
Abstract
Background
The methyl-CpG Binding Protein two gene (MECP2) encodes a multifunctional protein comprising two isoforms involved in nuclear organization and regulation of splicing and mRNA template activity. This gene is normally expressed in all tissues, with a higher expression level in the brain during neuronal maturation. Loss of MECP2 function is the primary cause of Rett syndrome (RTT) in humans, a dominant, X-linked disorder dramatically affecting neural and motor development.
Results
We investigated the molecular evolution of MECP2 in several primate taxa including 36 species in 16 genera of neotropical (platyrrhine) primates. The coding region of the MECP2_e2 isoform showed a high level of evolutionary conservation among humans and other primates, with amino acid substitutions in 14 codons and one in-frame insertion of a single serine codon, between codons 357 and 358, in Ateles paniscus. Most substitutions occurred in noncritical regions of MECP2 and the majority of the algorithms used for analyzing selection did not provide evidence of positive selection. Conversely, we found 48 sites under negative selection in different regions, 23 of which were consistently found by three different algorithms. Similar to an inverted Alu insert found previously in a lesser ape at a parallel location, one Alu insertion of approximately 300 bp in Cebus and Sapajus was found in intron 3. Phylogenetic reconstruction of the intron 3 data provided a topology that was coincident with the consensus arrangement of the primate taxa. RNAseq data in the neotropical primate Callimico goeldii revealed a novel transcript consisting of a noncontinuous region of the human-homologous intron 2 in this species; this transcript accounted for two putative polypeptides.
Conclusions
Despite the remarkable evolutionary conservation of MECP2, one in-frame codon insertion was observed in A. paniscus, and one region of intron 3 was affected by a trans-specific Alu retrotransposition in two neotropical primate genera. Moreover, identification of novel MECP2 transcripts in Callimico suggests that part of a homologous human intronic region might be expressed, and that the potential open reading frame in this region might be a subject of interest in RTT patients who carry an apparently normal MECP2 sequence.
Similar content being viewed by others
Background
A serious clinical entity with arrested development between 6 and 18 months of age, regression of acquired skills, loss of speech, stereotypical movements (classically of hands), microcephaly, seizures and mental retardation, as initially described by Rett [1], is presently known as Rett syndrome (RTT). Molecular studies later showed that association between the X-linked, methyl-CpG Binding Protein 2 gene (MECP2) involved in neural development and RTT accounts for the first known association between an epigenetic regulator and a human disease [2–4]. Loss of MECP2 function, resulting from mutations in 85 to 90 % of individuals affected by RTT [2], is the primary cause of this syndrome, a dominant disorder occurring almost exclusively in females [2, 3].
MECP2, located on Xq28, is closely associated with the epigenetic mechanisms of DNA methylation and gene inactivation. MECP2, encoded by MECP2, is a chromatin-associated nuclear protein member of the methyl binding domain (MBD) family capable of binding a single methyl-CpG [5]. MECP2 is a multifunctional protein involved in nuclear organization, chromatin architecture, chromatin template binding, chromatin compaction and fiber binding, heterochromatin rearrangement [6], loop domain chromatin organization, pericentromeric heterochromatin structure, and DNA methylation [7]. MECP2 also plays roles in regulating splicing and mRNA template activity [8] and in activating and repressing transcription [6, 9], and is normally expressed in all tissues, but has a higher expression level in the brain during neuronal maturation [10]. However, in mice, MECP2 has not been detected in microglial cells from the retina, cortex, cerebellum and spinal cord, in intestinal epithelial cells, or in erythropoietic lineage cells, hair matrix keratinocytes, mature oocytes and spermatozoids [11].
In addition to its C-terminal and N-terminal regions, MECP2 consists of two functional domains, the methylated DNA binding domain (MBD) and the transcriptional binding domain (TRD), along with other relevant structures. MBD, the first of these domains, is required for recognition of and binding to methylated dinucleotides in the CpG islands of promoter regions; binding requires the presence of an A/T-rich sequence adjacent to the methylated CpG regions [12, 13]. TRD, the second domain, is specifically required for transcriptional silencing via recruitment of the chromatin remodeling co-repressor Sin3A and histone deacetylases [14]. Three other domains have also been identified: (i) a WW domain binding region involved in interactions with the WW regions of splicing factors [15]; (ii) an arginine-glycine repetitive region known to mediate RNA-protein interactions [16]; and (iii) two nuclear localization signals, one of which is inside the TRD.
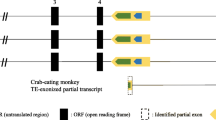
Through alternative splicing (Fig. 1), two mRNAs of different sizes are normally transcribed resulting in the following two isoforms: MECP2_e1, with a start codon in exon 1 and containing exon 1, 3 and 4 transcripts, and MECP2_e2, with a start codon in exon 2 and containing exon 1, 2, 3 and 4 transcripts. MECP2_e1 encodes a protein of 498 amino acids and MECP2_e2 a protein of 486 amino acids [17, 18]. MECP2_e1 is the major isoform found in the brain and throughout development [4, 19, 20]. MECP2_e1 has more relevance to the RTT phenotype [21], a finding also supported by studies on MeCP2-e1 deficient mice that developed forelimb stereotypy, hindlimb clasping, excessive grooming and hypo-activity at 7 to 31 weeks prior to death [22]. Conversely, selective deletion of MeCP2_e2 did not result in RTT-associated neurological phenotypes, but resulted in a survival disadvantage for embryos carrying a MeCP2_e2 null allele of maternal origin. A specific requirement for MeCP2_e2 function was found in extraembryonic tissue, where selective loss of MeCP2_e2 resulted in placental defects [23].

MECP2 structure showing exons (E) and introns (In). The figure shows transcripts resulting from alternative splicing with 1,734 and 10,241 nucleotides (nt). Transcripts from different regions of exon 4 are indicated as 4a and 4b. Translation of mature mRNA molecules results in proteins of 498 and 486 amino acids (aa), MECP2_e1 and MECP2_e2, respectively
Pathogenic and silent mutations, polymorphisms, and intronic variants have been identified in RTT patients [24, 25]. The most common mutation hotspots are reported to occur in the MBD and TRD domains and affect both MECP2 isoforms [18]. Additionally, the “RettBASE: IRSF MECP2 Variation Database” (available at http://mecp2.chw.edu.au), records 862 different MECP2 mutations. As seen in human males, lack of a functional MECP2 protein during embryonic development is fatal in early postnatal life [26]. In vivo experiments with transgenic mice carrying the functional gene and a mutant allele with inducible expression showed that several characteristics of the RTT phenotype were retrievable in adult life after inducing expression of the mutant allele [27]. Nevertheless, MECP2 duplication (MECP2 duplication syndrome) also affects neural and motor development in humans, with similar characteristics to RTT displayed in male patients, while women with MECP2 duplications exhibit normal cognitive abilities and the propensity for neuropsychiatric abnormalities (depression, anxiety, compulsions, and autism) [28].
Here, we investigated the MECP2 gene from humans and other primates to determine the evolutionary divergence of its functional regions and to identify the nucleotide (nt) sites that might be under selective pressure. The molecular analyses revealed that MECP2 is evolutionary conserved among the primates studied herein. We also report the presence of two independent Alu retrotranspositions in intron 3, and a new alternative exon that includes part of intron 2 in one neotropical (platyrrine) primate species.
Methods
DNA isolation, polymerase chain reaction (PCR) amplification and DNA sequencing
Blood and tissue samples were collected from 61 neotropical primates belonging to 16 genera and from Pan troglodytes (Additional file 1). The blood samples collected were obtained as part of a regular health checkup and disease control scheme for captive animals. Tissue samples were collected in the field or from dead animals (donated by Rio de Janeiro Zoo and São Paulo Zoo). All procedures followed the National Guidelines and Provisions of ICMBio (Instituto Chico Mendes de Biodiversidade Brazil; permanent license number 11375-1).
DNA was isolated from all the samples with the exception of Callimico goeldii. DNA quality was assessed by electrophoresis on 0.8 % agarose gels and quantification in a NanoDrop® 1.000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). Exons 2, 3 and 4 were amplified as reported previously [24]. Amplification of these regions allowed us to analyze in detail the MBD and TRD domains where most of the common mutation hotspots have been reported in humans [18]. To amplify a specific region of exon 4 a primer pair was designed (F = 5′-AAG GAG TCT TCT ATC CGA TCT GT-3′ and R = 5′-TGT CCA CAG GCT CCT CTC TG-3′) using PRIMER3 and In silico PCR (http://frodo.wi.mit.edu/primer3 and http://genome.ucsc.edu/cgi-bin/hgPcr, respectively). To analyze the noncoding regions we designed a primer pair to amplify intron 3 based on the genomic alignments from catarrhines and platyrrhines available in GenBank (Additional file 1): F = 5′-CAC GGA AGC TTA AGC AAA GG-3′ and R = 5′-CTG GGG ACT GTG AGG ACA AA-3′. PCR conditions were as follows: initial denaturation at 94 °C (2 min 30 s) followed by 35 cycles at 94 °C (30 s), 57 °C (45 s), and 72 °C (45 s) with a final elongation step at 72 °C (3 min). PCR products were purified using GFX™ PCR DNA and a Gel Band Purification kit® (Amersham Biosciences). Sequencing reactions were conducted with the same PCR primers and BigDye Terminator v3.1 Cycle Sequencing Kit® (Applied Biosystems) according to the manufacturer’s instructions. Reactions were run on an ABI3130/XL sequencing platform. Sequence data have been deposited in GenBank (see Additional file 1).
RNA isolation and RNAseq
RNA was isolated from the mononuclear blood cells of Brachyteles arachnoides CPRJ2160, C. goeldii CPRJ2033 and Sapajus robustus CPRJ2456 using RNeasy® (Qiagen) and then quantified with a NanoDrop spectrophotometer (Thermo Scientific). RNA quality and integrity were checked by electrophoresis on 1 % agarose gels. Libraries were prepared with a TruSeq RNA Sample Prep Kit v2® (Illumina) following the protocol recommended by the manufacturer. Such libraries were subsequently tested by quantitative (q)PCR using the KK4824 Quantification Kit (KAPA) for validation. Clusters were generated for at least one lane of a PE Flowcell v.3 following the manufacturer’s protocol, and the 2 × 100 bp reads were analyzed with an Illumina HiSeq2500 platform. Cycles registered at least 80 % base calls with a Q30 quality score.
Data were converted to Fastq files by CASAVA v1.8.2 (Illumina), and the contigs were assembled using Trinity software [29] with default settings. Contigs over 200 bp were subjected to Basic Local Alignment Search Tool analysis against human MECP2_e2 (NM_004992.3) using CLC Genomics Workbench 7 software (Qiagen). The reads were mapped using the contigs as the reference with Bowtie 2 [30], at an average coverage of 63X. Open reading frames were manually annotated with MEGA 5.1 [31]. Data have been deposited in GenBank (see Availability of Supporting Data).
cDNA synthesis and reverse transcriptase (RT)-PCR
RNA from B. arachnoides CPRJ2160, C. goeldii CPRJ2033 and S. robustus CPRJ2456 was treated to remove DNA with an RQ1 RNAse-Free DNAse Kit® (Promega) and cDNA was subsequently synthesized with Superscript™ II Reverse Transcriptase® (Life Technologies). The constitutively expressed beta-2-microglobulin (B2M) gene was RT-PCR amplified to check the quality of the cDNA using Platinum Taq DNA polymerase buffer 1X (Invitrogen), 1.5 mM MgCl2, 0,25 mM of each dNTP, 1 U of Platinum Taq DNA polymerase (Invitrogen), 1 pmol of B2M-F 5′-ATG AGT ATG CCT GCC GTG TGA-3′ and 1 pmol of B2M-R 5′-CGG CAT CTT CAA ACC TCC ATG-3′ using the following conditions: 94 °C (2 min) followed by 35 cycles at 94 °C (30 s), 55 °C (30 s) and 72 °C (1 min).
A novel C. goeldii CPRJ2033 splicing product identified by RNAseq was validated and homologous contigs in B. arachnoides and S. robustus were searched using RT-PCR with the primers designed using PRIMER3 and In Silico PCR based on the Callimico data (F = 5′-TCG GAG AGA GGG CTG TGG-3′ and R2 = 5′-AAT TTT GTT GAG GAA AGA AGG CA-3′). PCR conditions were as follows: an initial denaturation at 94 °C (5 min) followed by 35 cycles at 94 °C (15 s), 65 °C (15 s), 72 °C (15 s) and a final elongation at 72 °C (15 min). PCR products were sliced from low melting-point agarose gels and sequenced.
Molecular analyses
Three datasets were constructed, one from the MECP2 exons coding for MECP2_e2, another from the exon 1 data, and a third one from the intron 3 sequences. Only one sequence was used when shared sequences were identified between individuals. When the sequences differed in length because of missing data, the larger sequence was used for analysis.
The MECP2_e2 dataset contained 30 different sequences from 45 specimens (see Additional file 1). The MECP2_e2 dataset included the RNAseq data for B. arachnoides CPRJ2160, C. goeldii CPRJ2033, and S. robustus CPRJ2456. The following additional data from GenBank were included: Homo sapiens (NG_007107), Callithrix jacchus (NC_013918), Gorilla gorilla gorilla (NC_018447), Macaca fascicularis (NC_022292), Macaca mulatta (NW_001218202), Nomascus leucogenys (NC_019841), Pan paniscus (NW_003869946), P. troglodytes (NC_006491), Papio anubis (NC_018172), Pongo abelii (NC_012614), Saimiri boliviensis (NW_003943798) and Otolemur garnettii (NW_003852644). For P. abelii, the recommendation described in XM_009235414.1, that is, “the sequence of the model RefSeq transcript was modified relative to its source genomic sequence to represent the inferred CDS: deleted 1 base in 1 codon”, was followed.
The exon 1 dataset comprised 15 sequences and included data from GenBank and RNAseq from the three species studied herein (Additional file 2), while the intron 3 dataset comprised 44 different sequences from 70 specimens (Additional file 3). All the datasets were aligned with Mega 5 [31]. Amino acid sequences were deduced from the aligned data and were compared with the human reference sequence (NP_004983). Amino acid substitutions were analyzed with respect to the human mutations reported in RettBASE: IRSF MECP2 Variation Database (http://mecp2.chw.edu.au). Nucleotide sequences encoding MECP2_e2 were analyzed to identify codons under negative, neutral or positive selection through use of SLAC, FEL, REL, PARRIS and MEME (www.datamonkey.org); [32] and using a phylogenetic topology reported previously [33].
The MECP2 nt sequences of intron 3 (excluding gaps and Alu inserts) were used for phylogenetic reconstruction and for inferring the best model of evolution, as based on ModelGenerator v. 0.85 [34]. Kimura’s K80 model with gamma distributed rate heterogeneity was used for phylogenetic reconstructions with PHYML 3.0 [35] for maximum likelihood (ML), and with MrBayes 3.2.1 [36] for Bayesian analysis. In MrBayes, the Markov chain Monte Carlo algorithm was implemented using two independent runs with four chains each and the cold chains were sampled every 100th generation until 10,000 trees were obtained (with a burn-in of 1,000). Support for each node in the ML topology was estimated by aLRT [37], and by bootstrap analyses based on 1,000 replicates.
Alu elements were detected in Cebus, Sapajus and Nomascus capuchin genera by manual alignment and specific Alu families were identified with RepeatMasker (http://repeatmasker.org). Cebus and Sapajus consensus sequences, estimated using BioEdit [38], were compared with the N. leucogenys sequence using the Alu data available in RepBase (http://www.girinst.org).
Results and discussion
Amino acid substitution comparisons
Comparisons of the MECP2_e2 coding region of nonhuman primates revealed amino acid substitutions in 14 codons when compared with the human sequence (Fig. 2), and one in-frame insertion of a single serine codon, between codons 357 and 358 in A. paniscus, was identified. In terms of amino acid polarity, analysis of the substitutions identified three that occurred between nonpolar residues, followed by polar for nonpolar substitutions (n = 2), nonpolar for polar substitutions (n = 3), one negatively charged for another negatively charged substitution and substitution of one polar residue for another polar residue. We observed three different substitutions in three codons of the N-terminal region of MECP2_e2. Of these, E38D (substitution of a negatively charged residue by another negatively charged residue) occurred in all of the non-hominid primates. The exception to this was the Saguinus midas sequence, which shared the same residue to that of the human sequence. Our interpretation of these findings lends to the proposal that aspartate was the ancestral residue of codon 38 and that a D38E substitution must have occurred twice and independently in the phylogenetically distant lineages leading to the large hominoids and Saguinus. Another substitution was found in the N-terminal region of MECP2_e2 (S49P, which involves a nonpolar for a polar substitution); this was restricted to the Sapajus genus and was absent in all other taxa studied herein, including Cebus, a closely related genus. Two substitutions, T203V (in the interdomain) and V275A (in the TRD domain), were restricted to two species of Saimiri, and A277T (in the TRD domain) to Callicebus species. Three different substitutions were found in four species; these were in the interdomains or C-terminal regions (T196S, A358T and V380M) previously listed in RettBASE as being nonpathogenic in humans. Indeed, T196S and A358T are polymorphic variants and V380M is a variant with unknown effects. Finally, the O. garnettii alignment showed an isoleucine codon that matched the first methionine codon in the majority of the other primates, and that the first methionine codon in this species corresponded to codon five of human MECP2_e2.

Comparison of human MECP2_e2 amino acid sequences with those from other primates. Top bar indicates different MECP2 regions with codon numbers delimiting each region. ID = interdomain. Numbers inside parentheses indicate the number of specimens analyzed. Ins* indicates an insertion between codons 357 and 358
Amino acid substitutions did not occur in the MBD domain, a region of critical relevance in that it binds exclusively to DNA that contains one or more symmetrically methylated CpGs in promoter regions [12, 13]. In contrast, three amino acid substitutions were found in the TRD domain (T228A, V275A, and A277T) but these did not occur in the sites previously associated with RTT [39].
Identification of sites under selection
Our findings suggested that MECP2 is highly conserved in primates. Few amino acid substitutions occurred twice and independently according to the phylogenetic relationships of the primate lineages [33]. To evaluate whether specific amino acid residues were under positive or negative selection an analysis was carried out using Datamonkey [32]. Three codons (38, 358 and 380) were found to be under positive selection by REL only, the least stringent algorithm of the three we used for analyzing selection (Table 1). None of these sites occurred in critical regions. Indeed, residue 38 occurred in the N-terminal region, while two other, 358 and 380, occurred in the C-terminal region. Conversely, a total of 48 sites were found to be under negative selection in the different regions, 23 of which were found consistently by REL, FEL and SLAC (Table 1). Five of the sites were found in the N-terminal region, one in the MBD region, 10 in the TRD region, and seven in the C-terminal region. Furthermore, no evidence of positive selection on any codon was apparent by PARRIS analysis. These findings highlight the high evolutionary conservation in MECP2_e2 although the possibility that the restricted, interspecific variations might affect posttranslational modification of MECP2 cannot be ruled out.
Analysis of the exon 1 dataset
Exon 1 could not be amplified, a result probably caused by the high number of repetitive motifs at the 5′-UTR and intron 1 of the human MECP2 sequence (GenBank NG007107) and in other primate sequences with available data. For some primates, including P. paniscus (NW003869946), S. boliviensis (NW003943798), and Rhinopithecus roxellana (NW010801008), the complete genomic data do not contain information on MECP2 exon 1 and its flanking regions, confirming the difficulties involved in sequencing this genome region. However, Callimico, Sapajus and Brachyteles RNAseq data allowed for identification of exon 1 in these genera. Comparisons of exon 1 revealed remarkable evolutionary conservation and phylogenetic signal grouping the three cebids (Callithrix, Callimico and Sapajus). These genera also shared a codon deletion in a poly-alanine region. The lack of critical information on exon 1 for the majority of the species we studied stopped us from conducting a more comprehensive evolutionary analysis of a region of critical relevance in RTT. Further approaches, via whole genome sequencing and RNAseq analysis might provide the relevant information.
Phylogenetic analysis based on intron 3
Taking into account the limited sequence divergence of the MECP2 coding regions in the species analyzed herein, and the dearth of sequence data for several taxa, a phylogenetic analysis based on intron 3 was carried out to evaluate whether MECP2 evolution was in agreement with a previous phylogenetic proposal [33] based on a large dataset (~34,000 bp of aligned sequences) of autosomal genes and genes allocated to X and Y chromosomes. In total, 44 different sequences with a similar length to the human intronic region (ca. 756 bp) were found, except for all Sapajus and Cebus specimens in which we identified an Alu insert (in nt 418 or 419 of intron 3, respectively) of approximately 300 bp in length. Regardless of this insert, the majority of the intron 3 sequences were species-specific except for some sequences that were shared between some Sapajus, Callithrix and Leontopithecus species.
ML and Bayesian analyses of intron 3 generated very similar topologies to the proposed primate phylogeny [33], with three evolutionary lineages corresponding to the main families of the neotropical primates (Cebidae, Atelidae and Pitheciidae). The ML topology (Fig. 3), however, showed a different arrangement within the Cebidae, viz. (((Sapajus,Cebus)Callitrichini)(Aotus,Saimiri)), and within the Pitheciidae ((Pithecia,Chiropotes)Cacajao), although these discordant arrangements were supported by low bootstrap and aLRT estimates. The lack of a consensus arrangement for the Cebidae primates probably results from the short time span between their origin and the radiation of their derived lineages [33, 40, 41].

Maximum likelihood topology based on MECP2 intron 3. Values above the nodes indicate the aLRT support estimates. Values below nodes indicate the posterior probabilities of the Bayesian tree and bootstrap estimates. Red circles indicate Alu insertions
Independent Alu insertions
The intron 3 sequence alignments of neotropical species revealed the presence of an Alu insert shared by species of two closely related genera: Cebus and Sapajus. This insert was not found in any other primate except for a similar, albeit inverted Alu insert in nt 437 of intron 3 from N. leucogenys (NC_019841). RepeatMask analysis showed that the Cebus/Sapajus insert shared high similarity with the Alu subfamily S, while the N. leucogenys insert was found to be similar to the Alu Y subfamily (Additional file 3). These findings indicate that these insertions occurred twice and independently. The Alu insertion must have occurred in the common stock of Cebus and Sapajus, confirming the monophyly of these taxa [42]. The subsequent divergence of Alu inserts, resulting in Cebus-specific and Sapajus-specific lineages, points to their separation as is also evident by the presence of a derived S49P substitution in Sapajus.
Expression of MECP2 transcripts in blood cells
Because alternative splicing of MECP2 results in different protein isoforms and in view of their importance to normal development in humans, coverage of mRNAs coding for isoforms 1 and 2 was analyzed by RNAseq de novo assembly in the three platyrrhine species (S. robustus, B. arachnoides, and C. goeldii). Homologous contigs for each human MECP2 isoform were identified, while a third novel C. goeldii CPRJ2033 contig of 525 nt was also identified. This contig is homologous to a region that includes human exons 1 and 2 and a 270 bp region corresponding to a noncontiguous region of human intron 2 (from nt 12,518 to 12,788). RNAseq analysis showed a higher coverage of MECP2_e2 mRNA than MECP2_e1 in S. robustus and C. goeldii, with a MECP2_e2:MECP2_e1 ratio equal to 1.20 and 1.88 respectively. Conversely, in B. arachnoides, the ratio was 0.76. Differences in sequence coverage might reflect differences in expression or, alternatively, technical artifacts reflecting the efficiency of amplification of the different isoforms caused by the GC-rich composition of exon 1 and its flanking regions (Additional file 4).
Regardless of these findings, differential expression of MECP2 isoforms might also reflect the different epigenetic changes that can vary between nonhuman primate species. Some evidence has been reported supporting the role of methylation in RTT [43], in the expression of MeCP2 isoforms in different brain regions [20], and in differentiating neural stem cells [44]. Further studies should help to clarify the role of epigenetics in regulating expression of MECP2 isoforms in different species.
RT-PCR confirmed the presence of a third novel transcript of 525 nt. From this, there were two amplified products, one fragment of the expected size (403 bp) and an additional 279 bp product that was C. goeldii–specific. The larger product matched the 525 bp contig while the smaller one matched exon 1 and the noncontiguous fragment of intron 2 (Fig. 4).

RT-PCR products corresponding to novel transcripts in Callimico goeldii. a RT-PCR products following amplification of a 525 bp contig resulted in two fragments of 403 and 279 bp. b Transcript composition (E1 = exon 1; E2 = exon 2). Arrows indicate the primer positions. The new alternative exon, homologous to part of human intron 2, is shown in black. c Schematic representation of the novel exon and the alternative splicing resulting in the two new transcripts. Start and stop codons indicate three likely open reading frames. IN1 = intron 1; IN2 = intron 2; IN3 = intron 3; E1 = exon 1; E2 = exon 2; E3 = exon 3
Sequence analysis of the RT-PCR products showed that, excluding the primers, the 403 bp fragment contained part of exon 1 (76 bp), the complete exon 2 (124 bp) and part of the noncontiguous segment of intron 2 (162 bp) confirming the presence of the exon2–intron2 segment junction found by the RNAseq de novo assembly. The deduced amino acid sequence of this fragment, from the ATG initiation codon of exon 1 used for translation of human MECP2_e1 to an in-frame stop codon inside exon 2, corresponded to a 33 amino acid polypeptide. Alternatively, the possibility exists that the 403 bp fragment could produce a 26 amino acid polypeptide following translation from the ATG initiation codon at exon 2, and this polypeptide might be used for translation of human MECP2_e2 to an in-frame stop codon inside intron 2. The smaller 279 bp fragment contained the same part of exon 1 and the same part of the noncontiguous segment of intron 2. A sequence of 37 amino acids was deduced from the ATG initiation codon of exon 1 used for translation of human MECP2_e1 to the same in-frame stop codon inside intron 2.
Identification of a transcribed region in Callimico, which is homologous to part of human exon 2 and part of intron 2, points to evolutionary conservation of these regions regardless of their expression patterns. Why this region is transcribed in Callimico is an open question. Human intron 2 is not analyzed in conventional RTT genetic testing and many RTT patients appear to have a normal MECP2 sequence. Expression of this region in humans, where potentially deleterious mutations are expressed, might explain this paradox. Whole genomic analysis of Callimico, which is presently under way, will allow identification of the boundaries surrounding the new alternative exon and comparisons of splice donor/acceptor sites in humans. Future in silico analysis may provide molecular explanations for the novel exon, such as whether it is related to novel exonic splicing enhancers or splice donor/acceptor signals.
Conclusions
These studies show that despite the remarkable evolutionary conservation of MECP2_e2, one in-frame codon insertion occurred in Ateles paniscus, and one region of intron 3 was affected by trans-specific retrotransposition (Alu insertion). Moreover, identification of novel MECP2 transcripts in Callimico suggests that part of the homologous intronic region in humans might be expressed, and that the potential open reading frame in this region might be a subject of interest in RTT patients who carry an apparently normal MECP2 sequence. This is particularly relevant because alterations in this gene are associated with RTT in humans. Here, we found that evolutionary variation in MECP2 is not correlated with any pathological alteration previously associated with RTT.
Availability of supporting data
The following sequence data have been deposited in GenBank: MECP2 intron 3 (accession numbers KM206798 to KM206856), MECP2 exon 2, 3 and 4 (accession numbers KM206857 to KM207008), and MECP2 isoforms (accession numbers KR265897 to KR265903). Accession numbers of intron and exons are listed for each individual sample in Additional file 1. Intron 3 data and topology have been deposited in TreeBASE (Study Accession URL: http://purl.org/phylo/treebase/phylows/study/TB2:S17761) .
References
Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr. 1966;116(37):723–6.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–8.
Long SW, Ooi JY, Yau PM, Jones PL. A brain-derived MeCP2 complex supports a role for MeCP2 in RNA processing. Biosci Rep. 2011;31(5):333–43.
Zachariah RM, Rastegar M. Linking epigenetics to human disease and Rett syndrome: the emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012;2012:415825.
Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20(19):5085–92.
Hite KC, Adams VH, Hansen JC. Recent advances in MeCP2 structure and function. Biochem Cell Biol. 2009;87(1):219–27.
Weitzel JM, Buhrmester H, Stratling WH. Chicken MAR-binding protein ARBP is homologous to rat methyl-CpG-binding protein MeCP2. Mol Cell Biol. 1997;17(9):5656–66.
Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci U S A. 2005;102(49):17551–8.
Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88(4):471–81.
Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11(2):115–24.
Song C, Feodorova Y, Guy J, Peichl L, Jost KL, Kimura H, et al. DNA methylation reader MECP2: cell type- and differentiation stage-specific protein distribution. Epigenetics Chromatin. 2014;7:17.
Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21(21):4886–92.
Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. 2005;19(5):667–78.
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–9.
Buschdorf JP, Stratling WH. A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on Rett syndrome. J Mol Med (Berl). 2004;82(2):135–43.
Singh J, Saxena A, Christodoulou J, Ravine D. MECP2 genomic structure and function: insights from ENCODE. Nucleic Acids Res. 2008;36(19):6035–47.
Reichwald K, Thiesen J, Wiehe T, Weitzel J, Poustka WA, Rosenthal A, et al. Comparative sequence analysis of the MECP2-locus in human and mouse reveals new transcribed regions. Mamm Genome. 2000;11(3):182–90.
Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJ, et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet. 2004;36(4):339–41.
Dragich JM, Kim YH, Arnold AP, Schanen NC. Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. J Comp Neurol. 2007;501(4):526–42.
Olson CO, Zachariah RM, Ezeonwuka CD, Liyanage VR, Rastegar M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE. 2014;9(3), e90645.
Gianakopoulos PJ, Zhang Y, Pencea N, Orlic-Milacic M, Mittal K, Windpassinger C, et al. Mutations in MECP2 exon 1 in classical Rett patients disrupt MECP2_e1 transcription, but not transcription of MECP2_e2. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(2):210–6.
Yasui DH, Gonzales ML, Aflatooni JO, Crary FK, Hu DJ, Gavino BJ, et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum Mol Genet. 2014;23(9):2447–58.
Itoh M, Tahimic CG, Ide S, Otsuki A, Sasaoka T, Noguchi S, et al. Methyl CpG-binding protein isoform MeCP2_e2 is dispensable for Rett syndrome phenotypes but essential for embryo viability and placenta development. J Biol Chem. 2012;287(17):13859–67.
Buyse IM, Fang P, Hoon KT, Amir RE, Zoghbi HY, Roa BB. Diagnostic testing for Rett syndrome by DHPLC and direct sequencing analysis of the MECP2 gene: identification of several novel mutations and polymorphisms. Am J Hum Genet. 2000;67(6):1428–36.
Monnerat LS, Moreira AS, Alves MC, Bonvicino CR, Vargas FR. Identification and characterization of novel sequence variations in MECP2 gene in Rett syndrome patients. Brain Dev. 2010;32(10):843–8.
Moog U, Smeets EE, van Roozendaal KE, Schoenmakers S, Herbergs J, Schoonbrood-Lenssen AM, et al. Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2). Eur J Paediatr Neurol. 2003;7(1):5–12.
McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333(6039):186.
Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CM, Schaaf CP, et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009;66(6):771–82.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–512.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–9.
Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26(19):2455–7.
Perelman P, Johnson WE, Roos C, Seuanez HN, Horvath JE, Moreira MA, et al. A molecular phylogeny of living primates. PLoS Genet. 2011;7(3), e1001342.
Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 2006;6:29.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.
Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–4.
Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol. 2006;55(4):539–52.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–8.
Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013;16(7):898–902.
Hodgson JA, Sterner KN, Matthews LJ, Burrell AS, Jani RA, Raaum RL, et al. Successive radiations, not stasis, in the South American primate fauna. Proc Natl Acad Sci U S A. 2009;106(14):5534–9.
Schrago CG, Menezes AN, Moreira MA, Pissinatti A, Seuanez HN. Chronology of deep nodes in the neotropical primate phylogeny: insights from mitochondrial genomes. PLoS ONE. 2012;7(12), e51699.
Alfaro JW, Silva Jr JD, Rylands AB. How different are robust and gracile capuchin monkeys? An argument for the use of sapajus and cebus. Am J Primatol. 2012;74(4):273–86.
Aldinger KA, Plummer JT, Levitt P. Comparative DNA methylation among females with neurodevelopmental disorders and seizures identifies TAC1 as a MeCP2 target gene. J Neurodev Disord. 2013;5(1):15.
Liyanage VR, Zachariah RM, Rastegar M. Decitabine alters the expression of Mecp2 isoforms via dynamic DNA methylation at the Mecp2 regulatory elements in neural stem cells. Mol Autism. 2013;4(1):46.
Acknowledgments
This work was supported by Conselho Nacional de Desenvolvimento (CNPq grant 303306/2010-6) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ grant E26/103.012/2011). A.N. Menezes was a recipient of a FAPERJ/CAPES post-doctoral grant (E26/102.810/2011) from Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES-Brazil). We are grateful to Dr. Cibele R. Bonvicino for providing samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MCV carried out Sanger sequencing and RT-PCR analyses, ANM and MAM participated in the RNAseq experiments and analyses. AP provided samples from several neotropical primates kept under his care at the Centro de Primatologia do Rio de Janeiro. HNS contributed to planning and drafting the original version of the manuscript. All authors have read, commented on and approved the final manuscript.
Maria Carolina Viana and Albert Nobre Menezes contributed equally to this work.
Additional files
Additional file 1:
List of specimens, field numbers and GenBank accession numbers of sequence data.
Additional file 2:
Alignment of the available data for exon 1.
Additional file 3:
Alignment of Alu sequences.
Additional file 4:
Transcript coverage following de novo assembly of MECP2 in Sapajus , Brachyteles and Callimico . The number on the left side of each illustration indicates the highest level of coverage.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Viana, M.C., Menezes, A.N., Moreira, M.A.M. et al. MECP2, a gene associated with Rett syndrome in humans, shows conserved coding regions, independent Alu insertions, and a novel transcript across primate evolution. BMC Genet 16, 77 (2015). https://doi.org/10.1186/s12863-015-0240-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-015-0240-x