection favoring these most aggressive genotypes. This fact highlights the importance of integrated plant disease management strategies to prevent undesired selection of the most aggressive genotypes of F. culmorum. Consistently, plant breeding must then steadily increase the level of resistance in cultivars, e.g. by pyramiding effective quantitative disease resistance loci.
A suggested role of intronic regions in the expression of aggressiveness
The three SNPs found in HOG1 are located in introns three and five. The fact that SNPs located within a noncoding region of HOG1 are associated with the variation in aggressiveness and DON production could be explained by two hypotheses. Firstly, the detected SNPs are in linkage disequilibrium (LD) with causal polymorphisms in nearby genes or regulatory sequences that are responsible for the trait variance. Secondly, the nucleotide change within the intron is involved in post-transcriptional regulation, like alternative splicing, associated with the response to multiple stresses. Alternative splicing has been reported in fungi, including F. verticillioides [100] and F. graminearum [100,101,102,103]. In F. graminearum, grown under just one stress condition, 231 genes undergoing alternative splicing were found, but more genes are expected to follow post-transcriptional regulation if tested under different stresses [103], as shown for Arabidopsis thaliana [104]. Zhao et al. [103] demonstrated that for some genes in F. graminearum the alternative splicing events took place at different vegetative growth stages and suggested they might be important in adaptation of F. graminearum to changing environmental conditions. Furthermore, active functionality of intronic polymorphisms has been found in other organisms e.g. humans [105] and pigs [106].
Several SNPs in our study were discarded because of low calling rate, including some SNPs within HOG1 located both in exons (positions 505, 609 and 686) and introns (positions 476, 484 and 730). On the other hand, some other genes had polymorphic SNPs with MAF > 5% but showed no association or had p values just slightly higher than the significance threshold (TRI147 and CUT563 + 3; Additional file 7). It must be noted, that this does not rule out a contribution of these genes to the variation in aggressiveness or DON production, as the population size of 100 isolates used in this study may have not allowed a statistically significant association.
Conclusions
We phenotyped 100 F. culmorum isolates under field conditions and analyzed aggressiveness and DON production. Our results further support a quantitative pathogenicity model for the bread wheat - F. culmorum pathosystem. Variation in aggressiveness was largely explained by isolate genotype although the isolate-by-environment interaction was also significant, as expected for quantitative interactions. Of the 17 candidate genes ten showed polymorphisms that were tested for their association with the two traits. HOG1 was identified as a component of the quantitative pathogenicity of F. culmorum. To the best of our knowledge, this is the first report of a protein kinase regulator explaining differences in field aggressiveness and mycotoxin production in a natural population of a plant pathogen.
Abbreviations
- 15-ADON:
-
15-acetyl-deoxynivalenol
- 3-ADON:
-
3-acetyl-deoxynivalenol
- BH-MADR:
-
Bonferroni–Holm with re-scaled MAD standardized residuals
- BLUE:
-
Best linear unbiased estimators
- DON:
-
Deoxynivalenol
- FHB:
-
Fusarium head blight
- h 2 :
-
Heritability
- HOH:
-
Hohenheim
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- MAPK:
-
Mitogen-activated protein kinase
- NIV:
-
Nivalenol
- OLI:
-
Oberer Lindenhof
- PCoA:
-
Principal coordinate analysis
- REML:
-
Restricted maximum likelihood
- SNPs:
-
Single nucleotide polymorphisms
- SSRs:
-
Single sequence repeats
- ZEA:
-
Zearalenone
References
Mackay Trudy FC, Stone Eric A, Ayroles Julien F. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10:565–77.
Pariaud B, Ravigné V, Halkett F, et al. Aggressiveness and its role in the adaptation of plant pathogens. Plant Pathol. 2009;58:409–24.
Lannou C. Variation and selection of quantitative traits in plant pathogens. Annu Rev Phytopathol. 2012;50:319–38.
Zhu C, Gore M, Buckler ES, et al. Status and prospects of association mapping in plants. Plant Genome J. 2008;1:5–20.
Poland JA, Balint-Kurti PJ, Wisser RJ, et al. Shades of gray: the world of quantitative disease resistance. Trends Plant Sci. 2009;14:21–9.
Hogenhout SA, Van der Hoorn RAL, Terauchi R, et al. Emerging concepts in effector biology of plant-associated organisms. Mol Plant-Microbe Interact. 2009;22:115–22.
Le Fevre R, Evangelisti E, Rey T, et al. Modulation of host cell biology by plant pathogenic microbes. Annu Rev Cell Dev Biol. 2015;31:201–29.
Schulze-Lefert P, Panstruga R. A molecular evolutionary concept connecting nonhost resistance, pathogen host range, and pathogen speciation. Trends Plant Sci. 2011;16:117–25.
Miedaner T, Schilling AG, Geiger HH. Molecular genetic diversity and variation for aggressiveness in populations of Fusarium graminearum and Fusarium culmorum sampled from wheat fields in different countries. J Phytopathol. 2001;149:641–8.
Goswami RS, Kistler HC. Heading for disaster: Fusarium graminearum on cereal crops. Mol Plant Pathol. 2004;5:515–25.
Pestka JJ. Deoxynivalenol: mechanisms of action, human exposure, and toxicological relevance. Arch Toxicol. 2010;84:663–79.
D’Mello JPF, Placinta CM, Macdonald AMC. Fusarium mycotoxins: a review of global implications for animal health, welfare and productivity. Anim Feed Sci Technol. 1999;80:183–205.
EFSA: Deoxynivalenol in food and feed: occurrence and exposure European Food Safety Authority. EFSA J. 2013;11:3379–3435.
Touati-Hattab S, Barreau C, Verdal-Bonnin MN, et al. Pathogenicity and trichothecenes production of Fusarium culmorum strains causing head blight on wheat and evaluation of resistance of the varieties cultivated in Algeria. Eur J Plant Pathol. 2016;145:797–814.
Rebib H, Bouraoui H, Rouaissi M, et al. Genetic diversity assessed by SSR markers and chemotyping of Fusarium culmorum causal agent of foot and root rot of wheat collected from two different fields in Tunisia. Eur J Plant Pathol. 2014;139:481–95.
Pancaldi D, Tonti S, Prodi A, et al. Survey of the main causal agents of Fusarium head blight of durum wheat around bologna, northern Italy. Phytopathol Mediterr. 2010;49:258–66.
Scherm B, Balmas V, Spanu F, et al. Fusarium culmorum: causal agent of foot and root rot and head blight on wheat. Mol Plant Pathol. 2013;14:323–41.
Miedaner T, Caixeta F, Talas F. Head-blighting populations of Fusarium culmorum from Germany, Russia, and Syria analyzed by microsatellite markers show a recombining structure. Eur J Plant Pathol. 2013;137:743–52.
Pasquali M, Beyer M, Logrieco A, et al. A European database of Fusarium graminearum and F. culmorum Trichothecene genotypes. Front Microbiol. 2016;7:406.
Giraud F, Pasquali M, El JM, et al. Fusarium head blight and associated mycotoxin occurrence on winter wheat in Luxembourg in 2007/2008. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2010;27:825–35.
Cuomo CA, Güldener U, Xu JR, et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 2007;317:1400–2.
Urban M, King R, Andongabo A, et al. First daft genome sequence of a UK strain (UK99) of Fusarium culmorum. Genome Announc. 2016;4:15–6.
Van der Plank JE. Plant diseases: epidemics and control. Minnesota: Academic Press; 1963.
Miedaner T, Cumagun CJR, Chakraborty S. Population genetics of three important Hhead blight pathogens Fusarium graminearum, F. pseudograminearum and F. culmorum. J Phytopathol. 2008;156:129–39.
von der Ohe C, Gauthier V, Tamburic-Ilincic L, et al. A comparison of aggressiveness and deoxynivalenol production between Canadian Fusarium graminearum isolates with 3-acetyl and 15-acetyldeoxynivalenol chemotypes in field-grown spring wheat. Eur J Plant Pathol. 2010;127:407–17.
Bai GH, Shaner G. Variation in Fusarium graminearum and cultivar resistance to wheat scab. Plant Dis. 1996;80:975–9.
van Eeuwijk FA, Mesterhazy A, Kling Ch I, et al. Assessing non-specificity of resistance in wheat to head blight caused by inoculation with European strains of Fusarium culmorum, F. graminearum and F. nivale using a multiplicative model for interaction. Theor Appl Genet. 1995;90:221–8.
Voss H-H, Bowden RL, Leslie JF, et al.: Variation and transgression of aggressiveness among two Gibberella zeae crosses developed from highly aggressive parental isolates. Phytopathology. 2010;100:904–912.
Miedaner T, Schilling AG, Geiger HH. Competition effects among isolates of Fusarium culmorum differing in aggressiveness and mycotoxin production on heads of winter rye. Eur J Plant Pathol. 2004;110:63–70.
Proctor RH, Desjardins AE, McCormick SP, et al. Genetic analysis of the role of trichothecene and fumonisin mycotoxins in the virulence of Fusarium. In: Mycotoxins in plant disease. Dordrecht: Springer Netherlands; 2002. p. 691–8.
Walter S, Nicholson P, Doohan FM. Action and reaction of host and pathogen during Fusarium head blight disease. New Phytol. 2010;185:54–66.
Maier FJ, Miedaner T, Hadeler B, et al. Involvement of trichothecenes in fusarioses of wheat, barley and maize evaluated by gene disruption of the trichodiene synthase (Tri5) gene in three field isolates of different chemotype and virulence. Mol Plant Pathol. 2006;7:449–61.
Cumagun CJR, Miedaner T. Segregation for aggressiveness and deoxynivalenol production of a population of Gibberella zeae causing head blight of wheat. Eur J Plant Pathol. 2004;110:789–99.
Obanor F, Erginbas-Orakci G, Tunali B, et al. Fusarium culmorum is a single phylogenetic species based on multilocus sequence analysis. Fungal Biol. 2010;114:753–65.
Kazan K, Gardiner DM, Manners JM. On the trail of a cereal killer : recent advances in Fusarium graminearum pathogenomics and host resistance. Mol Plant Pathol. 2012;13:399–413.
Urban M, Irvine AG, Cuzick A, et al. Using the pathogen-host interactions database (PHI-base) to investigate plant pathogen genomes and genes implicated in virulence. Front Plant Sci. 2015;6:605.
Talas F, Würschum T, Reif JC, et al. Association of single nucleotide polymorphic sites in candidate genes with aggressiveness and deoxynivalenol production in Fusarium graminearum causing wheat head blight. BMC Genet. 2012;13:14.
Talas F, Kalih R, Miedaner T, et al. Genome-wide association study identifies novel candidate genes for aggressiveness, deoxynivalenol production, and azole sensitivity in natural field populations of Fusarium graminearum. Mol Plant-Microbe Interact. 2016;29:417–30.
Reid LM, Mather DE, Hamilton RI, et al. Genotypic differences in the resistance of maize silk to Fusarium graminearum. Can J Plant Pathol. 1992;14:211–4.
Bernal-Vasquez AM, Utz HF, Piepho HP. Outlier detection methods for generalized lattices: a case study on the transition from ANOVA to REML. Theor Appl Genet. 2016;129:787.
Patterson HD. Analysis of series of variety trials. In: Statistical methods for plant variety evaluation. Dordrecht: Springer Netherlands; 1997. p. 139–61.
Stram DO, Lee JW. Variance components testing in the longitudinal mixed effects model. Biometrics. 1994;50:1171.
Piepho HP, Möhring J. Computing heritability and selection response from unbalanced plant breeding trials. Genetics. 2007;177:1881–8.
Gilmour AR, Gogel BJ, Cullis BR, et al.: ASReml user guide release 3.0. VSN Int ltd. 2009.
R Development Core Team: R: a language and environment for statistical computing. 2016.
Untergasser A, Cutcutache I, Koressaar T, et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40:1–12.
King R, Urban M, Hammond-Kosack MCU, et al. The completed genome sequence of the pathogenic ascomycete fungus Fusarium graminearum. BMC Genomics. 2015;16:544.
Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Rogers JS. Measures of similarity and genetic distance, vol. VII. Austin: University of Texas Publication; 1972.
Maurer HP, Melchinger AE, Frisch M. Population genetic simulation and data analysis with Plabsoft. Euphytica. 2008;161:133–9.
Yu J, Pressoir G, Briggs WH, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–8.
Jiang Y, Zhao Y, Rodemann B, et al. Potential and limits to unravel the genetic architecture and predict the variation of Fusarium head blight resistance in European winter wheat (Triticum aestivum L.). Heredity. 2015;114:318–26.
Aulchenko YS, Ripke S, Isaacs A, et al. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–6.
Utz HF, Melchinger AE, Schön CC. Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics. 2000;154:1839–49.
Paradis E, Claude J, Strimmer K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics. 2004;20:289–90.
Pritchard JK, Przeworski M. Linkage disequilibrium in humans: models and data. Am J Hum Genet. 2001;69:1–14.
Bradbury PJ, Zhang Z, Kroon DE, et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Kikot GE, Hours RA, Alconada TM. Contribution of cell wall degrading enzymes to pathogenesis of Fusarium graminearum: a review. J Basic Microbiol. 2009;49:231–41.
Yang Fen EN, Jensen Jens D, Svensson B, et al. Secretomics identifies Fusarium graminearum proteins involved in the interaction with barley and wheat. Mol Plant Pathol. 2012;13:445–53.
Brown NA, Antoniw J, Hammond-Kosack KE. The predicted secretome of the plant pathogenic fungus Fusarium graminearum: a refined comparative analysis. PLoS One. 2012;7:e33731.
Rampitsch C, Day J. Subramaniam Rajagopal, et al.: comparative secretome analysis of Fusarium graminearum and two of its non-pathogenic mutants upon deoxynivalenol induction in vitro. Proteomics. 2013;13:1913–21.
Wang C, Zhang S, Hou R, et al. Functional analysis of the kinome of the wheat scab fungus Fusarium graminearum. PLoS Pathog. 2011;7:e1002460.
Yun Y, Liu Z, Yin Y, et al. Functional analysis of the Fusarium graminearum phosphatome. New Phytol. 2015;207:119–34.
Xiao BZ, Chen X, Xiang CB, et al. Evaluation of seven function-known candidate genes for their effects on improving drought resistance of transgenic rice under field conditions. Mol Plant. 2009;2:73–83.
Barrett RDH, Hoekstra HE. Molecular spandrels: tests of adaptation at the genetic level. Nat Rev Genet. 2011;12:767–80.
Talas F, Mcdonald BA. Genome-wide analysis of Fusarium graminearum field populations reveals hotspots of recombination. BMC Genomics. 2015:1–12.
Stefansson TS, McDonald BA, Willi Y. Local adaptation and evolutionary potential along a temperature gradient in the fungal pathogen Rhynchosporium commune. Evol Appl. 2013;6:524–34.
Stewart EL, Croll D, Lendenmann MH, et al. QTL mapping reveals complex genetic architecture of quantitative virulence in the wheat pathogen Zymoseptoria tritici. Mol Plant Pathol. doi:10.1111/mpp.12515.
Zhan J, Linde CC, Jürgens T, et al. Variation for neutral markers is correlated with variation for quantitative traits in the plant pathogenic fungus Mycosphaerella graminicola. Mol Ecol. 2005;14:2683–93.
Yang L, Gao F, Shang L, et al. Association between virulence and triazole tolerance in the phytopathogenic fungus Mycosphaerella graminicola. PLoS One. 2013;8:1–8.
Talas F, Kalih R, Miedaner T. Within-field variation of Fusarium graminearum isolates for aggressiveness and deoxynivalenol production in wheat head blight. Phytopathology. 2012;102:128–34.
Gang G, Miedaner T, Schuhmacher U, et al. Deoxynivalenol and nivalenol production by Fusarium culmorum isolates differing in aggressiveness toward winter rye. Phytopathology. 1998;88:879–84.
Cooper M, Hammer GL. Plant adaptation and crop improvement. Wallingford: CAB International; 1996.
Bernardo R. Breeding for quantitative traits in plants. Woodbury: Stemma Press; 2010.
Bustos-Korts D, Malosetti M, Chapman S, et al. Modelling of genotype by environment interaction and prediction of complex traits across multiple environments as a synthesis of crop growth modelling, genetics and statistics. In: Yin X, Struik PC, editors. Crop systems biology. Switzerland: Springer International; 2016. p. 55–82.
Miedaner T, Gang G, Geiger HH. Quantitative-genetic basis of aggressiveness of 42 isolates of Fusarium culmorum for winter rye head blight. Plant Dis. 1996;80:500–4.
Aoki T, O’Donnell K, Geiser DM. Systematics of key phytopathogenic Fusarium species: current status and future challenges. J Gen Plant Pathol. 2014;80:189–201.
Rep M, Kistler HC. The genomic organization of plant pathogenicity in Fusarium species. Curr Opin Plant Biol. 2010;13:420–6.
Sperschneider J, Gardiner DM, Thatcher LF, et al. Genome-wide analysis in three Fusarium pathogens identifies rapidly evolving chromosomes and genes associated with pathogenicity. Genome Biol Evol. 2015;7:1613–27.
Dangl JL, Jones JDG. Plant pathogens and integrated defence responses to infection. Nature. 2001;411:826–33.
Aguileta G, Refregier G, Yockteng R, et al. Rapidly evolving genes in pathogens: methods for detecting positive selection and examples among fungi, bacteria, viruses and protists. Infect Genet Evol. 2009;9:656–70.
Jiang Rays HY, Tripathy S, Govers F, et al. RXLR effector reservoir in two Phytophthora species is dominated by a single rapidly evolving superfamily with more than 700 members. Proc Natl Acad Sci U S A. 2008;105:4874–9.
Panstruga R, Dodds PN. Terrific protein traffic: the mystery of effector protein delivery by filamentous plant pathogens. Science. 2009;324:748–50.
Dodds PN. Genome evolution in plant pathogens. Science. 2010;330:1486–7.
de Wit Pierre JGM, van der Burgt A, Ökmen B, et al.: The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 2012;8:e1003088.
Jonkers W, Dong Y, Broz K, et al. The Wor1-like protein Fgp1 regulates pathogenicity, toxin synthesis and reproduction in the phytopathogenic fungus Fusarium graminearum. PLoS Pathog. 2012;8:e1002724.
Kim H-K, Lee S, Jo S-M, et al. Functional roles of FgLaeA in controlling secondary metabolism, sexual development, and virulence in Fusarium graminearum. PLoS One. 2013;8:e68441.
Park AR, Cho A-R, Seo J-A, et al. Functional analyses of regulators of G protein signaling in Gibberella zeae. Fungal Genet Biol. 2012;49:511–20.
Chen D, Wang Y, Zhou X, et al. The Sch9 kinase regulates conidium size, stress responses, and pathogenesis in Fusarium graminearum. PLoS One. 2014;9:e105811.
Baldwin TK, Urban M, Brown N, et al. A role for topoisomerase I in Fusarium graminearum and F. culmorum pathogenesis and sporulation. Mol Plant-Microbe Interact. 2010;23:566–77.
Bluhm BH, Zhao X, Flaherty JE, et al. RAS2 regulates growth and pathogenesis in Fusarium graminearum. Mol Plant-Microbe Interact. 2007;20:627–36.
Zheng D, Zhang S, Zhou X, et al. The FgHOG1 pathway regulates hyphal growth, stress responses, and plant infection in Fusarium graminearum. PLoS One. 2012;7:e49495.
Gu Q, Chen Y, Liu Y, et al. The transmembrane protein FgSho1 regulates fungal development and pathogenicity via the MAPK module Ste50-Ste11-Ste7 in Fusarium graminearum. New Phytol. 2015;206:315–28.
Hohmann S. Osmotic stress signaling and osmoadaptation in yeasts. Microbiol Mol Biol Rev. 2002;66:300–72.
Widmann C, Gibson S, Jarpe MB, et al. Mitogen-activated protein Kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–80.
Turrà D, Segorbe D, Di Pietro A. Protein kinases in plant-pathogenic fungi: conserved regulators of infection. Annu Rev Phytopathol. 2014;52:267–88.
Hamel LP, Nicole MC, Duplessis S, et al. Mitogen-activated protein kinase signaling in plant-interacting fungi: distinct messages from conserved messengers. Plant Cell. 2012;24:1327–51.
Rispail N, Soanes DM, Ant C, et al. Comparative genomics of MAP kinase and calcium-calcineurin signalling components in plant and human pathogenic fungi. Fungal Genet Biol. 2009;46:287–98.
Zhao X, Mehrabi R, Xu JR. Mitogen-activated protein kinase pathways and fungal pathogenesis. Eukaryot Cell. 2007;6:1701–14.
Brown DW, Butchko RAE, Proctor RH. Genomic analysis of Fusarium verticillioides. Food Addit Contam Part A. 2008;25:1158–65.
McGuire AM, Pearson MD, Neafsey DE, et al. Cross-kingdom patterns of alternative splicing and splice recognition. Genome Biol. 2008;9:R50.
Galagan JE, Henn MR, Ma LJ, et al. Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res. 2005;15:1620–31.
Zhao C, Waalwijk C, de Pierre JGM W, et al. RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum. BMC Genomics. 2013;14:21.
Ding F, Cui P, Wang Z, et al. Genome-wide analysis of alternative splicing of pre-mRNA under salt stress in Arabidopsis. BMC Genomics. 2014;15:431.
Millar DS, Horan M, Chuzhanova NA, et al. Characterisation of a functional intronic polymorphism in the human growth hormone (GH1) gene. Hum Genomics. 2010;4:289–301.
Muráni E, Ponsuksili S, Seyfert H-M, et al. Dual effect of a single nucleotide polymorphism in the first intron of the porcine secreted phosphoprotein 1 gene: allele-specific binding of C/EBP beta and activation of aberrant splicing. BMC Mol Biol. 2009;10:96.
Lysenko A, Urban M, Bennett L, et al. Network-based data integration for selecting candidate virulence associated proteins in the cereal infecting fungus Fusarium graminearum. PLoS One. 2013;8:e67926.
Nasmith CG, Walkowiak S, Wang L, et al. Tri6 is a global transcription regulator in the phytopathogen Fusarium graminearum. PLoS Pathog. 2011;7:1–12.
Scherm B, Orrú M, Balmas V, et al. Altered trichothecene biosynthesis in TRI6-silenced transformants of Fusarium culmorum influences the severity of crown and foot rot on durum wheat seedlings. Mol Plant Pathol. 2011;12:759–71.
Yu H-Y, Seo J-A, Kim J-E, et al. Functional analyses of heterotrimeric G protein G alpha and G beta subunits in Gibberella zeae. Microbiology. 2008;154:392–401.
Lanver D, Mendoza-Mendoza A, Brachmann A, et al. Sho1 and Msb2-related proteins regulate appressorium development in the smut fungus Ustilago maydis. Plant Cell. 2010;22:2085–101.
Perez-Nadales E, Di Pietro A. The membrane mucin Msb2 regulates invasive growth and plant infection in Fusarium oxysporum. Plant Cell. 2011;23:1171–85.
Voigt CA, Schäfer W, Salomon S. A secreted lipase of Fusarium graminearum is a virulence factor required for infection of cereals. Plant J. 2005;42:364–75.
Niu X-W, Zheng Z-Y, Feng Y-G, et al. The Fusarium graminearum virulence factor FGL targets an FKBP12 immunophilin of wheat. Gene. 2013;525:77–83.
Blümke A, Falter C, Herrfurth C, et al. Secreted fungal effector lipase releases free fatty acids to inhibit innate immunity-related callose formation during wheat head infection. Plant Physiol. 2014;165:346–58.
ENSEMBL fungi database. http://ensemblgenomes.org. Accessed 1 Aug 2016.
Acknowledgments
We highly appreciate the excellent technical support of the teams at the Experimental Stations of Heidfeldhof (Hohenheim) and Oberer Lindenhof and specially the dedicated and engaged work of Bärbel Lieberherr. We thank Prof. Dr. Camilo Lopez for critical reading of the manuscript and Philipp Boeven for fruitful discussions on data analysis.
Funding
This research was conducted with the financial support of the German Academic Exchange Service (DAAD) provided to V. Castiblanco and J.J. Marulanda in the frame of the program “PhD Scholarships for international students”. A part of the study was financially supported by the German Federal Ministry of Education and Research granted through the Projektträger Jülich within the FusResist consortium that is gratefully acknowledged (grant no. 031B0011A).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Authors’ contributions
TM designed the study, helped with analyses and interpretation of data, revised the manuscript and finally approved it. VC selected candidate genes, extracted DNA, designed primers, amplified gene regions and performed sequence analyses. VC developed inoculum production and inoculations on the field, data collection and biometrical analyses. JM assisted in data collection, biometrical analyses and improved the manuscript. VC wrote figures and tables and prepared the manuscript. TW assisted with association mapping, double checked the results, and improved the quality of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Number of isolates, sampling scheme, origin, and host characterizing the F. culmorum populations under study. (XLS 33 kb)
Additional file 2:
Primer sequences, PCR conditions, and the full amplified sequences for the candidate genes used in this study. (XLS 53 kb)
Additional file 3:
Best linear unbiased estimates (BLUEs) for mean aggressiveness and DON production calculated across four environments (location × year combinations) for 100 F. culmorum isolates. (XLSX 15 kb)
Additional file 4:
Genotyping information of 100 isolates of F. culmorum. (XLSX 21 kb)
Additional file 5:
Histograms for mean aggressiveness and DON production. Histograms of best linear unbiased estimates (BLUEs) for mean aggressiveness (top) and DON production (bottom) calculated across four environments (location × year combinations) for 100 F. culmorum isolates. (PDF 10 kb)
Additional file 6:
Principal coordinate analysis for 100 F. culmorum isolates. Population structure and familial relatedness based on 10 SSR markers. Principal coordinate analysis for 100 F. culmorum isolates, based on modified Rogers’ distance. Number in parentheses refer to the proportion of variance explained by the principal coordinate. (PDF 17 kb)
Additional file 7:
Characteristics of identified single nucleotide polymorphisms (SNPs) in the candidate genes and p values for association tests against mean aggressiveness and DON production. (LOG 402 bytes) (XLS 34 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Castiblanco, V., Marulanda, J.J., Würschum, T. et al. Candidate gene based association mapping in Fusarium culmorum for field quantitative pathogenicity and mycotoxin production in wheat. BMC Genet 18, 49 (2017). https://doi.org/10.1186/s12863-017-0511-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-017-0511-9