
Abstract
Background
Genetic interactions are keys to understand complex traits and evolution. Epistasis analysis is an effective method to map genetic interactions. Large-scale quantitative epistasis analysis has been well established for single cells. However, there is a substantial lack of such studies in multicellular organisms and their complex phenotypes such as development. Here we present a method to extend quantitative epistasis analysis to developmental traits.
Methods
In the nematode Caenorhabditis elegans, we applied RNA interference on mutants to inactivate two genes, used an imaging system to quantitatively measure phenotypes, and developed a set of statistical methods to extract genetic interactions from phenotypic measurement.
Results
Using two different C. elegans developmental phenotypes, body length and sex ratio, as examples, we showed that this method could accommodate various metazoan phenotypes with performances comparable to those methods in single cell growth studies. Comparing with qualitative observations, this method of quantitative epistasis enabled detection of new interactions involving subtle phenotypes. For example, several sex-ratio genes were found to interact with brc-1 and brd-1, the orthologs of the human breast cancer genes BRCA1 and BARD1, respectively. We confirmed the brc-1 interactions with the following genes in DNA damage response: C34F6.1, him-3 (ortholog of HORMAD1, HORMAD2), sdc-1, and set-2 (ortholog of SETD1A, SETD1B, KMT2C, KMT2D), validating the effectiveness of our method in detecting genetic interactions.
Conclusions
We developed a reliable, high-throughput method for quantitative epistasis analysis of developmental phenotypes.
Similar content being viewed by others


Background
A genetic interaction occurs when two mutations at different loci generate a phenotype that cannot be explained by the additive effect of the two single mutations [1]. Genetic interactions are defined as positive (alleviating) when the combination of mutations shows a phenotype that is milder than the expected additive effect from the two single mutations; and negative (aggravating) when the combined phenotype is more severe than expected [1, 2]. Negative interactions can result from the loss of compensatory pathways. Positive interactions may indicate that genes function within a common pathway. Genetic interactions play an important role in many evolutionary processes, such as evolution of sex and recombination [3, 4], robustness and canalization [5], genetic polymorphism [6], and speciation [7]. Genetic interactions are also keys to the complex human diseases [8].
Historically large-scale mapping of genetic interactions mainly focused on synthetic lethality [9,10,11]. Synthetic lethality occurs when mutations of two genes cause lethality yet mutation of either gene does not [8]. Synthetic lethality is only one type of negative genetic interaction. Synthetic lethality screens would miss non-lethal negative genetic interactions and all positive interactions.
Subsequently, large-scale quantitative epistasis analysis was developed to map the whole spectrum of genetic interactions [12]. In this method, phenotypes are quantitatively measured for double and single mutants. Expected double mutant phenotype is calculated from the single mutant phenotypes using one of the neutrality models [1]. The observed and expected double mutant phenotypes were compared to detect significant differences that indicate genetic interactions. Such method has been applied successfully in unicellular organisms such as yeast and Escherichia coli [13,14,15]. It has also been extended to study metazoan cell lines [16,17,18,19].
The next logical step is to extend large-scale quantitative epistasis analysis to intact multicellular organisms. Multicellular organisms have a variety of complex traits. To map the genetic interaction networks underlying those traits, several challenges must be resolved: 1) high-throughput inactivation of two genes in whole multicellular organisms; 2) rapid and quantitative scoring of various phenotypes of interest; and 3) statistical analysis to detect genetic interactions from different phenotypic data.
Here we present a method to resolve those challenges in the nematode Caenorhabditis elegans. We applied this method to map the genetic interaction networks regulating two different developmental phenotypes, body length and sex ratio. We validated our interaction results on brc-1, a worm ortholog of the human breast cancer gene BRCA1, and revealed new genes that regulate DNA damage response and interact with brc-1.
Results
Collection of quantitative phenotypic data at a large scale
We chose two developmental phenotypes, C. elegans sex ratio and body length, to apply quantitative epistasis analysis for the following reasons. First, instead of lethality or general sickness that may involve many biological processes, these are specific developmental traits. Second, these traits represent different types of phenotypic data: the sex ratio measures a population of a binary output from each animal (hermaphrodite/male); the body length measures an individual animal of a continuous variable. A valid quantitative epistasis method should be applicable to both data types and thus be adapted to a wide range of metazoan phenotypes. Third, these phenotypes are easy to score and have biological importance.
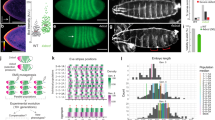
A research pipeline was developed to enable large-scale quantitative epistasis in C. elegans (Fig. 1a). As it is time consuming to generate double mutants, we used RNA interference (RNAi) by feeding [20] on mutant worm background to inactivate the functions of two genes at a high throughput. To enable quantitative phenotyping at a high throughput, we developed an automated imagining system [21, 22] to measure various phenotypes. Sex ratio was measured as percentage of hermaphrodites on a plate. Body length was measured for each worm in μm.

High-throughput method of acquiring quantitative epistasis data. a Flow chart showing the experimental process. b Reproducibility of raw measurements before (left panels) and after (right panels) quality control steps. c Reproducibility of S scores after quality control steps (left panels) and after applying a minimum standard deviation (SD) bound for the sex ratio phenotype (right panel). r, correlation; p < 0.001 for all r
We selected genes whose RNAi caused phenotypes in sex ratio or body length but not general lethality or sickness. For these genes, we obtained homozygous viable mutants that are publicly available. This resulted in 114 RNAi clones and 36 mutants for sex ratio, and 109 RNAi and 31 mutants for body length. Applying RNAi on mutants, 4104 gene pairs were tested for sex ratio, and 3379 gene pairs were tested for body length. For each gene pair, phenotypes of animals with two and one gene inactivated were measured. For each genotype, duplicates or more repeats were tested to measure over 100 animals for sex ratio, and over 40 animals for body length, to enable statistical analysis.
Data quality control
Quality control (QC) steps are critical for high-throughput large-scale experiments. We first flagged plates with too few worms for manual inspection. While some of these were valid data points indicating synthetic lethality, others were caused by issues such as unfocused images or overcrowded plates, and were thus invalidated.
Next we examined the variation of measurements of the same genotype among replicates tested in the same trial, as well as variation among different trials. A high variation would be flagged for re-testing. Often, one data point deviated from the majority and could not be reproduced in subsequent experiments. We removed such data points as they were likely caused by mistakes such as picking a wrong RNAi bacterial colony.
These QC steps improved data quality. Over 1000 gene pairs for each trait were tested in multiple independent trials, providing a good resource to evaluate the reproducibility of the phenotypic measurements. We split these animals into two groups of similar numbers of worms, and compared these two independent data sets for their consistency. For both sex ratio and body length, the QC steps increased the reproducibility of phenotypic measurements (Fig. 1b), demonstrating improved data quality.
Normalization of phenotypic measurements
In quantitative epistasis analysis, let f ab , f a , f b denote the survival rate (fitness) for the animals with two and one gene inactivated respectively, it is expected that f ab = f a f b if the two genes do not interact [23]. Let p ab , p a , p b denote the lethality rate (phenotypic severity) for the animals with two and one gene inactivated, then p ab = p a + p b − p a p b if the two genes do not interact. When p a and p b are low, for example, less than 0.1, then p ab calculation can be approximated to p ab = p a + p b − p a p b ≈ p a + p b . Therefore, either a multiplicative model or an additive model should be used depending on whether the fitness or the phenotypic severity was measured.
To determine whether our phenotypic measurements were fitness or phenotypic severity values, we reasoned that most mutations would reduce the fitness. In comparison with wild-type animals, most sex ratio mutants had reduced hermaphrodite rates; most body length mutants had reduced length. Therefore, we used hermaphrodite rates and length as fitness values. The mutant values were divided by wild-type values to obtain normalized fitness values. The normalized values were then used in the multiplicative model to compute the expected RNAi-on-mutant fitness values from single mutant and single RNAi values.
Calculation of genetic interaction scores
The next step in quantitative epistasis is using the phenotypic measurements to compute an interaction score. In general, a score of zero indicates no interactions. A score of negative values indicates negative interactions where the phenotype of RNAi-on-mutant animals is more severe than the additive effect of RNAi and mutation. A score of positive values indicates positive interactions where the phenotype of RNAi-on-mutant animals is milder than the additive effect of two single gene inactivation.
One challenge in extending quantitative epistasis analysis to metazoan is to develop interaction scores that can accommodate diverse types of phenotypic data. For example, the scoring method must be adaptable to data with different sample sizes. Our sex ratio data had a small sample size, with most genotypes having fewer than 10 plates tested. In contrast, the body length data had a big sample size, where the median sample size was over 100 worms for any genotype. In previous yeast studies, the median sample size was six colonies for any genotype; accordingly, t-score statistics was used to evaluate interactions because of the small sample size [12]. This would not be appropriate for the body length data with large sample size.
We adapted an S score to detect genetic interactions. The S score is defined as \( S=\frac{v_{obs}-{v}_{exp}}{\sigma} \), where v obs is the observed phenotype of RNAi-on-mutant animals, v exp is the expected phenotype of these animals if there is no genetic interactions, and σ is the standard deviation of the numerator. The S score is based on z-score statistics, and can thus readily accommodate data with a large sample size. To make it also applicable for data with small sample sizes, we placed a minimum bound [12] for the population standard deviation σ. If an unusually small standard deviation was calculated from the few plates, the minimum bound value was used instead of the calculated standard deviation for σ. This strategy improved the reproducibility of S scores for sex ratio data (Fig. 1c). For data with large sample size such as the body length data, S scores were directly computed without such estimation (Fig. 1c). Using this approach, the reproducibility of our S scores for both sex ratio and body length (correlation of 0.43, and 0.6, respectively, Fig. 1c) was comparable to previous yeast studies (correlation of 0.5) [12].
Comparison of different genetic interaction scoring methods
Three different interaction scores have been used in previous large-scale quantitative epistasis studies. In addition to the S score, the ε score [12] is defined as ε = v obs − v exp ; the π score [15] is defined as \( \pi ={log}_2\left(\frac{v_{obs}}{v_{exp}}\right) \). All three scores were highly correlated for both sex ratio and body length data (Fig. 2a), suggesting that they were in principle detecting the same interactions.

Comparison of different scoring methods for genetic interactions. a Correlation (r) between different genetic interaction scores, p < 0.001 for all r. b Histogram showing the distribution of ε, π, and S scores for sex ratio and body length phenotypes. c Enrichment of known interacting pairs over all gene pairs with high |S| scores and low p values
A key difference among the three scores was that S scores took variation into consideration, whereas the other two scores relied solely on mean values. As different traits have different variations, adjustment with variations enabled S scores to be comparable across different traits. For example, body length is more variable than sex ratio in C. elegans. Accordingly, the distribution of ε scores and π scores was more widely spread in body length than in sex ratio (Fig. 2b). In contrast, the distribution of S scores of the two traits appeared similar (Fig. 2b). Therefore, S scores are more versatile than the other scoring methods as it can provide a consistent scoring scale for different traits.
Comparison of different thresholds
Several thresholding methods can be applied to interaction scores to determine which genes interact. First, an empirically-determined cutoff can be directly applied to the values of S scores [18]. Second, assuming that S scores follow a normal distribution, Z scores of these S values can be used as a threshold. Thresholds for Z scores are commonly set to Z > 1.96 or Z > 4 [15, 24], corresponding to p < 0.05 and p < 0.0001, respectively. Third, if there are multiple independent trials of S score testing for each gene pair, then these S scores from different trials can be compared with zero using statistical tests such as t-test to calculate p-values of this gene pair having a non-zero S score. As such test is performed for each gene pair, p-values are usually adjusted for false discovery rate (FDR). Then adjusted p-value (usually p < 0.05) can be used to determine which scores are considered significant [19].
We tested the first method of thresholding S scores. The uniformity of S scores across different traits enabled us to use the same threshold value of S for both sex ratio and body length data. To determine a proper threshold, we queried WormBase for known genetic interactions, and found 31 sex ratio interactions and 41 body length interactions. Guided by these known interactions, we empirically determined the absolute value of S (|S|) over 0.8 as the threshold for interaction. Known interacting pairs were enriched with |S| values over 0.8: a 2.6-fold enrichment (23% vs. 9%) was observed for sex ratio and a 2.1-fold enrichment (15% vs. 7%) was observed for body length (Fig. 2c), suggesting that this |S| threshold can effectively capture interacting genes.
We also examined the second method of using Z scores of S scores as thresholds. Z scores of 1.96 corresponded in our study to S scores of 0.75 and −0.83 for sex ratio and 0.46 and −0.75 for body length (Additional file 1: Table S1). Therefore, the first method of |S| > 0.8 is in general more stringent than this method Z > 1.96.
Finally, we compared the performance of |S| and p values. As we did not have multiple biological replicates for each gene pair, we could not calculate p-values comparing S scores from multiple trials with the value of 0. Instead, we calculated p-values comparing the expected and observed fitness values of RNAi-on-mutant animals. Because sex ratio is based on counts of two types of animals (hermaphrodites/males) and body length is a continuous measurement, different statistical tests were required to analyze these data. Exact binomial test was used on sex ratio data to compare the observed sex ratio of RNAi-on-mutant animals with the expected one, whereas Student t-test was used on body length data to compare the observed body length of RNAi-on-mutant animals with the expected one [25]. Calculated p-values were then adjusted for multiple comparisons [26]. We found that known interacting pairs were mildly enriched with p < 0.05 for sex ratio with a 1.6-fold enrichment (29% vs. 18%), but not enriched for body length (86% vs. 88%, Fig. 2c). Therefore, p value alone was not a strong indicator of interactions. Combining both p values and |S| scores did not improve performance for the body-length data, but provided a slightly better performance than |S| alone for sex-ratio data: the enrichment was 3.3 fold (23% vs. 7%) among interacting genes in comparison with the 2.6 fold using |S| alone (Fig. 2c). Overall, these data suggested that those p values had limited effects on distinguishing interacting gene pairs in our data.
All these data suggested that the first threshold of |S| > 0.8 was the most conservative cutoff for genetic interactions, and that it was also most consistent in both traits. In addition, this threshold required no statistical assumptions. Considering these factors, we concluded that using the S score values |S| > 0.8 as a cutoff was the most effective thresholding method for interactions.
Discovery of new genetic interactions
Using |S| > 0.8 and p < 0.05 as the cutoffs, our analysis revealed 288 sex-ratio genetic interactions, including 132 synthetic lethality, and 228 body-length genetic interactions (Additional file 1: Table S1). 98% (504/516) of these interactions have not been reported previously, suggesting that our previous knowledge on genetic interactions was incomplete and that quantitative analysis could improve our understanding on genetic interactions.
Comparing these newly discovered interactions with previously known interactions, we noted that previously known interactions tend to involve genes with more severe phenotypes (Fig. 3a and b). Among the sex ratio mutants that were associated with previously known interactions, the median hermaphrodite rate was 95.5% of the wild-type value (Fig. 3a). Among the group of mutants without known interactions, many of them were close to wild-type, with the median hermaphrodite rate being 99.8% of the wild-type value (Fig. 3a). A similar trend was observed for the body length data: mutants with known interactions had more severe phenotypes (median being 69.5% of the wild-type value) than mutants without known interactions (median being 90.7% of the wild-type value) (Fig. 3b). As previously known interactions were detected using qualitative observations, these data suggested that quantitative epistasis analysis was particularly effective with genes of subtle phenotypes.

Quantitative epistasis can detect interactions for mutants with subtle phenotypes. a Sex ratio phenotypes. b Body length phenotypes. Box plots show phenotype distribution for mutants with previously known genetic interactions (Known) and for mutants with only interactions discovered in this quantitative study (New). Mutant phenotypes were divided by wild-type values to be normalized, so that 1 indicates wild-type
Computational validation of genetic interactions
Standard criteria were applied to validate our methodology. First, the method must generate reproducible data. We confirmed that our S scores were reproducible with a correlation between trials similar to that in yeast studies (Fig. 1c). Second, genetic interactions are expected to be sparse. We confirmed that the distributions of our S scores were centered at zero (Fig. 2b), and that only 6.9% (516/7483) of gene pairs were interacting. Such rate of interactions was similar to that in studies of E. coli (7%) [24] and S. pombe (6%) [14]. Finally, the method must recapture known interactions. We confirmed that known interacting pairs were enriched with high |S| scores (Fig. 2c). These data validated our methodology in detecting genetic interactions.
It is expected that interacting genes often share similar functions. We thus evaluated whether the interactions we discovered were consistent with known functional annotations. We partitioned the genetic interaction networks using the METIS software [27]. Three groups of densely connected genes were identified in sex ratio and body length networks (Additional file 2: Table S2). Many genes partitioned into the same groups shared similar functions, demonstrating the validity of the interactions. For example, among sex ratio genes, six dosage compensation genes [28], sdc-1, sdc-2, dpy-21, dpy-26, dpy-27, and dpy-28, were partitioned into the same group (Group 2, Additional file 2: Table S2). Another group (Group 3, Additional file 2: Table S2) contained seven genes involved in DNA damage response (GeneOntology.org), brc-1, brd-1, cep-1, F26B1.2, mre-11, rad-51, and rfs-1. Among them, brc-1, brd-1, cep-1 are known to interact with each other [29]. The consistency in biological functions suggested that our method is detecting true genetic interactions.
Discovery of new brc-1 interactions
One of the sex ratio genes we examined was brc-1, the ortholog of the human gene BRCA1. In humans, BRCA1 was associated with early onset of breast and ovarian cancer [30]. BRCA1 physically binds to BARD1 [31], another protein that was linked to breast cancer susceptibility [32]. BRCA1 functionally interacts with RAD51 in DNA damage repair [33]. In worms, mutants of brc-1 and the BARD1 ortholog brd-1 had elevated numbers of apoptotic germ cells before and after irradiation, and increased embryonic lethality after irradiation [29], suggesting that they function in DNA damage repair.
Our quantitative epistasis analysis on sex ratio revealed 38 genes interacting with brc-1 or brd-1 (Fig. 4a). Seven of them, cep-1, F26B1.2, mre-11, rad-50, rad-51, rfs-1, and smk-1, are involved in DNA repair (GeneOntology.org). We recaptured known brc-1 interactions with brd-1 and the P53 ortholog cep-1 [29]. The RAD51 ortholog rad-51 was also found to interact with brc-1. These results demonstrated the effectiveness of our method in detecting genetic interaction.

brc-1 interactions. a Genes interacting with brc-1 and brd-1 in sex ratio. Red circles indicate genes associated with DNA repair in Gene Ontology; Blue lines indicate negative interactions (S < −0.8); Yellow lines indicate positive interactions (S > 0.8). b brc-1 genetic interactions in embryonic survival with (+) and without (−) MMS exposure. Heat map displays S scores. Blue, S < 0; Black, S = 0; Yellow, S > 0. c brc-1 genetic interactions in apoptotic germ cell numbers with or without MMS exposure. Left panels display the number of apoptotic cells of various RNAi on wild-type background. Right panels display the increase of apoptotic cells in brc-1 mutant background. Bars and error bars indicate mean and standard deviation. *, p < 0.05 in comparison with the control RNAi group. d Images of apoptotic germ cells showing that brc-1 mutation interacts synergistically with RNAi of C34F6.1. WT, wild-type. Scale bar, 10 μm
In addition to recapturing known interactions, our analysis on sex ratio also revealed new brc-1 interactions. For example, 11 genes, C34F6.1, C41G7.3, cep-1, dyp-21, dpy-28, him-3, rad-51, sdc-1, sdc-2, set-2, and sex-1, were found to interact with both brc-1 and brd-1 (Fig. 4a). Among them, only cep-1 was known to interact with brc-1 in worms. To validate the new interactions, we tested these genes on a different phenotype, DNA damage response. We reasoned that if these genes interact with brc-1, then they are also likely to be involved in DNA damage response. To evaluate DNA damage response, we measured embryonic lethality and apoptotic germ cells with and without exposure to the DNA damaging reagent methyl methanesulfonate (MMS). Four genes were not included in the test for technical reasons: sdc-2 and sex-1 were not included because they had synthetic lethality with the brc-1 mutant; dyp-21 and dpy-28 were not included because these worms had egg laying defects that caused difficulties in scoring embryonic lethality, and their squeezed organs in the dumpy body caused difficulties in scoring apoptotic germ cells.
We applied RNAi of seven genes, C34F6.1, C41G7.3, cep-1, him-3, rad-51, sdc-1, and set- 2, on the brc-1 mutant, and scored the embryonic survival rate with and without MMS exposure. The interaction score S was calculated, and the threshold |S| > 0.8 and p < 0.05 was applied. Five out of the seven genes, C34F6. 1, him-3, rad-51, sdc-1 and set-2, were found to interact with brc-1 before or after MMS exposure (Fig. 4b, Additional file 3: Table S3).
RNAi of these five genes also caused a significant increase of apoptotic germ cells (Fig. 4c, Additional file 3: Table S3), confirming the function of these genes in DNA damage response. Our experiment also confirmed previously reported [29] interaction between brc-1 and cep-1 in regulating apoptotic cells (Fig. 4c). Interestingly, while brc-1 mutation caused a 4.5-cell increase of apoptotic germ cells on wild-type background (4.3 ± 1.9 vs. 8.8 ± 3.5), the same mutation caused an 8-cell increase in the sdc-1(RNAi) background (7.2 ± 2.3 vs. 15.2 ± 3.5) (Fig. 4c, Additional file 3: Table S3), suggesting a synergistic effect between sdc-1 and brc-1 inactivation in generating apoptotic cells. Similar effects with brc-1 were also observed for rad-51 and C34F6.1 RNAi after MMS exposure (Fig. 4c, d, Additional file 3: Table S3). Together, these data confirmed the brc-1 interactions with C34F6.1, rad-51, and sdc-1.
Overall, we tested six new brc-1 interactions, C34F6.1, C41G7.3, him-3, rad-51, sdc-1, and set-2, for their functions in DNA damage response. Five of them, C34F6.1, him-3, rad-51, sdc-1 and set-2, were found to interact with brc-1 in embryonic survival before or after MMS exposure. RNAi of all genes caused an increase of apoptotic germ cells. C34F6.1, rad-51, and sdc-1 were also found to interact with brc-1 in apoptotic germ cells before or after MMS exposure. While C34F6.1 and sdc-1 do not have a clear human ortholog, rad-51 is orthologous to human RAD51, him-3 is orthologous to human HORMAD1 and HORMAD2; set-2 is orthologous to human SETD1A, SETD1B, KMT2C, and KMT2D. One of those interactions, with rad-51, is known to be conserved in humans [34, 35]. Similar to the brc-1 ortholog BRCA1, HORMAD1 [36], SETD1A [37], KMT2C [38, 39], and KM2D/MML4 [40, 41] have all been linked to breast cancer, suggesting that these genetic interactions may be conserved in human cells.
Discussion
We describe here a methodology to extend quantitative epistasis analysis to metazoan. We validated our results both computationally and experimentally. Computationally, we evaluated the reproducibility of S scores and the consistency with known interactions. Experimentally, we tested the brc-1 sex-ratio interactions with an orthogonal phenotype: DNA damage response. Our results demonstrated that the principles of quantitative epistasis analysis remain the same from single cells to whole animals. While C. elegans has some unique advantages such as feeding RNAi, genome editing tools enable inactivation of two genes in many species. Therefore, it is possible to extend this quantitative epistasis analysis to other phenotypes in other multicellular organisms.
Conclusions
We have developed and validated a method for quantitative epistasis analysis on different developmental traits. Application of the method on C. elegans sex ratio and body length has enabled us to detect over 500 new genetic interactions including genes interacting with the breast cancer gene ortholog brc-1. This method can be used to study genetic interactions of phenotypes in C. elegans and it provides guidelines for developing similar methods in other metazoans.
Methods
Gene selection
Genes were selected based on WormBase (version 225) RNAi phenotype annotation of high incidence of males (him) for sex ratio phenotype, and reported RNAi phenotype of dumpy, small or long for body length [42]. RNAi bacterial clones were taken from the Ahringer RNAi library [42], or the ORFeome-RNAi library [43]. HT115 bacteria with the empty RNAi vector L4440 was used as a negative control.
Homozygous viable mutants were obtained from the Caenorhabditis Genetics Center (CGC). Unoutcrossed strains were outcrossed six times (all WWZ strains). Bristol N2 strain was used as wild-type. The following 36 sex-ratio mutants were used: CB1256 him-3(e1256) IV, CB1416 unc-86(e1416) III, CB1489 him-8(e1489) IV, CB4088 him-5(e1490) V, CB428 dpy-21(e428) V, CB5380 fox-1(e2643) X, CB541 unc-71(e541) III, CV138 sgo-1(tm2443) IV, DW102 brc-1(tm1145) III, DW103 brd-1(dw1) III, JK3101 fbf-2(q738) II, MH801 sur-7(ku119) X, MT1080 sdc-1(n485) X, MT1446 her-1(n695) V, MT14851 set-2(n4589) III, MT2244 sel-10(n1077) V, SP488 smk-1(mn156) V, WWZ238 htp-3(gk26) I, WWZ239 gpr-2(ok1179) III, WWZ240 rfs-1(ok1372) III, WWZ241 klp-10(ok704) IV, WWZ242 cki-2(ok2105) II, WWZ243 skr-1(ok1696) I, WWZ244 atl-1(ok1063) V, WWZ245 hil-4(ok1945) V, WWZ246 C30G12.6(ok2389) II, WWZ247 ess-2(ok3569) III, WWZ249 pme-3(gk120) IV, WWZ250 F52H3.4(ok2692) II, WWZ251 T08D2.7(ok431) X, WWZ252 zhp-3(ok1993) I, WWZ255 hcp-2(ok1757) V, WWZ256 coh-3(gk112) V, WWZ257 lipl-5(ok3581) V, WWZ258 W02D9.3(ok2857) I, XY1054 cep-1(lg12501) I. The following 31 body-length mutants were used: BE93 dpy-2(e8) II, CB1166 dpy-4(e1166) IV, CB12 dpy-9(e12) IV, CB128 dpy-10(e128) II, CB130 dpy-8(e130) X, CB14 dpy-6(e14) X, CB1482 sma-6(e1482) II, CB187 rol-6(e187) II, CB224 dpy-11(e224) V, CB27 dpy-3(e27) X, CB491 sma-3(e491) III, CB88 dpy-7(e88) X, CT11 hbl-1(mg285) X, DA2154 phb-2(ad2154) II, DR1369 sma-4(e729) III, DR933 dpy-13(m399) IV, FF41 unc-116(e2310) III, FX1053 ins-11(tm1053) II, JJ1237 mex-6(pk440) II, JK2729 dpy-18(ok162) III, JR2370 egl-18(ok290) IV, MT5998 sem-5(n2195) X, NG41 sex-1(gm41) X, RB1093 C08H9.2(ok1071) II, TN322 dpy-20(cn322) IV, WWZ127 M195.2(ok1503) II, WWZ130 sams-5(gk147) IV, WWZ131 fat-3(ok1126) IV, WWZ136 F42A8.1(ok2579) II, WWZ137 F54C4.3(ok2037) III, VC2428 sams-1(ok2946) X.
Animals were grown at 20 °C on standard nematode growth media (NGM) seeded with the OP50 strain of Escherichia coli as described in Stiernagle [44].
RNAi
RNAi was performed on solid media as described [20]. 6-well NGM plates with 50 μg/ml carbenicillin and 1 mM IPTG were used. Bacteria were cultured overnight at 37 °C in 96-well deep-well plates in L-broth with 50 μg/ml carbenicillin. Seventy microliter of bacteria were seeded onto each well of RNAi plates, and incubated at room temperature overnight.
For sex ratio, depending on the fecundity of each worm strain, 5 to 15 L4 hermaphroditic larvae were picked onto each well of 6-well plates seeded with RNAi bacteria, cultured at 25 °C for 20 to 30 h, and removed, so that each well had ~100 eggs. The eggs were cultured for 2 days at 25 °C so that the animals reached adulthood. The animals were then scored for the sex ratio phenotype.
For body length, worms were synchronized by bleaching gravid adults to obtain eggs and cultured in M9 buffer with 5 μg/ml cholesterol at 20 °C overnight to obtain synchronized L1 larvae [44]. About 100 L1 s were dropped to each well of the 6-well seeded RNAi plates. Animals were grown for 48 to 56 h at 25 °C until animals on control bacteria reached adulthood.
Automatic phenotyping
For sex ratio, adult animals were washed off from RNAi plates with S Basal solution [44], and transferred to unseeded scanning plates (modified NGM plates that do not contain peptone or cholesterol). The scanning plates were left without lids for about 30 min to air dry. Ten microliter of 1 M sodium azide was then added to each well to kill the animals.
For body length, 20 μl of 1 M sodium azide was added to each well of the RNAi plates to kill the animals. All RNAi on the same strain of worms were killed at the same time, and scanned immediately afterwards.
Quality control
Several quality control steps were employed to detect potential experimental errors. 1) A well was flagged for manual examination if it had fewer than 10 animals in sex ratio experiments [22] or 20 animals in body length experiments. 2) Wells with standard deviation of body length over 250 μm were re-tested. Empirically, we found these wells often had experimental issues such as worm crowding, or poor focusing during scanning. 3) As we tested at least duplicates for each genotype in each experiment, consistency between the replicates in the same experiment was examined using coefficient of variation (CV) of hermaphrodite percentage or mean length from each well. All genotypes with CV > 15% were re-tested in another experiment. 4) Consistency of the same genotype tested in different experiments was examined using the absolute difference (AD) among measured phenotypes. If the maximum AD was over 12% of wild-type values, then the genotype was re-tested. 5) The minimal number of animals tested for each genotype must be at least 100 for sex ratio and 40 for body length. Additional testing was conducted if the total number of animals were below this threshold. All thresholds were chosen empirically.
Calculation of genetic interaction scores
For sex ratio, male and hermaphrodite counts from all experiments were summed up to calculate the percentage of hermaphrodites for a given genotype. The percentage of hermaphrodites was then divided by the wild-type value to obtain the normalized fitness measurement. Let w ab , w a , and w b denote the normalized fitness for RNAi-on-mutant, RNAi, and mutant animals, respectively, then the observed fitness of RNAi-on-mutant animals is v obs = w ab ; the expected fitness of RNAi-on-mutant animals is v exp = w a w b . To compute \( S=\frac{v_{obs}-{v}_{exp}}{\sigma} \), the standard deviation σ was calculated as \( \upsigma =\sqrt{\sigma_{a b}^2+{\sigma}_a^2{w}_b^2+{\sigma}_b^2{w}_a^2} \), where σ ab , σ a , σ b denote the standard deviation for animals with two and one gene inactivated, respectively. Standard deviation was calculated using the hermaphrodite percentage of each well as a data point, normalizing it using wild-type values, and combining data points from all experiments to compute the standard deviation. If there are fewer than six wells available for σ ab calculation, then the median standard deviation from all RNAi on this mutant was used for σ ab . The minimal bound for σ was set to 0.04. This threshold was based on the average σ calculated from all genotypes with at least six wells available for σ ab calculation.
For body length data, the growth time of the same genotype from different experiments could vary up to 4 h, while the growth time for all RNAi on the same worm strain had the exact same growth time in a given experiment. Consequently, we chose not to combine the length measurement directly from different experiments, but instead computed and combined RNAi impact from different experiments. The RNAi impact for mutant a on b RNAi was \( {r}_{a b}=\frac{L_{a b}}{\overline{L_a}} \), where L ab denote the length of an animal with two genes inactivated, and \( \overline{L_a} \) denote the mean length of the single mutant a tested in the same experiment. Similarly, let L b denote the length for a wild-type worm fed with RNAi bacteria b, \( \overline{L_{wt}} \) denote the mean length of all wild-type worms fed with control bacteria in the same experiment, then the RNAi impact for a wild-type worm on b RNAi was \( {r}_b=\frac{L_b}{\overline{L_{wt}}} \). The observed and expected fitness values of RNAi-on-mutant animals were calculated as \( {v}_{obs}=\overline{r_{ab}} \), \( {v}_{exp}=\overline{r_b} \), where \( \overline{r_{ab}} \) and \( \overline{r_b} \) denote the mean values of r ab and r b combined from all experiments. To compute \( S=\frac{v_{obs}-{v}_{exp}}{\sigma} \), the standard deviation σ was calculated as \( \upsigma =\sqrt{\sigma_{ab}^2+{\sigma}_b^2} \), where σ ab , σ b denote the standard deviations for r ab and r b combined from all experiments.
DNA damage experiment
The strain ZH231 unc-76(e911) V; enIs7 [Pced-1::ced-1::GFP + unc-76(+)] was used as wild-type control. This strain was kindly provided by Dr. Zheng Zhou at Baylor College of Medicine, Houston, TX. In this strain, apoptotic cells were highlighted by green fluorescence, allowing easy visualization of apoptotic cells [45]. This strain was crossed with the strain DW102 to generate WWZ125 brc-1(tmll45); enls7[pced-1 ced-1::gfp and punc-76(+)], which was used as brc-1 mutant.
L4 worms were fed with different RNAi clones and removed after they reached adulthood and laid eggs. Those eggs were left on RNAi plates to develop to the L4 larval stage and then transferred onto new RNAi plates with or without 0.05 mg/ml methyl methanesulfonate (MMS) added to the agar. After 24 h, 10 to 20 worms were examined under a Zeiss AxioImager M2 m microscope for green fluorescence of apoptotic cells. Another 10 worms were moved to fresh RNAi plates with or without MMS for egg laying. Eggs were transferred to fresh RNAi plates. After 24 h, unhatched eggs were counted to evaluate embryonic lethality. Over 120 embryos were scored for each genotype. Minimum nine worms for each genotype were scored for apoptotic cells. S score calculation for embryonic survival was the same as that for the sex ratio phenotype.
Statistical analysis
The exact binomial test [25] was used to compare observed and expected values for sex ratio and survival rate of RNAi-on-mutant animals. The student t-test [25] was used on body length data to compare the observed and expected RNAi impact on mutants. Apoptotic cell numbers were compared using the ANOVA test followed by contrast analysis [25]. The p-values from all statistical tests were adjusted for multiple comparison (separately for each phenotype) using Benjamini-Hochberg correction with family-wise error set to 5% [26] to control for false discovery rate. All tests were performed in R [46].
Abbreviations
- AD:
-
Absolute difference
- CGC:
-
Caenorhabditis Genetics Center
- CV:
-
Coefficient of variation
- FDR:
-
False discovery rate
- him :
-
High incidence of males
- MMS:
-
Methyl methanesulfonate
- NGM:
-
Nematode growth media
- QC:
-
Quality control
- RNAi:
-
RNA interference
- SD:
-
Standard deviation
- WT:
-
Wild-type
References
Mani R, St Onge RP, Hartman JL 4th, Giaever G, Roth FP. Defining genetic interaction. Proc Natl Acad Sci U S A. 2008;105(9):3461–6.
Segrè D, Deluna A, Church GM, Kishony R. Modular epistasis in yeast metabolism. Nat Genet. 2005;37(1):77–83.
Charlesworth B. Mutation-selection balance and the evolutionary advantage of sex and recombination. Genet Res. 1990;55:199–221.
Azevedo RB, Lohaus R, Srinivasan S, Dang KK, Burch CL. Sexual reproduction selects for robustness and negative epistasis in artificial gene networks. Nature. 2006;440:87–90.
Rice SH. The evolution of canalization and the breaking of von Baer’s laws: modeling the evolution of development with epistasis. Evolution. 1998;52:647–56.
Zhivotovsky LA, Gavrilets S. Quantitative variability and multilocus polymorphism under epistatic selection. Theor Popul Biol. 1992;42(3):254–83.
Presgraves DC. Speciation Genetics: Epistasis, Conflict and the Origin of Species. Curr Biol. 2007;17(4):R125–7.
Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat Rev Genet. 2007;8(6):437–49.
Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303(5659):808–13.
Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet. 2006;38(8):896–903.
Byrne AB, Weirauch MT, Wong V, Koeva M, Dixon SJ, Stuart JM, et al. A global analysis of genetic interactions in Caenorhabditis elegans. J Biol. 2007;6(3):8.
Collins SR, Schuldiner M, Krogan NJ, Weissman JS. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 2006;7(7):R63.
Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123(3):507–19.
Roguev A, Bandyopadhyay S, Zofall M, Zhang K, Fischer T, Collins SR, et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science. 2008;322(5900):405–10.
Butland G, Babu M, Díaz-Mejía JJ, Bohdana F, Phanse S, Gold B, et al. eSGA: E. coli synthetic genetic array analysis. Nat Methods. 2008;5(9):789–95.
Bakal C, Linding R, Llense F, Heffern E, Martin-Blanco E, Pawson T, et al. Phosphorylation networks regulating JNK activity in diverse genetic backgrounds. Science. 2008;322(5900):453–6.
Laufer C, Fischer B, Billmann M, Huber W, Boutros M. Mapping genetic interactions in human cancer cells with RNAi and multiparametric phenotyping. Nat Methods. 2013;10(5):427–31.
Roguev A, Talbot D, Negri GL, Shales M, Cagney G, Bandyopadhyay S, et al. Quantitative genetic-interaction mapping in mammalian cells. Nat Methods. 2013;10(5):432–7.
Fischer B, Sandmann T, Horn T, Billmann M, Chaudhary V, Huber W, et al. A map of directional genetic interactions in a metazoan cell. Elife. 2015;4:e05464.
Ahringer J. ed. Reverse genetics. 2006 WormBook, ed. The C. elegans Research Community, WormBook, doi: 10.1895/wormbook.1.47.1, http://www.wormbook.org.
Jung SK, Aleman-Meza B, Riepe C, Zhong W. QuantWorm: a comprehensive software package for Caenorhabditis elegans phenotypic assays. PLoS One. 2014;9(1):e84830.
Labocha MK, Jung SK, Aleman-Meza B, Liu Z, Zhong W. WormGender - Open-Source Software for Automatic Caenorhabditis elegans Sex Ratio Measurement. PLoS One. 2015;10(9):e0139724.
Baryshnikova A, Costanzo M, Myers CL, Andrews B, Boone C. Genetic interaction networks: toward an understanding of heritability. Annu Rev Genomics Hum Genet. 2013;14:111–33.
Babu M, Arnold R, Bundalovic-Torma C, Gagarinova A, Wong KS, Kumar A, et al. Quantitative genome-wide genetic interaction screens reveal global epistatic relationships of protein complexes in Escherichia coli. PLoS Genet. 2014;10(2):e1004120.
Sokal RR, Rohlf FJ. Biometry: the principles and practice of statistics in biological research. San Francisco: Freeman; 1995.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300.
Karypis G, Kumar VA. Fast and Highly Quality Multilevel Scheme for Partitioning Irregular Graphs. SIAM J Sci Comput. 1999;20:359–92.
Meyer BJ. X-Chromosome dosage compensation. 2005. WormBook, ed. The C. elegans Research Community, WormBook, doi:10.1895/wormbook.1.8.1, http://www.wormbook.org.
Boulton SJ, Martin JS, Polanowska J, Hill DE, Gartner A, Vidal M. BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr Biol. 2004;14(1):33–9.
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71.
Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev. 2002;12(1):86–91.
Karppinen SM, Heikkinen K, Rapakko K, Winqvist R. Mutation screening of the BARD1 gene: evidence for involvement of the Cys557Ser allele in hereditary susceptibility to breast cancer. J Med Genet. 2004;41(9):e114.
Cousineau I, Abaji C, Belmaaza A. BRCA1 regulates RAD51 function in response to DNA damage and suppresses spontaneous sister chromatid replication slippage: implications for sister chromatid cohesion, genome stability, and carcinogenesis. Cancer Res. 2005;65(24):11384–91.
Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88(2):265–75.
Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–74.
Watkins J, Weekes D, Shah V, Gazinska P, Joshi S, Sidhu B, et al. Genomic Complexity Profiling Reveals That HORMAD1 Overexpression Contributes to Homologous Recombination Deficiency in Triple-Negative Breast Cancers. Cancer Discov. 2015;5(5):488–505.
Salz T, Deng C, Pampo C, Siemann D, Qiu Y, Brown K, et al. Histone Methyltransferase hSETD1A Is a Novel Regulator of Metastasis in Breast Cancer. Mol Cancer Res. 2015;13(3):461–9.
Fusco N, Geyer FC, De Filippo MR, Martelotto LG, Ng CK, Piscuoglio S, et al. Genetic events in the progression of adenoid cystic carcinoma of the breast to high-grade triple-negative breast cancer. Mod Pathol. 2016;29(11):1292–305.
Manso L, Mourón S, Tress M, Gómez-López G, Morente M, Ciruelos E, et al. Analysis of Paired Primary-Metastatic Hormone-Receptor Positive Breast Tumors (HRPBC) Uncovers Potential Novel Drivers of Hormonal Resistance. PLoS One. 2016;11(5):e0155840.
Ansari KI, Hussain I, Kasiri S, Mandal SS. HOXC10 is overexpressed in breast cancer and transcriptionally regulated by estrogen via involvement of histone methylases MLL3 and MLL4. J Mol Endocrinol. 2012;48(1):61–75.
Rabello Ddo A, de Moura CA, de Andrade RV, Motoyama AB, Silva FP. Altered expression of MLL methyltransferase family genes in breast cancer. Int J Oncol. 2013;43(2):653–60.
Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421(6920):231–7.
Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, et al. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 2004;14(10B):2162–8.
Stiernagle T. Maintenance of C. elegans. 2006. WormBook. ed. The C. elegans Research Community, WormBook, doi:10.1895/wormbook.1.101.1, http://www.wormbook.org.
Venegas V, Zhou Z. Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol Biol Cell. 2007;18(8):3180–92.
R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2016. URL https://www.R-project.org/
Acknowledgements
We thank Joaquina Nunez and all members in the Zhong lab for technical assistance, Dr. Zheng Zhou and the Caenorhabditis Genetics Center (CGC) for strains.
Funding
This research was funded by the National Institutes of Health [HG004724, DA018341] and by a Searle Scholar grant to WZ.
Availability of data and materials
The datasets used and/or analysed during the current study and not included in Additional files are available from the corresponding author on reasonable request.
Authors’ contributions
MKL, WY, and WZ performed experiments. MKL, BA-M, WY, and WZ analyzed data. MKL and WZ wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Genetic interactions with S scores, Z scores and p values. (XLSX 456 kb)
Additional file 2: Table S2.
Partition of genetic interaction networks showing functional groups. (XLSX 11 kb)
Additional file 3: Table S3.
DNA damage data for brc-1 interactions. (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Labocha, M.K., Yuan, W., Aleman-Meza, B. et al. A strategy to apply quantitative epistasis analysis on developmental traits. BMC Genet 18, 42 (2017). https://doi.org/10.1186/s12863-017-0508-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-017-0508-4