
Abstract
Background
Foxtail millet (Setaria italica) is a diploid C4 panicoid species. Because of its prominent drought resistance, small genome size, self-pollination, and short life cycle, foxtail millet has become an ideal model system for studying drought tolerance of crops. MicroRNAs (miRNAs) are endogenous, small RNAs that play important regulatory roles in the development and stress response in plants.
Results
In this study, we applied Illumina sequencing to systematically investigate the drought-responsive miRNAs derived from S. italica inbred An04-4783 seedlings grown under control and drought conditions. Degradome sequencing was applied to confirm the targets of these miRNAs at a global level. A total of 81 known miRNAs belonging to 28 families were identified, among which 14 miRNAs were upregulated and four were downregulated in response to drought. In addition, 72 potential novel miRNAs were identified, three of which were differentially expressed under drought conditions. Degradome sequencing analysis showed that 56 and 26 genes were identified as targets of known and novel miRNAs, respectively.
Conclusions
Our analysis revealed post-transcriptional remodeling of cell development, transcription factors, ABA signaling, and cellar homeostasis in S.italica in response to drought. This preliminary characterization provided useful information for further studies on the regulatory networks of drought-responsive miRNAs in foxtail millet.
Similar content being viewed by others

Background
Foxtail millet (Setaria italica), a diploid C4 panicoid species with a small genome of ~ 510 Mb [1], is an important food and fodder grain crop in arid and semi-arid regions of Asia, especially in China and India. The majority of foxtail millet varieties are abiotic stress tolerant, particularly to drought. The water use efficiency (WUE) of foxtail millet is higher than that of maize, wheat, and sorghum given that maize requires 470 g of water, wheat requires 510 g, and foxtail millet requires only 257 g for 1 g of dry biomass [2]. Because of its drought tolerance, foxtail millet was described as the “Oasis of Arid Agriculture” [3]. The prominent drought resistance, small genome size, self-pollination, and short life cycle has made foxtail millet an ideal model system for studying drought tolerance in plants. Many drought-inducible genes with various functions have been identified by molecular and genomic analyses in foxtail millet [4–7]. Recently, with the discovery of small RNAs, post-transcriptional regulation of drought response by miRNAs has been examined [8–10].
MicroRNAs (miRNAs) are endogenous, 20–24 nt noncoding RNAs that play important regulatory roles in eukaryotes by targeting mRNAs for cleavage or translational repression [11]. In plants, primary miRNAs are transcribed by RNA polymerase II [12, 13] and then processed by Dicer-like (DCL) into precursors (pre-miRNA) with stem-loop structures. Subsequently, these pre-miRNAs are cleaved into miRNA::miRNA* duplexes and exported to the cytoplasm by the HASTY protein [14, 15]. The mature miRNAs are incorporated into the RNA-induced silencing complex (RISC) to target specific mRNAs and downregulate the expression of target mRNAs [16]. Increasing evidence indicates that miRNAs have an influential role in numerous processes in plants, including development, abiotic stress tolerance, nutrient starvation, and metabolism [17–20].
Drought is a major abiotic stress factor that limits crop productivity. Plants respond to drought stress through complex mechanisms that allow them to adapt to water-deficit conditions by synthesizing or suppressing specific drought-related proteins. Recent studies have shown that miRNAs play important roles in drought tolerance by regulating the expression of drought-responsive genes [21, 22]. Many miRNAs associated with abiotic stress responses have been identified in plant species, including Arabidopsis thaliana [23, 24], Oryza sativa [25–28], Zea mays [29, 30], Populus [31], and Medicago truncatula [32]. Li et al. (2008) reported that miR169 was downregulated by drought stress through an ABA-dependent pathway, and miR169-overexpressing plants showed enhanced leaf water loss and were more sensitive to drought stress than wild-type plants [33]. Zhou et al. (2013) found that transgenic Creeping Bentgrass overexpressing Osa-miR319a showed morphological changes and enhanced drought and salt tolerance [34].
Next-generation sequencing and bioinformatics prediction provide effective methods for plant miRNA discovery and analysis. In foxtail millet, Yi et al. (2013) characterized the miRNA repertoire by deep sequencing and identified 43 known miRNAs and 172 novel miRNAs [35]. Khan et al. (2014) identified 355 mature miRNAs through computer analysis [36], and Han et al (2014) identified 271 foxtail millet miRNAs belonging to 44 families using a bioinformatics approach [37]. These results are useful for miRNA studies in foxtail millet. However, there has been no study on the differential expression of miRNAs in foxtail millet under drought stress, and most microRNA targets in previous studies were predicted by bioinformatics, which require confirmation.
Various studies have indicated that different genotypes of plant showed different gene-expression profiles in response to drought, and more genes were significantly drought regulated in the sensitive compared with the tolerant cultivars [38]. Thus, in this study a drought-sensitive cultivar was used to study potential drought-responsive miRNAs and their targets in foxtail millet. We constructed two libraries of sRNAs from foxtail millet under control and water-deficit conditions, which were sequenced using the Illumina sequencing platform. Degradome sequencing was applied to directly detect cleaved miRNA targets at a global level in foxtail millet.
Methods
Plant materials and stress treatment
To evaluate drought resistance at the seedling stage, 10 varieties of foxtail millet were subjected to repeated drought treatments [39], and the results are shown in Additional file 1. Among them, An04-4783 was identified to be more sensitive to drought stress. An04-4783 is a mordern cultivar of S. italica, which was developed a decade ago in Anyang academy of agriculture sciences, Henan, China, and is publicly available as germplasm resource in Chinese Crop Germplasm Information System (CGRIS). The An04-4783 seedlings were grown in the greenhouse (28 °C day/20 °C night and 16 h day/8 h night) with compound media, including vermiculite and sand. Plants were well irrigated according to evaporation demand and watered with 1 × Hoagland nutrient solution. After 14 days, deficit irrigation treatments were applied by withholding watering on the stressed pots while control pots were well-watered. Leaf water potential (LWP) was measured using the psypro WP data logger (Wesco) as an indicator of stress level. Fresh leaves were sampled from control plants (CL, well watered, Ψwp = -0.5 MPa) and moderately drought-stressed plants (DT, Ψwp = -1.4 MPa), and frozen in liquid nitrogen for RNA extraction.
Small RNA library and degradome library preparation and sequencing
Total RNAs was extracted from mixed leaf tissues using the Tri-Reagent kit (Sigma, USA) according to the manufacturer’s instructions. The 5’ and 3’ adaptors were ligated sequentially to the RNAs and amplified by RT-PCR. Small RNAs from 140 bp to 160 bp were selected by polyacrylamide gel electrophoresis and sequenced on an Illumina HiSeq 2000 sequencer.
Degradome libraries were constructed as described previously [40] with some modification. Total RNAs from control plant leaves was sent to BGI (Beijing, China) to prepare the degradome sequencing libraries. The protocol is as below: 1. Approximately 150 ng of mRNA was used to anneal with biotinylated random primers (BPRs). 2. Streptavidin capture of RNA fragments through BPRs. 3. 5’ adaptor was ligated to only those RNAs containing 5’-monophosphates, followed with reverse transcription and PCR. 4. Libraries were sequenced using the 5’ adapter only, resulting in the sequencing of the first 50 nucleotides of the inserts that represented the 5’ ends of the original RNAs. Single-end sequencing (50 bp) were then performed on an Illumina Hiseq 2000 [41].
Small RNA data analysis
Raw sequences were processed by removing adapters and discarding low-quality sequences, and clean small reads were obtained and aligned against the S. italica genome (Phytozome v10.0) using bowtie software v1.01 [42] with perfect match. The matched reads were then used as queries to search against the Rfam database [43] to remove rRNA, tRNA, snRNA, and snoRNA, and the remaining reads were search against the miRBase database (Release 21) [44] and evaluated using miRcheck [45]. Only miRNAs matched to known miRNAs with no more than two mismatches in the miRBase database and whose precursors could fold into stem-loop structures were considered to be known miRNAs of S. italica. The unannotated sRNAs were subsequently analyzed for potential novel miRNAs using miRCat software with default plant parameters [46] and psRobot software [47]. Secondary structures of potential miRNAs were checked using RNAfold software [48]. The criteria we used to identify miRNAs were referred to previous research Meyer et al (2008) [49] and Li et al (2011) [50] and listed as follows: (1) precursor sequence could form a marked stem-loop hairpin secondary structure, (2) there are no more than four mismatches between miRNA and miRNA*, (3) asymmetric bulges are minimal in size no more than 2 bases in mature sequence, and (4) the maximum free energy allowed for a miRNA precursor was -18 kcal mol-1. To exclude the contaminative effect of small interfering RNA (siRNA) on miRNA identification, we mainly considered two main factors: 1. Structural feature of siRNA. siRNA is a class of short double-stranded RNA molecules, each strand of which is 2 nt longer than the other at the 3’end [50]. 2. Limit the number of loci of identified miRNAs. The number of loci of known miRNAs in plant genome is mostly less than 24 [51]. Higher number of loci often occurs in repeat-rich regions from which siRNAs are produced.
Degradome analysis
After adapter trimming and removing low quality reads, clean reads perfectly matching the S. italica genome were collected for further analysis using PAREsnip software [52]. The cleaved target transcripts were categorized into five classes based on the abundance of degradome tags indicative of miRNA-mediated cleavage. Category 0 comprised the sequences whose abundance at the cleavage site was the only maximum on the transcript; in category 1, the reads abundance at the cleavage site was the maximum but not unique; category 2 consisted of sequences whose abundance at the cleavage site was higher than the median but not the maximum; category 3 included sequences whose abundance at the cleavage site was equal to or below the median; the remaining sequences, which were the only raw reads at the cleavage site, were classified as category 4.
Differential expression analysis of miRNAs
The reads of each library were normalized by TPM (Transcript per million), normalized expression = (actual miRNA count/total count of clean reads) × 1,000,000 [49, 50]. Differential expression between drought and control conditions was calculated using IDEG6 software [53] (http://telethon.bio.unipd.it/bioinfo/IDEG6_form/). Audic and Claverie, Fisher’s exact test, and general chi-square statistical methods were applied. miRNAs with absolute value log2 A fold-change (DT/CK) ≥ 1 and a significance threshold ≤ 0.01 were considered to reflect significantly differentially expressed (DE) miRNAs.
miRNA validation
To validate the results of miRNAs from high-throughput sequencing, qRT-PCR was performed using SYBR Green PCR Master Mix on qTOWER 2.2 (Analytik Jena AG). The small RNAs were extracted from leaves of foxtail millet using the miRNA pure Mini kit (Beijing ComWin Biotech). The miRNA cDNA kit (Beijing ComWin Biotech) was used in the reverse transcription reaction. Quantitative real time PCR was performed using the miRNA Real-Time PCR Assay kit (Beijing ComWin Biotech). Each PCR reaction consisted of 2 μl of product from the diluted reverse transcription reaction, 0.5 μl sequence-specific forward primer, 0.5 μl universal reverse primer, 12.5 μl of 2 × miRNA qPCR premix (with SYBR and ROX), and 9.5 μl of nuclease-free water. The U6 gene was used as an internal control. The reaction conditions were set as follows: 95 °C for 10 min followed by 40 cycles of 95 °C for 15 seconds and 60 °C for 1 minute, with a final dissociation curve analysis. All reactions were performed with three biological replicates for each sample (primer list in Additional file 2). Real-time PCR data analysis was performed by qPCR soft 3.0 (Analytik Jena AG).
Drought-related miRNA-mRNA network construction
Based on the analysis of small RNA data and degradome sequencing, drought-related miRNAs and Arabidopsis genes homologous to drought-related miRNA targets were used to construct the miRNA–mRNA interaction network. The functional relationship between two genes was retrieved from STRING database v10 [54]. If two genes were annotated to be related, we added an edge between them in the network drawn using the Cytoscape software [55].
Results
Physiological response of foxtail millet to drought stress
Seedlings of the drought-treated group (DT) were subjected to a natural soil drying process, whereas the control group (CL) was irrigated to field water capacity daily. Pre-dawn leaf water potential (Ψwp) was measured throughout the process to monitor the intensity of drought stress. On the third day of drought treatment, Ψwp decreased by about one-half in the DT compared with the CL group (Fig. 1a). The drought-stressed seedlings showed yellow and rolling leaves as well as reduced plant growth (Fig. 1b), which indicated that drought stress significantly affected plants in the DT group during this time.

Effects of drought stress on phenotypic alterations and changes in leaf water potential (WP) in foxtail millet seedlings. a After drought treatment for 3 days, the plants were smaller compared with control plants, and the leaves changed color. b Leaf water potential (LWP) of control and drought treatment plants. After drought treatment, LWP decreased from -0.5 Mp (CL) to -1.4 Mp (DT)
High-throughput sequencing of small RNAs in foxtail millet
To identify miRNAs from foxtail millet under water-deficit conditions, two small RNA libraries were constructed based on sequencing data from CL and DT groups of foxtail millet leaves. Raw sRNA sequencing reads have been deposited at EMBL (https://www.ebi.ac.uk/) under accession number ERP014347. After removing the low-quality sequences, adapter, and sequences smaller than 16 nt and larger than 30 nt, 14,124,084 clean reads (1,487,858 unique sequences) in CL and 10,374,842 clean reads (1,579,854 unique sequences) in DT were obtained. Analysis of the length distribution of unique sRNAs reads showed that the two libraries contain similar data, with the 24 nt sRNAs being the most abundant (Fig. 2) and this result was consistent with previous studies in foxtail millet [35] and maize [56, 57]. The common and different sequences in the CL and DT libraries were analyzed for unique and total sRNAs (Table 1), and 52.41 % of total sequences appeared in two libraries. However, only 11.85 % percent of unique sequences overlapped between the two libraries. This limited overlap indicated that there was diversity in the sRNAs of foxtail millet in response to drought.

Sequence length distribution of sRNA in the library of control and drought versions of foxtail millet
To obtain a comprehensive view of the sequence distribution of all sRNA reads, clean reads were divided into different categories of exon-sense, exon-antisense, intron antisense, intron sense, and sRNAs, including non-coding RNAs (tRNA, rRNA, snRNA, snoRNA and miRNA) through mapping against the Rfam database and miRBase (Table 2). Our results showed that the proportion of miRNA sequences represented a very small fraction (< 1 %) of the total and unique sequences. The majority of unique sequences (> 90 %) were classified as other, which could not be mapped to any known reference database. The largest fraction of unannotated sequences may represent novel miRNAs and other classes of small ncRNAs. Similar results have been found in other plant species [58].
Identification of known miRNAs
To identify the known miRNAs of foxtail millet (conserved and species-specific), clean reads of two libraries were searched against mature plant miRNAs from the miRNA database. After filtering miRNAs whose pre-miRNA could not form hairpin secondary structures, 81 miRNAs were identified in the CL and DT libraries, which were clustered into 28 families based on the similarity of the mature miRNA sequence. Among them, 29 miRNA* were identified based on sequence alignment. The length of pre-miRNA ranged from 66 to 222 nt and negative MFEs (minimum free energies) ranged from -32.1 to -98.9 kcal/mol (Additional file 3). Compared with the 48 foxtail millet miRNA families from a previous report by Bennetzen et al. [59], the results showed that these miRNA families are common. Analysis of all known miRNA family reads of two libraries showed that the number of reads varied significantly, ranging from 14 to 20,970 (1484.7 TPM) in the CL library and from 4 to 22,500 (2168.7 TPM) in the DT library. MIR166 was the most abundant miRNA family in both the CL and DT libraries. In contrast, MIR397 and MIR2118 showed low expression levels (Fig. 3). Based on analysis of location of precursor, we found that in foxtail millet, more than 87 % of known miRNAs are derived from intergenic regions, and others originate from coding sequence regions (Additional file 3). This result was consistent with previous studies [60].

Expression levels of known miRNA families in CL and DT libraries
Identification of potential novel miRNAs in foxtail millet
After identifying exons, rRNA, tRNA, snoRNA, snRNA, and known miRNAs, we pooled the remaining unannotated sRNA sequences of two libraries and predicted novel miRNAs using miRcat software with default plant parameters and psRobot software. A total of 72 novel miRNA candidates were obtained. The length of precursor miRNA sequences varied from 61 to 208 nt, and the negative MFEs of the identified foxtail millet miRNA precursors varied from -18.0 to -111.8 kcal/mol (Additional file 4). The secondary structures of novel miRNA precursors shown in Additional file 5. Among these potential miRNAs, eight miRNAs with complementary miRNA* were identified, which supported their role as novel miRNAs of foxtail millet (Table 3). The majority of these miRNAs had relatively low expression, which was consistent with previous studies in other plants [58, 59]. The low abundance of novel miRNAs suggests that the majority of foxtail millet-specific miRNAs are expressed at low levels. Characteristics of novel miRNA precursor location were similar to known miRNA, about 72 % miRNAs were from intergenic regions, 20 % miRNAs were derived from intronic and 8 % originated from coding sequences.
Differential expression analysis of known and novel miRNAs of foxtail millet under drought stress
To identify drought-associated miRNAs of foxtail millet, we removed miRNAs whose expression levels were too low to be analyzed for differential expression (sequencing frequency < 10 in CL and DT libraries) and compared the normalized expression of miRNAs between the CL and DT libraries. A total of 18 known miRNAs belonging to 16 families were significantly expressed with more than one log2 fold change (Additional file 6). Among these DE miRNAs, 14 miRNAs (sit-miR1432-3p, sit-miR156a-5p, sit-miR156b-5p, sit-miR164a-5p, sit-miR167b-5p, sit-miR171c-3p, sit-miR2118-3p, sit-miR390-5p, sit-miR394-5p, sit-miR395-3p, sit-miR408-3p, sit-miR529a-3p, sit-miR529b-3p, and sit-miR827) were upregulated and 4 miRNAs (sit-miR159b-3p, sit-miR319c-5p, sit-miR528-5p and sit-miR535-5p) were downregulated; some of these miRNA families have been associated with drought stress in previous studies: miR156 [31, 61], miR159 [23], miR167 [23, 61], miR395 [62], and miR408 [63]. We also identified three potential novel miRNAs considered to be drought-response miRNAs based on the differential expression between the CL and DT libraries. Of these miRNAs, two (sit-novel-miR10, sit-novel-miR56) were upregulated, and one (sit-novel-miR18) was downregulated (Additional file 7).
To verify the results of miRNA sequencing and bioinformatics analysis, six known miRNAs (sit-miR159b, sit-miR167b, sit-miR390, sit-miR394, sit-miR396a, and miR408) and four novel miRNAs (sit-novel-miR15, sit-novel-miR18, sit-novel-miR53, and sit-novel-miR56) were selected randomly for validation by qRT-PCR. The results showed that the fold change of expression obtained by qRT-PCR was not completely consistent with bioinformatics analysis results, but the expression trend was similar (Fig. 4). The stem-loop secondary structure of four novel miRNAs is shown in Fig. 5. These results suggested that Solexa sequencing was successfully applied to identify drought-related miRNAs in foxtail millet.

Differential expression analysis of conserved and novel drought-responsive miRNAs. a Fold change (log2) in control library relative to drought library detected by solexa small RNA sequencing. b The relative expression level of miRNAs measured by RT-qPCR. * means significant difference between control and drought stress at P ≤ 0.01

Secondary structure prediction of novel foxtail millet miRNA precursors. The blue colored sequences represent mature miRNA, and the green colored sequences represent the miRNA* (a, sit-novel-15; b, sit-novel-18; c, sit-novel-53; d, sit-novel-56).
Target prediction of miRNAs and validation by degradome sequencing
In foxtail millet, numerous miRNA targets have been predicted previously [35, 36], but few miRNA targets have been validated experimentally. To identify miRNA targets in foxtail millet at the global level, we employed the degradome sequencing approach to identify target genes for known miRNAs and candidate novel miRNAs. Raw sequencing data generated by degradome sequencing are available at EMBL with the accession number ERP014368. After removing adapter sequences and low-quality tags, we obtained a total of 11,762,879 clean reads (3,528,168 unique reads) representing the 5’ uncapped ends, of which 7,239,426 (2,433,599 unique reads) were perfectly matched to the S. italica genome. The reads that perfectly mapped to the genome were subjected to further analysis using PAREsnip software [52].
In this study, 56 target genes for 12 known miRNA families were identified. Based on the abundance of degradome tags at the target sites, these cleaved targets were classified into five categories; 42 target genes were classified into category 0, 4 target genes into category 1, 6 target genes into category 2, 2 target genes into category 3, and 2 target genes into category 4 (Table 4). The detailed information is provided in Additional file 8, and the t-plots for targets are illustrated in Additional file 9. The majority of known miRNAs regulated multiple target genes (ranging from 1 to 11). Among them, the sit-miR156 family, with 11 unique target genes, had the largest number of target genes; the sit-miR172 and sit-miR393 families had only one target gene, and the others had two to eight targets. Functional analysis of these target genes showed that they were enriched in transcription factors, such as SBP-box transcription factor (sit-miR156), MYB (sit-miR159), ARF (sit-miR160), NAC (sit-miR164), HD-zip transcription factor (sit-miR166), GRAS (sit-miR171), and GRF (sit-miR396). These results were consistent with a previous study in S. italica and other species [8, 35].
Furthermore, we identified a total of 26 target genes for 9 novel miRNAs (Additional file 8, Additional file 10). Unlike the targets of known miRNAs, most targets of novel miRNAs fell into category 2. Of these 26 target genes, 10 were in category 2, 6 were in category 3, four were in category 4, three were in category 0 and 1. Descriptions of the target gene showed that the target genes of novel miRNAs had more diverse functions, including hydroxyproline-rich glycoprotein, dirigent-like protein, ubiquitin conjugating enzyme protein, and some unknown genes.
Pervious researches suggested that most miRNA targets are located in protein coding regions in plants, which is different from animals [64]. Our results also support this point of view. In this study, among 56 target genes for known miRNAs, 51 target sites located in coding region, and only 5 target sites located in UTR region. Similar results were found in targets for novel miRNAs, 20 out of 26 target sites located in coding region, and others located in UTR region (Additional file 8).
At the same time, we predicted targets of known and novel miRNAs using the psRNA Target program with default parameters [65]. The S. italica (foxtail millet) CDS library (provide by the JGI Genomic Project) was selected as the transcript/genomic library for the target search (Additional file 11 and Additional file 12). Compared with the results of degradome sequencing, 28 known miRNAs targets and 6 novel miRNAs targets could be detected using two methods. Additionally, some targets that were not detected by degradome sequencing, such as the targets of miR2118 and miR408, were predicted using psRNATarget.
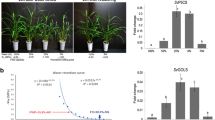
Combined with the results of transcriptome sequencing (data not published), we analyzed the relationship between DE miRNAs and their target’s expression. A total of 15 known miRNA–target pairs showed negative correlations (Fig. 6) and were composed of 10 miRNAs and 14 target genes. Functional annotation showed that some target genes, such as auxin response factor (ARF), NB-ARC domain-containing disease-resistance protein (NB-ARC), leucine-rich repeat receptor-like protein kinase (LRPK), F-box family protein, MYB transcript factor, and laccase, respond to stress. The sulfate transporter showed a strong negative association with miR395 and is known to play an important role in the response to abiotic stress [66]. In addition, Si030478m is homologous to NAD(P)-binding Rossmann-fold superfamily protein and involved in dehydrogenases of metabolic pathways, and Si031053m is homologous to copper ion-binding protein and involved in photorespiration.

A combined heat map of the negative correlation between a miRNA and its target in foxtail millet under drought stress. The red represents upregulated expression, and the green represents downregulated expression
Drought-related miRNAs network
To increase our understanding of the regulatory role of drought-related miRNAs, we constructed a miRNA-mediated interaction network based on targets and protein interaction data from the STRING database (Fig. 7). This network contains 14 DE miRNAs and 129 genes. The yellow diamond represents the miRNAs of foxtail millet that were DE in response to drought, the pink rectangle represents the target identified by degradome sequencing, the predicted target was labelled with a green ellipse, and other proteins are shown as a gray circle. The network revealed that many nodes were connected through protein–protein interaction data from the STRING database and form a complex network. In this network, most of these targets identified by degradome sequencing were transcription factors, including MYB, NAC, SPL, ARF, and GRAS. Targets predicted by in silico analysis included diverse important enzymes, such as NB-ARC, F-box, LAC, and copper ion-binding protein, which are believed to play an important role in the stress response.

microRNA-mediated regulatory networks. Targets of DE miRNAs homologous to Arabidopsis and the constructed network based on the Protein–Protein Interaction data from the STRING database. Pink round rectangle represents the target identified by degradome sequencing, green ellipse represents the predicted target by psRNA Target, and other proteins are shown as a gray circle
Discussion
As an important drought-tolerant crop, foxtail millet provides an ideal system to study drought tolerance. Increasing evidence has indicated that miRNAs play an important role in plant in response to drought. Considering the importance of miRNAs, many miRNAs of foxtail millet have been identified by high-throughput sequencing and bioinformatics approaches [35–37]. However, these studies focused on whole genome scales, which cannot reveal regulatory roles at the transcriptional level. Moreover, compared with identified miRNAs from other species, such as Arabidopsis, maize, and rice, there were fewer miRNAs in foxtail millet. The majority of foxtail millet-specific miRNAs, especially drought-related miRNAs, remain unidentified. In the present study, we constructed two sRNA libraries (control and drought treatment) and identified conserved, novel miRNAs, as well as drought-related miRNAs in foxtail millet.
Drought-responsive miRNA
Comparisons of the expression levels of miRNAs in the control and drought libraries revealed that 18 miRNAs belonging to 16 miRNA families changed significantly. Of these miRNA families, some are thought to be associated with drought in other species, such as miR159, miR167, and miR390. During the response to drought, miR167 was upregulated in Arabidopsis [23] and P. euphratica [50]. In this study, sit-miR167b was significantly upregulated under drought stress, and two target genes (Si021157m and Si000404m) encoding ARF genes were identified based on degradome sequencing. Recently, a study in soybeans showed that miR167 positively regulates nodules and lateral roots by repressing the target genes GmARF8a and GmARF8b (homologous genes of Arabidopsis AtARF8) [67], which indicated that miR167 modulates root adaptation to drought stress. miR390 is another miRNA known to be involved in drought stress. In the present study, miR390 was upregulated, which was consistent with the results in cowpeas [68] and Brachypodium distachyon [69]. It was reported that miR390 targets the TAS genes, which generates ta-siRNAs (trans-acting small interfering RNA) and regulates Auxin Response Factor (ARF) to modulate lateral root emergence and organ polarity establishment. These results indicated that some miRNAs are conserved in response to drought across plants.
However, as reported in previous studies, some drought-related miRNAs show different expression patterns in response to drought stress. For example, miR156 was upregulated in cowpeas and barley in response to drought stress [68, 70], but it was downregulated in rice under conditions of drought. [27] Our results showed that two members of miR156 (sit-miR156a and sit-miR156b) were significantly upregulated, with more than one log2 fold change. Furthermore, several studies have shown that the expression of miR398 was induced by drought stress. It was reported that miR398 was downregulated under drought stress in M. truncatula [32], maize, and peach [71]. However, upregulation was observed in Triticum dicoccoides [62] and M. truncatula [63]. In this study, miR398 showed no significant change in response to drought. This is another example of a discrepancy in the miRNA expression patterns across different studies. Previous studies indicated that some miRNAs respond differently in different tissues under conditions of drought; for example, in barley, miR166 was upregulated in leaves but downregulated in roots, whereas miR156a, miR171, and miR408 were induced in leaves but unchanged in roots [61]. In addition, some studies have suggested that the pattern of miRNA expression differ in different genotypes within the same species; for example, the majority of miRNAs were upregulated during water-deficit stress in the sensitive soybean. However, for the tolerant genotype, the majority of miRNAs were downregulated [72]. The similar results were found in foxtail millet [60], after drought treated with PEG6000, majority of drought-related miRNAs in tolerant cultivar were up-regulated, whereas in sensitive cultivar showed down-regulated. These conflicting results require more detailed research to characterize drought-responsive miRNAs in plants.
In addition to the known miRNAs, we also identified 72 novel miRNAs; 3 of these miRNAs changed significantly after drought stress. Previous reports have suggested that highly conserved miRNAs are widespread with high expression, whereas less conserved miRNAs are often species-specific with weak expression [71]. Our results were also consistent with previous reports. In this study, most predicted novel miRNAs had very low counts compared with known miRNAs, and only eight novel miRNAs had more than 100 TPM. This result may be due to the evolutionary conservation of plant miRNAs, and these conserved miRNAs may be involved in key metabolic processes; thus, their expression may be higher than non-conserved miRNAs [71]. It is possible that these miRNAs play a species-specific role in drought responses in foxtail millet. Although these novel miRNAs have low expression, they may have an effect similar to that of miRNAs with high expression. For all 153 miRNAs we identified, 55 of them were also found in Yadav’s study [60]. As we expected, majority of these common miRNAs (92.7 %) show the similar expression patterns in response to stresses in both studies (Additional file 13).
Target genes of foxtail millet miRNAs
In foxtail millet, the majority of miRNA targets were predicted using bioinformatics, and very few miRNA targets were identified experimentally. Degradome sequencing technology provides a powerful tool with which to study miRNA–target interactions at the transcriptome level. In this study, we identified 56 targets for 12 known miRNA families using degradome sequencing. Based on our analysis, the majority of these targets have a conserved function with miRNA targets in other species. Most of the identified targets of the known miRNAs belong to transcription factors, such as miR156 targeting the squamosal promoter-binding family (SPB), miR159 targeting MYB, miR160 targeting several auxin response factors (ARF), and miR164 targeting no apical meristem protein (NAC). It has been indicated that conserved miRNAs play a crucial role in post-transcriptional regulation in plant species [64]. These results suggest that degradome sequencing can be successfully applied to identify miRNA targets in foxtail millet with high accuracy and efficiency.
Gene function annotation of conserved miRNAs targets showed that most of them were classified into transcription factors. For example, in Arabidopsis and Populus, 95 conserved miRNAs targets were identified, 68 % of those encode transcription factor. In soybean, 82 % of miRNA targets were transcription factors [73]. Same results were also reported in rice [74], maize [75], and grapevine [76]. There are also a few miRNAs target to genes encoding enzymes of basic biochemical pathways. For example, miR397 targets laccase and miR398 targets copper superoxide dismutases. However, those were only constitute a minor portion of all identified target genes in plants [64].
In the present study, 26 targets for 9 novel miRNAs were identified using degradome sequencing. Compared with the targets of known miRNAs, the targets of novel miRNA had a wide variety of functions, including those involving glycoprotein, dehydrogenase, oxidoreductase, transcription factor, and unknown proteins. Another difference between the targets of conserved and novel miRNAs is that the majority of novel miRNA targets belonged to categories 2 and 3, and similar results were also found in Brassica juncea [77] and grapevine [76], which suggest these novel miRNAs are young and not fully stabilized evolutionarily.
Three potential novel miRNAs were considered drought-response miRNAs based on the DE between the CL and DT libraries. Only target of sit-novel-miR10 was identified based on degradome sequencing, and others were not identified based on degradome sequencing, possibly because these miRNAs regulate target genes by repressing translation. Further studies are required to increase our understanding of the regulatory mechanism of these miRNAs.
miRNA role in the drought-stress responses of foxtail millet
Our study identified 16 known miRNA families and 3 novel miRNAs that were DE in foxtail millet under drought conditions (Additional file 6 and Additional file 7). Among known miRNAs, 34 unique targets of 6 DE miRNA families were validated using degradome sequencing (Table 4 and Fig. 7). As expected, the majority of these DE miRNAs were related to drought-stress responses in previous studies [78, 79]. For example, our study found that miR167b-5p was enriched and significantly upregulated in response to drought stress in foxtail millet. Similar results were shown in Arabidopsis [23], rice, and maize [80, 81], which indicates that miR167 can respond to ABA and control stomatal movement. miR390 was reported to be upregulated under drought stress in Brachypodium distachyon and Vigna unguiculata [69]. In Arabidopsis, miR390 mediated the miR390–tasiRNA–ARF regulatory pathway and regulated lateral root growth. In foxtail millet, a similar expression pattern and the same target gene of miR390 was identified via SL-qPCR and degradome sequencing, respectively. These results suggest that numerous miRNAs have conserved functions in regulating abiotic stress responses in various plant species. Combined with the miRNA expression patterns, target prediction, and degradome sequencing results (Figs. 6 and 7), our study increases our understanding about how foxtail millet An04 responded to water deficiency:
-
i.
Growth repression under drought conditions. Morphology and physiology experiments have shown that foxtail millet An04 has the lowest germination rates and significant growth repression when lacking a water supply. Several DE miRNAs and their targets involved in cell growth and development regulation were identified in drought-treated plants. miR2118 were induced up to ~ two-fold in An04 under water-deficient conditions, whereas its predicted target genes, Si001402m, were repressed markedly. Si001402m encoded a galacturonosyl transferase, and the homologous gene in Arabidopsis plays an important role in glucuronoxylan accumulation and secondary cell wall development [82]. Another putative mechanism crucial for cell growth remodeling in foxtail millet in response to drought is the miRNA397–laccase regulatory pathway. We found that miRNA397 and its target laccase 7 show an inverse correlation in gene expression, indicative of classical miRNA-mediated mRNA degradation. It has been shown that modulation of laccase by miR397 can affect plant biomass production in both Arabidopsis and Populus [83, 84].
-
ii.
Drought-responsive transcription factors regulated by miRNAs. In our study, several miRNA families were shown to regulate transcription factors and control the plant response to drought stress. The NAC transcription factor (TFs) family is one of the TFs that regulate drought tolerance in plants. In Arabidopsis, miR164 mediates the cleavage of NAC1, which further downregulates auxin signals and reduces lateral root growth [19]. In rice, the miR164-targeted NAC genes were shown to be negative regulators of drought tolerance [85]. Our study showed that miR164a was induced by water treatment; six target genes (Si006975m, Si017567m, Si010553m, Si017570m, Si022747m, and Si017931m), all of which belong to the NAC transcription factor family, were identified by degradome sequencing. It is possible that the enhanced expression of miR164a represses NAC gene expression and regulates drought responses in foxtail millet. Another example of the miRNA–TFs co-regulatory pathway found in our study is miR156-targeted squamosal-promoter binding protein-like (SPL) networks. Stief et al. (2014) demonstrated that miR156 can target SPL transcription factor genes and regulate tolerance to recurring environmental stress [86]. Our results showed that miR156 was induced by drought, and SPL4, 9, 10, and 11 were identified as its targets, indicating that miR156-SPLs play a role in response to drought in foxtail millets.
-
iii.
miRNAs mediate Abscisic acid (ABA) signaling. ABA is a phytohormone critical for drought-stress signaling in plants. miRNA159 is known to be a negative regulator of ABA responses [87]. In Arabidopsis, ABA increases miR159 accumulation through an ABI3-dependent pathway and suppresses MYB33 and MYB101 expression, which eventually induced plants stress responses. The same genes and their expression patterns were identified in foxtail millet, suggesting that the mechanism by which miR159 regulated ABA signaling through MYB33 and MYB101 transcript was conserved in foxtail millet An04. miR394 is involved in ABA-dependent Arabidopsis salt and drought stress responses [88], and upregulation of miR394 in drought-treated An04 plants may initiate ABA- and stress-responsive gene expression to allow for plant adaption to water-deficient conditions.
-
iv.
miRNAs involved in cellar homeostasis. It is important for plants to sustain cellar homeostasis under environmental stress. Two DE miRNAs (miR528 and miR395) were shown to be involved in cellar homeostasis maintenance. In our study, miR528 was significantly downregulated by drought, and the same result was reported in maize. The target gene of miR528 encoded a peroxidase (POD), which is an important component of the antioxidative enzyme system in plants. The downregulation of miR528 may promote the removal of excessive reactive oxygen species (ROS) and help maintain cellar homeostasis in foxtail millet under drought conditions. miR395 was enriched in the drought-treated group, and its putative target was predicted to be a sulfate transporter, SULTR2;1. Regulation of the sulfate assimilation process through the miR395–SULTR2;1 interaction had been confirmed in Arabidopsis [89]. Moreover, sulfate transporters play a role in re-equilibrating the flux of sulfate between and within different tissues and improving drought tolerance in plants [66].
Conclusions
In the present study, we detected 18 miRNA members in 16 families and predicted 3 novel miRNAs in response to drought in foxtail millet. Furthermore, 56 targets for 12 known miRNA families and 26 target genes for 9 novel miRNAs were identified by degradome sequencing at the global level. These results provide useful information for the study of drought-responsive miRNAs in foxtail millet. Further studies are required to identify these miRNAs and their targets using techniques such as RLM-RACE. On the other hand, it is important to determine whether these genes enhance drought tolerance in transgenic plants.
Ethics (and consent to participate)
Not applicable.
Consent to publish
Not applicable.
Availability of data and materials
Raw data supporting our findings can be found at EMBL-EBI European Nucleotide Archive (https://www.ebi.ac.uk/) under accession numbers ERP014347 and ERP014368.
Abbreviations
- miRNA:
-
microRNA
- sRNA:
-
small RNA
- siRNA:
-
small interfering RNA
- ta-siRNAs:
-
trans-acting small interfering RNA
- WUE:
-
water use efficiency
- pre-miRNA:
-
precursors of microRNA
- LWP:
-
leaf water potential
- TPM:
-
Transcript per million
- DT:
-
drought-treated group
- CL:
-
control group
- MFEs:
-
minimum free energies
- DE:
-
differentially expressed
References
Zhang G, Liu X, Quan Z, Cheng S, Xu X, Pan S, et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat Biotechnol. 2012;30(6):549–54.
Shantz HL, Piemeisel LN. The water requirement of plants at Akron Colorado. J Agri Res. 1927;34:1093–89.
He Z, Bonjean A P A. Cereals in China. CIMMYT; 2010.
Li C, Yue J, Wu X, Xu C, Yu J. An ABA-responsive DRE-binding protein gene from Setaria italica, SiARDP, the target gene of SiAREB, plays a critical role under drought stress. J Exp Bot. 2014;65(18):5415–27.
Peng Y, Zhang J, Cao G, Xie Y, Liu X, Lu M, et al. Overexpression of a PLDalpha1 gene from Setaria italica enhances the sensitivity of Arabidopsis to abscisic acid and improves its drought tolerance. Plant Cell Rep. 2010;29(7):793–802.
Lata C, Sahu PP, Prasad M. Comparative transcriptome analysis of differentially expressed genes in foxtail millet (Setaria italica L.) during dehydration stress. Biochem Biophys Res Commun. 2010;393(4):720–7.
Muthamilarasan M, Bonthala VS, Mishra AK, Khandelwal R, Khan Y, Roy R, et al. C2H2 type of zinc finger transcription factors in foxtail millet define response to abiotic stresses. Funct Integr Genomics. 2014;14(3):531–43.
Ding Y, Tao Y, Zhu C. Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot. 2013;64(11):3077–86.
Covarrubias AARJ. Post-transcriptional gene regulation of salinity and drought responses by plant microRNAs. Plant, Cell Environ. 2010;33(4):481–9.
Shuai P, Liang D, Zhang Z, Yin W, Xia X. Identification of drought-responsive and novel Populus trichocarpa microRNAs by high-throughput sequencing and their targets using degradome analysis. BMC Genomics. 2013;14(1):1–14.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281.
Zhang B, Pan X, Cobb GP, Anderson TA. Plant microRNA: a small regulatory molecule with big impact. Dev Biol. 2006;289(1):3–16.
Lee Y, Kim M, Han J, Yeom K, Lee S, Baek SH, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23(20):4051–60.
Bollman KM, Aukerman MJ, Park MY, Hunter C, Berardini TZ, Poethig RS. HASTY, the Arabidopsis ortholog of exportin 5/MSN5, regulates phase change and morphogenesis. Development. 2003;130(8):1493–504.
Park MY, Wu G, Gonzalez-Sulser A, Vaucheret H, Poethig RS. Nuclear processing and export of microRNAs in Arabidopsis. Proc Natl Acad Sci U S A. 2005;102(10):3691–6.
Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136(4):669–87.
Kim J, Jung JH, Reyes JL, Kim YS, Kim SY, Chung KS, et al. microRNA-directed cleavage of ATHB15 mRNA regulates vascular development in Arabidopsis inflorescence stems. Plant J. 2005;42(1):84–94.
Aukerman MJ, Sakai H. Regulation of flowering time and floral organ identity by a MicroRNA and its APETALA2-like target genes. Plant Cell. 2003;15(11):2730–41.
Guo HS, Xie Q, Fei JF, Chua NH. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell. 2005;17(5):1376–86.
Lauter N, Kampani A, Carlson S, Goebel M, Moose SP. microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc Natl Acad Sci U S A. 2005;102(26):9412–7.
Jeong D, Green PJ. The role of rice microRNAs in abiotic stress responses. Journal of Plant Biology. 2013;56(4):187–97.
Chen H, Li Z, Xiong L. A plant microRNA regulates the adaptation of roots to drought stress. FEBS Lett. 2012;586(12):1742–7.
Liu H, Tian X, Li Y, Wu C, Zheng C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA (New York, NY). 2008;14(5):836–43.
Sunkar R. Novel and Stress-Regulated MicroRNAs and Other Small RNAs from Arabidopsis. Plant Cell. 2004;16(8):2001–19.
Jian X, Zhang L, Li G, Zhang L, Wang X, Cao X, et al. Identification of novel stress-regulated microRNAs from Oryza sativa L. Genomics. 2010;95(1):47–55.
Zhao B, Liang R, Ge L, Li W, Xiao H, Lin H, et al. Identification of drought-induced microRNAs in rice. Biochem Biophys Res Commun. 2007;354(2):585–90.
Zhou L, Liu Y, Liu Z, Kong D, Duan M, Luo L. Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J Exp Bot. 2010;61(15):4157–68.
Ding Y, Chen Z, Zhu C. Microarray-based analysis of cadmium-responsive microRNAs in rice (Oryza sativa). J Exp Bot. 2011;62(10):3563–73.
Kong Y. Differential Expression of microRNAs in Maize Inbred and Hybrid Lines during Salt and Drought Stress. Am J Plant Sci. 2010;1(2):69–76.
Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot. 2009;103(1):29–38.
Lu S, Sun YH, Chiang VL. Stress-responsive microRNAs in Populus. Plant J. 2008;55(1):131–51.
Wang T, Chen L, Zhao M, Tian Q, Zhang WH. Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genomics. 2011;12(1):1–11.
Li WX, Oono Y, Zhu J, He XJ, Wu JM, Iida K, et al. The Arabidopsis NFYA5 Transcription Factor Is Regulated Transcriptionally and Posttranscriptionally to Promote Drought Resistance. Plant Cell. 2008;20(8):2238–51.
Zhou M, Li D, Li Z, Hu Q, Yang C, Zhu L, et al. Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol. 2013;161(3):1375–91.
Yi F, Xie S, Liu Y, Qi X, Yu J. Genome-wide characterization of microRNA in foxtail millet (Setaria italica). BMC Plant Biol. 2013;13(1):1–15.
Khan Y, Yadav A, Bonthala VS, Muthamilarasan M, Yadav CB, Prasad M. Comprehensive genome-wide identification and expression profiling of foxtail millet [Setaria italica (L.)] miRNAs in response to abiotic stress and development of miRNA database. Plant Cell, Tissue and Organ Culture (PCTOC). 2014;118(2):279–92.
Han J, Xie H, Sun Q, Wang J, Lu M, Wang W, et al. Bioinformatic identification and experimental validation of miRNAs from foxtail millet (Setaria italica). Gene. 2014;546(2):367–77.
Degenkolbe T, Do PT, Zuther E, Repsilber D, Walther D, Hincha DK, et al. Expression profiling of rice cultivars differing in their tolerance to long-term drought stress. Plant Mol Biol. 2009;69(1-2):133–53.
Qie L, Jia G, Zhang W, Schnable J, Shang Z, Li W, et al. Mapping of quantitative trait locus (QTLs) that contribute to germination and early seedling drought tolerance in the interspecific cross Setaria italica x Setaria viridis. PLoS One. 2014;9(7):e101868.
German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008;26(8):941–6.
Fang X, Zhao Y, Ma Q, Huang Y, Wang P. Identification and Comparative Analysis of Cadmium Tolerance-Associated miRNAs and Their Targets in Two Soybean Genotypes. PLoS One. 2013;8(12):e81471.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, et al. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2012;gks1005.
Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42(D1):D68–73.
Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14(6):787–99.
Stocks MB, Moxon S, Mapleson D, Woolfenden HC, Mohorianu I, Folkes L, et al. The UEA sRNA workbench: a suite of tools for analysing and visualizing next generation sequencing microRNA and small RNA datasets. Bioinformatics. 2012;28(15):2059–61.
Wu HJ, Ma YK, Chen T, Wang M, Wang XJ. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Res. 2012;gks554.
Hofacker IL. Vienna RNA, secondary structure server. Nucleic Acids Res. 2003;31(13):3429–31.
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, et al. Criteria for annotation of plant MicroRNAs. Plant Cell. 2008;20(12):3186–90.
Li B, Qin Y, Duan H, Yin W, Xia X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J Exp Bot. 2011;62(11):3765–79.
Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol. 2008;8(1):1–17.
Folkes L, Moxon S, Woolfenden HC, Stocks MB, Szittya G, Dalmay T, et al. PAREsnip: a tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res. 2012;40(13):e103.
Romualdi C, Bortoluzzi S, D’Alessi F, Danieli GA. IDEG6: a web tool for detection of differentially expressed genes in multiple tag sampling experiments. Physiol Genomics. 2003;12(2):159–62.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2014;gku1003.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Kang M, Zhao Q, Zhu D, Yu J. Characterization of microRNAs expression during maize seed development. BMC Genomics. 2012;13(1):1–11.
Wang X, Elling AA, Li X, Li N, Peng Z, He G, et al. Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell. 2009;21(4):1053–69.
Yin F, Qin C, Gao J, Liu M, Luo X, Zhang W, et al. Genome-wide identification and analysis of drought-responsive genes and microRNAs in tobacco. Int J Mol Sci. 2015;16(3):5714–40.
Bennetzen JL, Schmutz J, Wang H, Percifield R, Hawkins J, Pontaroli AC, et al. Reference genome sequence of the model plant Setaria. Nat Biotechnol. 2012;30(6):555–61.
Yadav A, Khan Y, Prasad M. Dehydration-responsive miRNAs in foxtail millet: genome-wide identification, characterization and expression profiling. Planta. 2016;243(3):749–66.
Kantar M, Unver T, Budak H. Regulation of barley miRNAs upon dehydration stress correlated with target gene expression. Funct Integr Genomics. 2010;10(4):493–507.
Kantar M, Lucas SJ, Budak H. miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta. 2011;233(3):471–84.
Trindade I, Capitao C, Dalmay T, Fevereiro MP, Santos DM. miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta. 2010;231(3):705–16.
Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57:19–53.
Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39 suppl 2:W155–9.
Gallardo K, Courty PE, Le Signor C, Wipf D, Vernoud V. Sulfate transporters in the plant’s response to drought and salinity: regulation and possible functions. Front Plant Sci. 2014;5:580.
Wang Y, Li K, Chen L, Zou Y, Liu H, Tian Y, et al. microRNA167-directed regulation of the auxin response factors, GmARF8a and GmARF8b, is required for soybean (Glycine max L.) nodulation and lateral root development. Plant Physiol. 2015;168(3):984–99.
Barrera-Figueroa BE, Gao L, Diop NN, Wu Z, Ehlers JD, Roberts PA, et al. Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC Plant Biol. 2011;11(1):1–11.
Budak H, Akpinar A. Dehydration stress-responsive miRNA in Brachypodium distachyon: evident by genome-wide screening of microRNAs expression. OMICS. 2011;15(11):791–9.
Hackenberg M, Gustafson P, Langridge P, Shi BJ. Differential expression of microRNAs and other small RNAs in barley between water and drought conditions. Plant Biotechnol J. 2015;13(1):2–13.
Eldem V, Celikkol AU, Ozhuner E, Bakir Y, Uranbey S, Unver T. Genome-wide identification of miRNAs responsive to drought in peach (Prunus persica) by high-throughput deep sequencing. PLoS One. 2012;7(12):e50298.
Kulcheski FR, de Oliveira LF, Molina LG, Almerao MP, Rodrigues FA, Marcolino J, et al. Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genomics. 2011;12(1):1–17.
Song QX, Liu YF, Hu XY, Zhang WK, Ma B, Chen SY, et al. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011;11(1):1–16.
Li YF, Zheng Y, Addo-Quaye C, Zhang L, Saini A, Jagadeeswaran G, et al. Transcriptome-wide identification of microRNA targets in rice. Plant J. 2010;62(5):742–59.
Zhang B, Pan X, Anderson TA. Identification of 188 conserved maize microRNAs and their targets. FEBS Lett. 2006;580(15):3753–62.
Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, et al. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62(6):960–76.
Yang J, Liu X, Xu B, Zhao N, Yang X, Zhang M. Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genomics. 2013;14(1):1–16.
Xie F, Stewart CN, Taki FA, et al. High-throughput deep sequencing shows that microRNAs play important roles in switchgrass responses to drought and salinity stress. Plant Biotechnol J. 2014;12(3):354–66.
Ferdous J, Hussain SS, Shi BJ. Role of microRNAs in plant drought tolerance. Plant Biotechnol J. 2015;13(3):293–305.
Liu Q, Zhang YC, Wang CY, Luo YC, Huang QJ, Chen SY, et al. Expression analysis of phytohormone-regulated microRNAs in rice, implying their regulation roles in plant hormone signaling. FEBS Lett. 2009;583(4):723–8.
Wei L, Zhang D, Xiang F, Zhang Z. Differentially expressed miRNAs potentially involved in the regulation of defense mechanism to drought stress in maize seedlings. Int J Plant Sci. 2009;170(8):979–89.
Persson S, Caffall KH, Freshour G, Hilley MT, Bauer S, Poindexter P, et al. The Arabidopsis irregular xylem8 mutant is deficient in glucuronoxylan and homogalacturonan, which are essential for secondary cell wall integrity. Plant Cell. 2007;19(1):237–55.
Wang CY, Zhang S, Yu Y, Luo YC, Liu Q, Ju C, et al. MiR397b regulates both lignin content and seed number in Arabidopsis via modulating a laccase involved in lignin biosynthesis. Plant Biotechnol J. 2014;12(8):1132–42.
Lu S, Li Q, Wei H, Chang MJ, Tunlaya-Anukit S, Kim H, et al. Ptr-miR397a is a negative regulator of laccase genes affecting lignin content in Populus trichocarpa. Proc Natl Acad Sci U S A. 2013;110(26):10848–53.
Fang Y, Xie K, Xiong L. Conserved miR164-targeted NAC genes negatively regulate drought resistance in rice. J Exp Bot. 2014;65(8):2119–35.
Stief A, Altmann S, Hoffmann K, Pant BD, Scheible WR, Baurle I. Arabidopsis miR156 Regulates Tolerance to Recurring Environmental Stress through SPL Transcription Factors. Plant Cell. 2014;26(4):1792–807.
Reyes JL, Chua NH. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007;49(4):592–606.
Song JB, Gao S, Sun D, Li H, Shu XX, Yang ZM. miR394 and LCR are involved in Arabidopsis salt and drought stress responses in an abscisic acid-dependent manner. BMC Plant Biol. 2013;13(1):1–16.
Kawashima CG, Matthewman CA, Huang S, Lee BR, Yoshimoto N, Koprivova A, et al. Interplay of SLIM1 and miR395 in the regulation of sulfate assimilation in Arabidopsis. Plant J. 2011;66(5):863–76.
Acknowledgements
This work was supported by Beijing Natural Science Foundation (6142019), and funding from the Chinese Academy of Agricultural Science (2014ZL002) and The Agricultural Science and Technology Innovation Program of CAAS, National High Technology Research and Development Program of China (863 Program) (2013AA102603), National Natural Science Foundation of China (31301328, 31171560), Fundamental Research Funds of ICS-CAAS (Grant to Guanqing Jia, 2013007), and China Agricultural Research System (CARS07-13.5-A02).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XD, GJ, HZ conceived the experiment. YW, LL, ST designed the experiment. YW, LL conducted the experiments, YW, ST conducted data analysis, drafted and revised the manuscript. JL and HZ contributed to the experiments and data analysis. All authors have read and approved the final manuscript.
Additional files
Additional file 1:
The survival rate of 10 varieties of foxtail millet under repeated drought treatments. (TIF 166 kb)
Additional file 2:
Primers used for RT-PCR in this study. (DOC 35 kb)
Additional file 3:
Detailed information of known miRNAs in foxtail millet. (XLS 72 kb)
Additional file 4:
Detailed information of novel miRNAs in foxtail millet. (XLS 58 kb)
Additional file 5:
The secondary structures of novel miRNA precursors (PDF 324 kb)
Additional file 6:
Differential expression analysis of known miRNAs under drought stress. (XLS 21 kb)
Additional file 7:
Differential expression analysis of novel miRNAs under drought stress. (XLS 38 kb)
Additional file 8:
Targets of miRNAs identified by degradome sequence. (XLS 46 kb)
Additional file 9:
Target plots of identified miRNA targets using degradome sequencing. (PDF 762 kb)
Additional file 10:
Targets of novel miRNAs identified by degradome sequencing. (DOC 57 kb)
Additional file 11:
Targets of known miRNAs predicted by the web tool psRNATarget. (XLS 69 kb)
Additional file 12:
Targets of novel miRNAs predicted by the web tool psRNATarget. (XLS 187 kb)
Additional file 13:
miRNA comparison data among different foxtail millet varieties. (XLSX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, Y., Li, L., Tang, S. et al. Combined small RNA and degradome sequencing to identify miRNAs and their targets in response to drought in foxtail millet. BMC Genet 17, 57 (2016). https://doi.org/10.1186/s12863-016-0364-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-016-0364-7