
Abstract
The African buffalo, Syncerus caffer, is a key species in African ecosystems. Like other large herbivores, it plays a fundamental role in its habitat acting as an ecosystem engineer. Over the last few centuries, African buffalo populations have declined because of range contraction and demographic decline caused by direct or indirect human activities. In Mozambique, historically home to large buffalo herds, the combined effect of colonialism and subsequent civil wars has created a critical situation that urgently needs to be addressed. In this study, we focused on the analysis of genetic diversity of Syncerus caffer caffer populations from six areas of Mozambique. Using genome-wide SNPs obtained from ddRAD sequencing, we examined the population structure across the country, estimated gene flow between areas under conservation management, including national reserves, and assessed the inbreeding coefficients. Our results indicate that all studied populations of Syncerus caffer caffer are genetically depauperate, with a high level of inbreeding. Moreover, buffaloes in Mozambique present a significant population differentiation between southern and central areas. We found an unexpected genotype in the Gorongosa National Park, where buffaloes experienced a dramatic population size reduction, that shares a common ancestry with southern populations of Catuane and Namaacha. This could suggest the past occurrence of a connection between southern and central Mozambique and that the observed population structuring could reflect recent events of anthropogenic origin. All the populations analysed showed high levels of homozygosity, likely due to extensive inbreeding over the last few decades, which could have increased the frequency of recessive deleterious alleles. Improving the resilience of Syncerus caffer caffer in Mozambique is essential for preserving the ecosystem integrity. The most viable approach appears to be facilitating translocations and re-establishing connectivity between isolated herds. However, our results also highlight the importance of assessing intraspecific genetic diversity when considering interventions aimed at enhancing population viability such as selecting suitable source populations.
Similar content being viewed by others

Introduction
Since the 19th century, many African mammal species have experienced a decline due to human population expansion, overexploitation, habitat loss and degradation, wars, and the spread of livestock-borne diseases [1–2]. Currently, several populations of endangered species survive in protected areas that are often poorly connected, not ensuring a sufficient gene flow among isolated populations [3].
Like other mammal species, the African buffalo (Syncerus caffer) has experienced shifts in its distribution range since the last phases of the Pleistocene, resulting in the expansion and contraction of suitable habitat [4–5]. These changes have left a distinctive signature in its genome. However, over the past 10,000 years, there has been evidence of a drop in the effective population size (Ne) of S. c. caffer populations, coinciding with the increase in human populations [6]. Moreover, during the last two centuries, S. caffer has undergone a significant numerical reduction and shrinking of its distribution range due to war, disease, poaching [7], and expansion of livestock activity [8]. This species is now listed as “Near Threatened” due to population decline over extensive areas [9] (IUCN 2019). In Mozambique, where the nominal subspecies S. c. caffer is present, after the outbreak of civil war (1976–1992), the African buffalo populations experienced a significant decline both within and outside protected areas [10]. For instance, in the emblematic Gorongosa National Park, no buffalo were sighted during aerial counts in 2000, with the surviving population estimated to be less than 100 individuals at that time. This period also witnessed a decline of over 90% in the populations of large mammals [10–11]. Although the biomass of large mammalian herbivores in the country has substantially recovered since 1994, the species composition has shifted dramatically, with formerly dominant large herbivores, including elephant (Loxodonta africana), hippo (Hippopotamus amphibius), buffalo (Syncerus caffer), zebra (Equus quagga), and wildebeest (Connochaetes taurinus), now outnumbered by waterbuck (Kobus ellipsiprymnus) and other small to mid-sized antelopes [11]. However, large and megaherbivores play a crucial functional role in terrestrial ecosystems by acting as ecosystem engineers [12–13]. They prepare the habitat for small and mid-sized herbivores [14,15,16], as well as mitigate the impact of large uncontrolled fires [17]. Thus, a reduction in the number of large herbivores could have an enormous ecological cost [12]. Therefore, the decline of S. caffer populations in many areas of Africa is particularly concerning as it has negative implications for the whole savanna ecosystem.
Between 2006 and 2011 the original buffalo population of Gorongosa was reinforced with 186 individuals translocated from the Kruger National Park in South Africa and the adjacent Limpopo National Park in Mozambique (Lopes Pereira, pers. Com.) in an attempt to increase the genetic diversity of S. c. caffer in the iconic Gorongosa National Park. Translocations are rarely followed up by monitoring activities, which means that the outcomes of these efforts are often unknown, and the eventual causes of failures are rarely understood [18]. While measuring an increase in population growth and size is often used to judge the success of reintroduction programs [19], it is important to note that the ability of a population to persist in the long term is also strongly influenced by the levels of genetic diversity. Therefore, simply achieving a demographic increase does not necessarily mean that genetic diversity has been restored, which is indispensable for the long-term persistence of the target populations [20].
Despite the clear and urgent necessity of planning actions to restore the buffalo population in Mozambique, including translocations, there is a lack of information regarding the current status of relict herds, particularly in terms of connectivity and genetic diversity. Furthermore, it is important to avoid genetic dilution during the process of translocation, as this carries the risk of losing the best characteristics of both the original and receiving populations. Therefore, the biogeographical aspects of populations must be considered before initiating the translocation process.
In the last decades, genetic assessments and monitoring are increasingly used for wildlife conservation and management [21,22,23]. Particularly, population genomics has provided key insights into ecological and evolutionary processes in natural and managed populations [24]. Population genomic data provide new opportunities for detecting natural selection and quantifying adaptive genetic variation in natural populations [25], while simultaneously increasing the accuracy and precision of estimates of genome-wide diversity [26], population structure, and demographic parameters [27].
The overarching aim of this research is to estimate the genetic diversity of African buffaloes in Mozambique, and to evaluate the population structure and connectivity among different areas using restriction-site-associated DNA (RAD) sequencing. Among the molecular methods aimed at genotyping a dense set of markers in a large set of individuals, RAD sequencing has become the preferred method of choice in molecular ecology and conservation studies [28, 29]. The RAD-based approaches are particularly valuable and widely used when applied to non-model organisms, where screening a large number of SNPs (Single Nucleotide Polymorphisms) might be cost- and time-prohibitive.
Despite the critical role of African buffaloes in Mozambique, there has been limited research that included individuals from this area [7, 30]. More recent studies that employ new genomic tools also did not include individuals from Mozambique [4, 5]. Therefore, in this study, we used genome-wide data of S. c. caffer to gain a deeper understanding of the population structure and connectivity between protected areas in Mozambique and to have an insight on the level of inbreeding of the focal populations with special attention to Gorongosa National Park buffaloes. Our emphasis on Gorongosa NP is due to the dramatic bottleneck experienced by this protected area and the recent restocking program. To our knowledge, there has been no previous assessment of the pre-restocking genetic diversity of African buffaloes in this area. Comparing the genetic diversity and inbreeding coefficients of the original Gorongosa population to other areas of Mozambique can provide a baseline for evaluating the effectiveness of current conservation efforts and assisting national and local authorities in developing effective conservation policies in Mozambique. An excessive numerical reduction and the consequent inbreeding are expected to increase the occurrence of recessive alleles, potentially making buffalo populations more vulnerable to environmental stressors [31]. Therefore, effective management of buffalo populations is critical for the survival of the species in Mozambique, and genetic monitoring is widely recognized as an essential tool to inform intervention plans [22].
Materials and methods
Sample collection and ethics statement
The blood samples used in this study were collected during past veterinary operations within the Gorongosa National Park (research permit: PNG/DSCi/C156/2019) and from animals immobilized under the mandate granted by the National Administration of Conservation Areas (ANAC) to the vet team of the Mozambique Wildlife Alliance (MWA). Samples are currently preserved at the Natural History Museum of Maputo (NHM-UEM) and the Biotechnology Centre of the Eduardo Mondlane University (CB-UEM) BioBank.
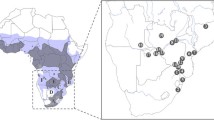
A total of 70 S. c. caffer blood samples, stored in ethylenediaminetetraacetic acid (EDTA), were collected from six localities: four protected areas of Central Mozambique (Gorongosa National Park, Marromeu National Reserve, Gilè National Park, and Coutada 9) and two in free areas in the south of Mozambique (Namaacha and Catuane). Details of sampling localities are shown in Fig. 1 and in Supplementary Table S1. Genomic DNA extraction was performed in the Natural History Museum of Maputo (NHM-UEM) and the Biotechnology Centre (CB-UEM) laboratories. The DNeasy Blood & Tissue Kit (Qiagen, Valencia, USA) was used for DNA extraction following the manufacturer’sguidelines. The DNA extracts were stored at − 80 °C in the NHM-UEM and CB-UEM Biobank.
Library preparation and sequencing
Sequencing libraries were constructed using the quaddRAD protocol developed by [32]. This protocol follows the general principles of the original double-digest RAD-Seq (ddRAD) approach developed by [33], with modifications that allow marking PCR duplicates, increasing multiplexing capacity, and minimise hands-on time. Genomic DNA concentration and quality were assessed using a Qubit v4.0 fluorometer (Life Technologies, Darmstadt, Germany) and a Bioanalyzer 2100 (Agilent Technologies, Palo Alto, USA), respectively. Approximately 200 ng of DNA per individual sample was digested using the restriction enzymes PstI (rare cutter) and MspI (frequent cutter). A single quaddRAD library including 70 barcoded individuals was size-selected from 450 to 550 bp in a Pippin Prep system (Sage Science, Beverly, USA) and paired-end sequenced (2 × 150 bp) in an Illumina HiSeq X-Ten platform at the Beijing Genomics Institute (BGI, Hong Kong).
Sequence alignment and genotyping
PCR clone removal, quality filtering, and demultiplexing of raw sequence data were performed using the clone_filter and process_radtags modules implemented in the Stacks v2.59 package [34]. Clean sequences of each individual were independently aligned to the S. caffer reference genome (NCBI GenBank assembly accession: GCA_902825105.1) by using the Burrows-Wheeler Aligner bwa-mem v0.7.17 [35]. Variant calling was carried out using the Stacks’s gstacks program using default settings. Once the loci have been identified and genotyped, gstacks phases the SNPs at each locus for each individual into a series of haplotypes using a Bayesian algorithm [36, 37].

Map of Mozambique showing the six sampling localities of the African Buffalo (Syncerus c. caffer) analysed in this study. The map was created by utilizing a Google Maps image as a template (https://www.google.com/maps/place/Mozambico) and modified using Adobe Illustrator v2021 to highlight the sampling areas
In a second step, the output assembled by gstacks was processed by the Stacks’s Populations program that processes the dataset based on various parameters. Specifically, rare variants, often resulting from PCR artifacts, were removed by setting a minor allele frequency (MAF) of 5% (--min-maf 0.05). The maximum observed heterozygosity required to process a nucleotide site was set to 70% (--max-obs-het 0.70) and all variants exceeding this heterozygosity threshold were eliminated; only the alleles present in at least 90% of the individuals of the total sample were retained (-R 0.9). The resulting dataset included 100,646 variants. To reduce the effect of non-independence of markers due to physical linkage and linkage disequilibrium (LD), and thus satisfying one of the main assumptions of model-based algorithms used in this study, a second dataset was produced by PLINK 1.9 to filter for LD the original dataset (parameters: --indep-pairwise 50 5 0.2), resulting in a new dataset including 53,056 variants.
Population structure, admixture and gene flow
For each population, we used Stacks to estimate standard diversity indexes such as heterozygosity, homozygosity and nucleotide diversity (π), and to assess population differentiation measured with Fst and Dxy. Population structure and admixture were estimated using the dataset pruned for LD. We computed a principal component analysis (PCA) using the R package SNPRelate v1.30.1 [38], whereas we estimated individual ancestry and admixture using the software Admixture v1.3.0 [39, 40]. Admixture uses a maximum likelihood estimation of individual ancestries from multilocus SNP genotype datasets. The best number of underlying populations was assessed by using the cross-validation procedure implemented in Admixture. A good value of K, i.e. the number of predefined genetic clusters or ancestral populations, is expected to exhibit a low cross-validation error compared to other K values. We then tested values of K ranging from 1 to 10. Because the right choice of K is a long-standing known issue with admixture modeling [41], we also performed a visual inspection of the results and presented more K values to describe population structure and substructures.
In addition, a network-based approach implemented in the program NetView v.1.1 [42, 43] using an Identity by State (IBS) distance matrix was performed to show relationships between genetic clusters inferred by Admixture. To visualize the population network connection for all the individuals, we used a k-NN value, a parameter determining the number of mutual nearest neighbors, selected by visual inspection of multiple k-NN values (k-NN = 10–60) plotted against the number of communities detected with a “Fast-greedy”, an “Infomap” and a “Walktrap” clustering algorithm (Supplementary Fig. S1).
Finally, relative migration between locations was estimated using the divMigrate function in the diveRsity package [44, 45]. Nei’s GST distances were calculated with 1,000 bootstrap replicates to detect significant asymmetrical gene flow between the sampled locations.
Homozygosity and inbreeding
We assessed inbreeding using the estimate of the heterozygosity-fitness correlation (HFC). HFC relies on the general assumption that genome-wide heterozygosity is a good proxy for inbreeding because it reduces heterozygosity and generates identity disequilibrium (ID), i.e., correlation of heterozygosity across loci [46]. This is because inbred individuals are expected to be less heterozygous at all loci than outbred ones. We evaluated the ID by calculating the parameter g2 [47] using the inbreedR package [48]. The parameter g2 is an estimate of ID that measures the excess of standardised double heterozygosity at two loci relative to the expectation under random association (i.e., g2 = 0) [49]. Specifically, g2 estimates the variance in inbreeding and if it is significantly different from 0 it can be interpreted as a signature of HFC, thus inbreeding. The probability that g2 differs from zero (i.e., the null hypothesis of no variance in inbreeding) was assessed with 1,000 permutations and the standard error with 1,000 bootstraps.
Successively, to quantify autozygosity and inbreeding in the African buffalo in Mozambique, we estimated runs of homozygosity (ROH) using a sliding window method implemented in the R package DetectRuns [50]. For this analysis the following parameters were used: windowSize = 50, minSNP = 50, minDensity = 1/10,000, maxMissWindow = 1, maxGap = 1,000,000, minLengthBps = 1,000,000, maxOppWindow = 0. We explored the ROH pattern for each individual by plotting the number of ROH versus the total and mean ROH length (in Mbps) respectively. Successively we estimated the genome-wide inbreeding coefficient for each population based on the ROH (FROH).
Effective population size
We estimated historical and recent effective population size (Ne) trajectories using two different approaches. First, we employed folded Site Frequency Spectrum (SFS) to infer the demographic history of African buffalo in Mozambique. We used Stairway Plot v2 [51], which employs a method that calculates the composite likelihood of the observed SFS under several possible size change scenarios across the history of a population. Stairway Plot estimates the most likely timing and magnitude of changes in Ne at various epochs, with the transition between epochs corresponding to coalescent events in the population. Stairway Plot calculates the population-scaled mutation rate θ, from which Ne can be derived. To do this, we use the µ (mutation rate) of ∼1.5 × 10− 9 estimated for ruminants [52] and a generation time of 7.5 years following previous studies of S. c. caffer historical demography [4, 5].
Subsequently, recent trends in Ne (below ∼1000 generations) were estimated using the program SNeP v1.1 [53], a multithreaded tool that assesses Ne based on linkage disequilibrium (LD). The method relies on the fact that LD between SNPs at different genetic distances provides information on Ne at different times in the recent past. We performed SNeP by using the standard PLINK file as input file format (.ped and.map files), considering all chromosomes in the analysis and by using Sved & Feldman [54] as recombination rate modifier. We also selected a MAF threshold of 0.05 because it has been demonstrated that accounting for MAF results in unbiased r2 estimates regardless of sample size [55].
Results
Genetic differentiation, population structure, admixture and gene flow
The average observed heterozygosity was lower than the expected heterozygosity in all the studied populations except for Gilè and Coutada 9 (Table 1), for which the estimate is based on only one individual each and thus cannot be considered sufficiently reliable. The remaining four populations show a comparable level of heterozygosity deficit and nucleotide diversity (Table 1). In contrast, the average inbreeding coefficient was higher in Marromeu and Gorongosa compared to the other populations. These two populations also exhibit the lowest values of genetic differentiation and absolute divergence when compared to Namaacha (Table 2).
According to the PCA (Fig. 2), individuals from Marromeu cluster together showing low intrapopulation variability on the first two axes compared to other populations. In the same portion of the PCA space, we can find individuals from Gorongosa, Gilè, and Coutada 9. The remaining individuals from Gorongosa form a separate cluster. It is interesting to note that this second cluster includes two individuals from Catuane and one from Namaacha. Finally, the remaining individuals from Namaacha and Catuane form a distinguished group.

Principal Component Analysis (PCA) showing the presence of different genetic clusters within Mozambique. Marromeu is clearly distinct but largely overlaps with some individuals from Gorongosa, and it is also closely related to Gilè and Coutada 9. By contrast, individuals from Gorongosa form a second cluster that shows some relatedness with individuals from Namaacha and Catuane. Lastly, the remaining individuals from the southern locality form a third cluster
In the Admixture analysis, the cross-validation (CV) shows an increasing error value from K = 1 to K = 10 (Supplementary Fig. S2). However, at K = 2 we observe just a very sharp increase of CV error compared to K = 1. By visually inspecting the admixture plots (Fig. 3) at K = 2, we observe a clear distinction between specimens from Namaacha and Catuane from specimens collected in Marromeu, Gilè, Coutada 9, and Gorongosa. However, nine individuals from Gorongosa are assigned to the other cluster or show a clear signature of admixture between the two clusters. At K = 3 (Fig. 3) individuals from Namaacha and one from Catuane form a new distinct genetic cluster. Three individuals from the southern clade still show admixture with the northern genetic clusters. This observation shows a congruence with the results depicted by the PCA (Fig. 2). Higher K values show a progressive increase of the cross-validation error (Supplementary Figure S2) and just a few differences can be observed within the Marromeu-Gorongosa cluster (data not shown).

Admixture and network analysis. We reported the genetic clustering for K = 2 and K = 3, putative ancestral genotypes. For both K values, we can observe a clear structure of southern and central-northern individuals. Network analysis based on Identity by State distance matrix shows relationships between genetic clusters. Circles represent individuals whereas color represents cluster membership according to the Admixture analysis (K = 2 and K = 3 respectively)
The relationships among the clusters found by the Admixture analysis (K = 2 and K = 3) are summarised by the network shown in Fig. 3. The network shows that the admixed individuals found at K = 2 are effectively in an intermediate position. Interestingly, when cluster membership at K = 3 is plotted on the network, the third cluster identified by the Admixture analyses, mainly including individuals from Gorongosa and three other individuals from southern localities, occupies an intermediate position between the two main clusters.
As expected, the relative migration was found to be high and symmetrical between Gorongosa NP and Marromeu, whereas gene flow from south to north is very low in both directions (Fig. 4).

Among-population gene flow, estimated using Nei’s GST distances. The arrow’s color intensity is proportional to the level of gene flow. Only Gorongosa National Park and Marromeu Reserve show a high and symmetrical gene flow
Genome-wide homozygosity and inbreeding
Identity disequilibrium is low but positive and significantly different from 0 for all the analysed populations, giving a clear indication of HFC in all the populations. Catuane shows the lowest value (g2 = 4.89E-06; p = 0.001), followed by Gorongosa (g2 = 1.51E-04; p = 0.001) and Marromeu (g2 = 5.56E-04; p = 0.001), and by Namaacha (g2 = 3.70E-04; p = 0.001) with the highest values.
The distribution patterns of regions of homozygosity (ROH) show that most of the analysed individuals show a high number (> 600) of long ROH regions (mean up to 3 Mbps, total length up to 1500 Mbps). Only a few individuals from Marromeu show a few short ROHs (Fig. 5).

Distribution pattern of regions of homozygosity (ROH) length and number. Only a few individuals, mainly from Marromeu, show the presence of a ROH sum < 1000 Mb. The mean length of ROH ranges approximately from 2 to 3.5 Mb
These long stretches of homozygosity are reflected in a high level of inbreeding observed in all the populations (Fig. 6).

FROH coefficients (left) and distribution of ROH classes (right). All the populations analysed show a high FROH suggesting a large genome-wide homozygosity resulting from inbreeding. The lower and upper hinges correspond to the 25th and 75th percentiles. Lower and upper whiskers give a roughly 95% confidence interval around the median represented as a horizontal bold line. Pairwise t-test (with Bonferroni correction) reported no significant divergence between means (for summary statistics see supplementary Table S3). ROH graphs suggest the occurrence of a high percentage of short ROH (length < 2 Mb) in all populations, but also the presence of a significant fraction of ROHs > 4Mbp in length can be observed, indicating that most ROHs are the result of recent inbreeding events. Individual inbreeding coefficients are reported in Supplementary Table S2
The frequency of ROH classes (Fig. 6) suggests the prevalence of large regions of long and very long ROH.
Historical demography and effective population size estimation
Finally, the plot of Ne over time (Fig. 7) shows a clear decline for all the populations analysed (Catuane, Gilè, and Coutada 9 were excluded due to the very low sample size). Following a stationary phase that started approximately 300–350 thousand years ago (ka), all three populations show a Ne decrease starting approximately between 30 and 50 ka (Fig. 7). For all three populations the Ne decrease was constant until recent time, without any signature of population expansion in recent times. According to SNeP estimates, the last 1000 generations were characterised by a steep decrease (Fig. 7). Both methods provide coherent final estimates with a Ne a few decades of individuals (Table 3).

Effective population size estimates obtained for the Gorongosa, Marromeu, and Namaacha populations. Left panel: median Ne and 95% confidence intervals based on Site Frequency Spectrum (SFS). The three populations show a similar shape of Ne through time. Notably, all three populations show a Ne decrease starting at approximately around 50 ka. All three populations show a constant decrease of Ne until recent times without any signature of population expansion in recent times. Right panel: the recent Ne trend obtained using linkage disequilibrium in SNeP software is coherent with what we observed using the SFS method. All the populations show a Ne decrease also in recent times reaching a very low Ne (see Table 3)
Discussion
Our findings provide new data on the conservation status and genetic diversity of some African buffalo populations from Mozambique. As expected, we observed evidence of genetic variability loss, reduced gene flow, and excess of homozygosity in a large part of the genome. Additionally, we found evidence of past connections between southern and northern populations. This information may aid in the planning of further studies and assist in the management of surviving buffalo herds.
Population structure and admixture in Mozambique African buffaloes
Historical data indicates that buffalo populations in Mozambique, as well as other large herbivores, were abundant at the onset of the colonial period [2]. However, since the end of the 19th century the increase of human settlement, the hunting pressure, and the expansion of cattle have resulted in the demographic decline of many large herbivores including S. caffer [2]. More recently, buffalo populations in Mozambique have undergone a dramatic numerical reduction due to persecution during the civil war that ended in 1992. Currently, the African buffalo primarily survives in protected areas with herds ranging from a few dozen to a few thousand individuals. Our study found these areas are weakly connected by gene flow, with the only exception of Marromeu and Gorongosa. Consequently, S. c. caffer in Mozambique exhibits a significant population structure, which is consistent with previous studies based on microsatellite data [7]. An interesting finding in our study is the unexpected population substructure observed in Gorongosa. While most individuals from Gorongosa showed genetic similarity to nearby Marromeu, a few individuals exhibited clear attribution to the southern cluster or evidence of admixture between southern and northern clusters. At K = 3 (three genetic clusters), these individuals are clearly distinct from both southern and northern clades, and according to the network analysis, they represent an intermediate genotype that connects southern and northern Mozambique buffaloes. Therefore, the Gorongosa population displays an unexpected population substructure with one of the two genotypes closely related to Marromeu, due to their geographical proximity, while the second cluster is apparently more related to southern individuals (as evidenced by the presence of two individuals from Catuane). Differently from previous studies conducted on buffaloes from the same area [7], we analysed autochthonous individuals from Gorongosa that survived human persecution during the civil war and were not recently introduced from South Africa. Based on this, one possible explanation of the presence of an unexpected genetic cluster in Gorongosa is that in the past the genetic connectivity between southern and northern herds was higher than it is now, and Gorongosa served as a biogeographical hinge between southern and northern Mozambique. In contrast, all the other populations analysed appear to be genetically homogeneous, likely due to the range restriction of all herds in Mozambique to a few protected and managed areas, resulting in minimal gene flow, especially in the last century.
Historical and contemporary demographic trends
In our analysis, the three distinct populations exhibit remarkably similar trends in effective population size (Ne), implying the occurrence of comparable drivers of demographic decline across Mozambique. This decline started roughly between 40 and 50 ka and persisted until the present era determining a very low Ne. These findings display partial alignment with recent literature. Indeed, De Jager et al. [4], employing a Sequentially Markovian Coalescent (PMSC) method, identified a signature of Ne decline within the South African S. c. caffer population around 50 ka, a pattern corroborated by our results from Stairway Plot 2. Notably, De Jager et al. [4] documented an expansion event around 30 ka, which contrasts with our observations. Subsequently, Quinn et al. [5], adopting an Approximate Bayesian Computation (ABC) simulation, identified a continuous Ne decline in South African population since roughly 10 ka, mirroring our recent demographic trajectory estimate obtained through SNeP and Stairway Plot 2. Presuming that both the Mozambique and South African buffalo populations were subject to analogous or at least similar drivers of demographic changes, the absence of a detected expansion around 30 ka could be partly attributed to methodological disparities. It is worth noting that the methodologies employed in our study, along with the ABC simulation method used by Quinn et al. [5], exhibit better performance in capturing recent historical demography when compared to the PMSC method [51, 56, 57]. Consequently, we are inclined to assert that our estimation of a recent and sustained decline is a reliable representation.
Our estimation of the demographic decline finds parallelism with a significant reduction in population and concurrent species extinctions documented in Africa since the late Pleistocene, where approximately 24 large mammal species are known to have disappeared from the continent during the last ∼100,000 years [58]. The precise causal factors behind these extinctions remain a subject of ongoing debate [58, 59]. However, a mounting body of evidence suggests that the disappearance of numerous large herbivores, likely preceded by declines in population density, can be explained by an interplay of natural and anthropogenic variables. These include shifts in climate, alterations in the environment, species interactions, and the expansion of human populations [60,61,62]. Hence, we argue that the observed decline in the population of S. c. caffer started from the latter stages of the Pleistocene could similarly be attributed to these phenomena. Certainly, for more recent population declines, human impact emerges as the principal cause. In the case of buffalo populations within the southern African region, historical events such as European colonization increased hunting pressure but also facilitated the transmission of diseases from domesticated cattle to wild herbivores [62]. In the specific context of Mozambique, the buffalo populations were dramatically affected by the civil war. Before this conflict, the Marromeu and Gorongosa area harbored some of the largest buffalo herds in southern Africa, with an estimated population of around 50,000 and 14,000 individuals. However, in the aftermath of the civil war, this population experienced an impressive contraction, leaving an estimated 2,500 buffaloes remaining in the Marromeu region and approximately less than 100 individuals in Gorongosa [11, 63]. Although the demographic crisis in Marromeu was not as severe as in Gorongosa, it still resulted in a significant erosion of genetic diversity. This is evident from low heterozygosity and PCA and Admixture analysis, which show that Marromeu individuals are genetically homogeneous. Thus, all the past and recent perturbations discussed above may have had a profound influence on the survival of buffalo populations in Mozambique and collectively, the convergence of these factors could explain our estimation of a continuous Ne decline since 50 ka and the remarkably low effective population size estimate observed in this study.
Genome-wide homozygosity and inbreeding
As expected, the observed Ne trends were accompanied by a heterozygosity deficit and a pervasive presence of ROH stretches in all the studied populations. Our sample revealed that only a few individuals had a low number of short ROHs, while the majority had many long ROHs. It is well established that the occurrence of long and large ROHs is related to the demographic history of populations, and the ROH pattern observed in our sample is expected for populations that have experienced recent inbreeding and bottleneck [64, 65]. Elevated levels of inbreeding have been previously documented within S. c. caffer populations in South Africa [4, 5]. Specifically, Quinn et al. [5] identified pronounced inbreeding within the southernmost populations, namely Hluhluwe-iMfolozi Park and Addo Elephant National Park, characterised by a notable abundance of long ROH. These extended ROH segments were attributed to recent inbreeding events, particularly impacting these two populations. In contrast, within Mozambique, all buffalo populations exhibit approximately 30% of ROH segments exceeding 4 megabases (Mb) in length, with the cumulative length of individual ROHs exceeding that observed in many South African populations but more closely aligned with the high levels of inbreeding noted in the inbred populations of Hluhluwe-Imfolozi Park and Addo Elephant National Park [5]. Instances of high levels of the fraction of the genome in runs of homozygosity (measured as FROH) have been recently reported for the Indian tiger [66] and in African for black rhinos from South Africa [67], as well as the plain zebra populations in East Africa [68]. In these cases, the shared characteristics are small population sizes, geographic isolation, and the relatively recent occurrence of inbreeding, culminating in an excessive frequency of extended ROH segments. Therefore, the pronounced prevalence of ROHs observed within Mozambique’s buffalo population, which exceeds the levels observed in South African counterparts and mirrors those seen in other populations of critically endangered species, indicates recent and pervasive inbreeding in Mozambique. Both the utilised metrics, g2, and FROH, consistently indicate a comparable degree of inbreeding across all populations. The co-occurrence of such elevated FROH values and statistically significant g2 signals points towards a scenario wherein an excess burden of recessive deleterious alleles is likely present. This, in turn, could trigger a fitness depression within the population.
Implication for management and conservation
From a conservation genetic perspective, the widespread presence of large and numerous ROHs is a cause for concern as it can have a dramatic impact on the survival of S. c. caffer in Mozambique. Studies on the wild Soay sheep have shown that individuals with long ROHs are more likely to succumb during their first winter, owing to the high frequency of deleterious mutations in these regions [69]. In the African buffalo, van Hooft et al. [31] highlighted the occurrence of a latitudinal cline of recessive alleles, which might negatively affect male body condition and bovine tuberculosis resistance. This suggests that a massive loss of animals in Mozambique cannot be ruled out, especially in the face of new diseases or environmental changes. In addition, the possibility of pathogens being introduced along with translocated individuals is a real risk [70], and the effect on a genetically depauperate population is hard to predict. Furthermore, due to the lack of corridors for gene flow, the progeny of introduced individuals could be susceptible to genetic diversity loss due to founder effects and small population size [71]. All these factors could lead to inbreeding depression and, ultimately, jeopardize the success of any translocation effort in the long run. Finally, it is interesting to note that van Hooft et al. [31, 72] also found that male-deleterious alleles could be epigenetically suppressed in a large fraction of animals. Therefore, future work focused on the management and conservation of buffalo populations should also explore epigenetic mutations as potential markers for past and present environmental stress events, as well as indicators of individual physiological conditions [73]. This information has the potential to enhance conservation plans by considering the capacity of organisms to rapidly adapt to environmental changes, thus improving the conservation of wild populations [73].
Conclusions
We found evidence of S. c. caffer decline in Mozambique dating back 100,000 years. This decline is congruent with previous studies focusing on the African buffalo in the South African region. According to our data, this decline persisted until recent times, culminating in a contemporary Ne estimate that is very low, as expected for populations that have experienced conflict with human activities and habitat loss. The observed loss of genetic diversity could expose the species to vulnerability to pathogens and possibly local extinction. The African buffalo plays a crucial role as an ecosystem engineer by preparing habitat for other herbivores. Therefore, preserving robust and resilient buffalo populations is vital to maintain a functional ecosystem. One potential solution to reduce long genomic regions in homozygosity is to introduce animals from compatible populations to increase genetic diversity. Despite successful reintroduction in Mozambique, such as in Gorongosa National Park, our findings suggest that buffaloes from Mozambique, including those from southern localities that likely originated from South Africa [74], have suffered from genetic erosion and possess long and abundant ROHs. The widespread inbreeding within S. c. caffer, also outside Mozambique [5], with potential source populations characterised by a high level of homozygosity, hence possibly a carrier of genetic load of recessive alleles, represents a significant issue that can determine the success or failure of any translocation program and should be taken seriously into consideration. We recommend that future translocation efforts should consider this aspect and conduct a survey of genome-wide homozygosity in all potential donor populations before initiating a restocking intervention. This will increase the likelihood of success in preserving genetic diversity and maintaining a functional ecosystem.
Data availability
All sequence data presented here (fastq format) have been deposited into the NCBI SRA database (accession number: PRJNA971211).
References
Daskin JH, Pringle RM. Warfare and wildlife declines in Africa’s protected areas. Nature. 218;553:328–32.
Roque DV, Macandza VA, Zeller U, Starik N, Göttert T. Historical and current distribution and movement patterns of large herbivores in the Limpopo National Park, Mozambique. Front Ecol Evol. 2022;10:978397.
Pacifici M, Di Marco M, Watson JEM. Protected areas are now the last strongholds for many imperiled mammal species. Conserv Lett. 2020;13:e12748.
de Jager D, Glanzmann B, Möller M, Hoal E, van Helden P, Harper C, Bloomer P. High diversity, inbreeding and a dynamic pleistocene demographic history revealed by African Buffalo Genomes. Sci. Rep. 2021; 11(1):4540 (2021).
Quinn L, Garcia-Erill G, Santander C, Brüniche-Olsen A, Liu X, Sinding MS, et al. Colonialism in South Africa leaves a lasting legacy of reduced genetic diversity in Cape buffalo. Mol Ecol. 2023;32:1860–74.
Talenti A, Wilkinson T, Cook EA, Hemmink JD, Paxton E, Mutinda M et al. Continent-wide genomic analysis of the African buffalo (Syncerus caffer). bioRxiv 2023.11.12.566748; doi: https://doi.org/10.1101/2023.11.12.566748.
Smitz N, Cornélis D, Chardonnet P, Caron A, de Garine-Wichatitsky M, Jori F, et al. Genetic structure of fragmented southern populations of African Cape buffalo (Syncerus caffer caffer). BMC Evol Biol. 2014;14:203.
Mavhunga C, Spierenburg MA. Finger on the pulse of the fly: hidden voices of colonial anti-tsetse science on the rhodesian and Mozambican borderlands, 1945–1956. S Afr Hist J. 2007;58:117–41.
IUCN SSC Antelope Specialist Group. 2019. Syncerus caffer. In: The IUCN Red List of Threatened Species 2019: e.T21251A50195031. https://dx.doi.org/10.2305/IUCN.UK.2019-1.RLTS.T21251A50195031.en. Accessed on 13 April.
Hatton J, Oglethorpe J, Couto M. Biodiversity and war: a case study of Mozambique. Washington, D.C.: Biodiversity Support Program; 2001.
Stalmans ME, Massad TJ, Peel MJ, Tarnita CE, Pringle RM. War-induced collapse and asymmetric recovery of large-mammal populations in Gorongosa National Park, Mozambique. PLoS ONE. 2019;14:e0212864.
Ripple WJ, Newsome TM, Wolf C, Dirzo R, Everatt KT, Galetti M, et al. Collapse of the world’s largest herbivores. Sci Adv. 2015;1:e1400103.
Lacher TE, Davidson AD, Fleming TH, Gómez-Ruiz EP, McCracken GF, Owen-Smith N, et al. The functional roles of mammals in ecosystems. J Mammal. 2019;100:942–64.
Venter JA, Prins HH, Balfour DA, Slotow R. Reconstructing grazer assemblages for protected area restoration. PLoS ONE. 2014;9:e90900.
Rouet-Leduc J, Pe’er G, Moreira F, Bonn A, Helmer W, Shahsavan Zadeh SAA, et al. Effects of large herbivores on fire regimes and wildfire mitigation. J Appl Ecol. 2021;58:2690–702.
Estes RD. The behavior guide to African mammals, including hoofed mammals, carnivores and primates. University of California Press; 1991.
Prins HHT. Ecology and behaviour of the African buffalo: social inequality and decision making. Chapman & Hall; 1996.
Fischer J, Lindenmayer DB. An assessment of the published results of animal relocations. Biol Conserv. 2000;96:1–11.
Morris SD, Brook BW, Moseby KE, Johnson CN. Factors affecting success of conservation translocations of terrestrial vertebrates: a global systematic review. Glob Ecol Conserv. 2021;28:e01630.
Weeks AR, et al. Conserving and enhancing genetic diversity in translocation programs. Advances in reintroduction biology of Australian and New Zealand Fauna. 1st ed. Csiro Publishing; 2015. pp. 127–40.
Luikart GS, Amish J, Winnie J, Beja-Pereira A, Godinho R, Allendorf FW, et al. High connectivity among argali sheep from Afghanistan and adjacent countries: inferences from neutral and candidate Gene Microsatellites. Conserv Genet. 2011;12:921–31.
Hohenlohe PA, Funk WC, Rajora OP. Population Genomics for Wildlife Conservation and Management. Mol Ecol. 2020;30:62–82.
Theissinger K, Fernandes C, Formenti G, Bista I, Berg PR, Bleidorn C, et al. How genomics can help biodiversity conservation. Trends Genet. 2023;39(7):545–59.
Luikart G, Kardos M, Hand BK, Rajora OP, Aitken SN, Hohenlohe PA. Population Genomics: advancing understanding of Nature. In: Rajora O, editor. Population Genomics. Population Genomics. Cham: Springer; 2018. pp. 3–79.
Schoville SD, Bonin A, François O, Lobreaux S, Melodelima C, Manel S. Adaptive genetic variation on the landscape: methods and cases. Annu Rev Ecol Evol Syst. 2012;43(1):23–43.
Väli Ü, Einarsson A, Waits L, Ellegren H. To what extent do microsatellite markers reflect genome-wide genetic diversity in natural populations? Mol. Ecol. 2008;17:3808–17.
Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet. 2003;4:981–94.
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet. 2016;17:81–92.
Puritz JB, Matz MV, Toonen RJ, Weber JN, Bolnick DI, Bird CE. Demystifying the RAD fad. Mol Ecol. 2014;23(24):5937–42.
Smitz N, Berthouly C, Cornélis D, Heller R, Van Hooft P, Chardonnet P, et al. Pan-african genetic structure in the African buffalo (Syncerus caffer): investigating intraspecific divergence. PLoS ONE. 2013;8(2):e56235.
van Hooft P, Getz WM, Greyling BJ, Zwaan B, Bastos ADS. A continent-wide high genetic load in African buffalo revealed by clines in the frequency of deleterious alleles, genetic hitchhiking and linkage disequilibrium. PLoS ONE. 2021;16(12):e0259685.
Franchini P, Monné Parera D, Kautt AF, Meyer A, Quaddrad. A new high-multiplexing and PCR duplicate removal ddRAD protocol produces novel evolutionary insights in a nonradiating cichlid lineage. Mol Ecol. 2017;26:2783–95.
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest radseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE. 2012;7:e37135.
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: an analysis tool set for population genomics. Mol Ecol. 2013;22:3124–40.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinform. 2009;25:1754–60.
Maruki T, Lynch M. Genotype calling from population-genomic sequencing data. G3-Genes. Genome Genet. 2017;7:1393–404.
Rochette NC, Rivera-Colón AG, Catchen JM. Stacks 2: Analytical methods for paired‐end sequencing improve radseq‐based population genomics. Mol Ecol. 2019;28:4737–54.
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C. Weir BS a high-performance computing toolset for relatedness and principal component analysis of SNP Data. Bioinform. 2012;28(24):3326–8.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 1009;19:1655–64.
Alexander DH, Lange K. Enhancements to the admixture algorithm for individual ancestry estimation. BMC Bioinform. 2011;12:246.
Liu CC, Shringarpure S, Lange K, Novembre J. Exploring population structure with admixture models and principal component analysis. Methods Mol. Biol. 2020; 2090:67–86.
Neuditschko M, Khatkar MS, Raadsma HW, NetView:. A high-definition network-visualization Approach to Detect Fine-Scale Population structures from genome-wide patterns of variation. PLoS ONE. 2012;7(10):e48375.
Steinig EJ, Neuditschko M, Khatkar MS, Raadsma HW, Zenger KR. Netview p: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol Ecol Resour. 2016;16(1):216–27.
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA. Diversity: an r package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol. 2013;4:782–88.
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D. Directional genetic differentiation and relative migration. Ecol Evol. 2016;6:3461–75.
Weir BS, Cockerham CC. Mixed self and random mating at two loci. Genet Res. 1973;21:247–62.
David P, Pujol B, Viard F, Castella V, Goudet J. Reliable selfing rate estimates from imperfect population genetic data. Mol Ecol. 2007;16:2474–87.
Stoffel MA, Esser M, Kardos M, Humble E, Nichols H, David P, et al. INBREEDR: an R package for the analysis of inbreeding based on genetic markers. Methods Ecol Evol. 2016;7:1331–9.
Szulkin M, Bierne N, David P. Heterozygosity-fitness correlations: a time for reappraisal. Evol. 2010;64:1202–17.
Biscarini F, Cozzi P, Gaspa G, Marras G, detectRUNS. Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. R package version 0.9.6, https://CRAN.R-project.org/package=detectRUNS (2019).
Liu X, Fu YX. Stairway plot 2: demographic history inference with folded SNP frequency spectra. Genome Biol. 2020;21:280.
Chen L, Qiu Q, Jiang Y, Wang K, Lin Z, Li Z, et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science. 2019;364(6446):eaav6202.
Barbato M, Orozco-terWengel P, Tapio M, Bruford MW. SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front Genet. 2015;6:109.
Sved JA, Feldman MW. Correlation and probability methods for one and two loci. Theor Popul Biol. 1973;4:129–32.
Sved JA, McRae AF, Visscher PM. Divergence between human populations estimated from linkage disequilibrium. Am J Hum Genet. 2008;83:737–43.
Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475(7357):493–6.
Boitard S, Rodríguez W, Jay F, Mona S, Austerlitz F. PLoS Genet. 2016;12(3):e1005877. Inferring Population Size History from Large Samples of Genome-Wide Molecular Data - An Approximate Bayesian Computation Approach.
Faith JT. Late pleistocene and holocene mammal extinctions on continental Africa. Earth-Sci Rev. 2013;128:105–21.
Koch PL, Barnosky AD. Late Quaternary extinctions: state of the debate. Annu. Rev. Ecol. Syst. 2006; 37(1):215–50 (2006).
Bibi F, Cantalapiedra JL. Plio-Pleistocene African megaherbivore losses associated with community biomass restructuring. Science. 2023;380:1076–80.
Andermann T, Faurby S, Turvey ST, Antonelli A, Silvestro D. The past and future human impact on mammalian diversity. Sci Adv. 2020;6:eabb2313.
Winterbach HEK. Research review: the status and distribution of Cape buffalo Syncerus caffer caffer in southern Africa. S Afr J Wildl Res. 1998;28:82–8.
Cumming DHM, Mackie C, Magane S, Taylor RD. Aerial census of large herbivores in the Gorongosa National Park and the Marromeu area of the Zambezi Delta in Mozambique: June, 1994. IUCN, DNFFB and WWF. In: Direcção Nacional de Florestas e Fauna Bravia 1994. Gorongosa– Marromeu: management plan for integrated conservation and development, 1995–1999. Part 2. DNFFB, Maputo, Mozambique.
Brüniche-Olsen A, Kellner KF, Anderson CJ, DeWoody JA. Runs of homozygosity have utility in mammalian conservation and evolutionary studies. Conserv Genet. 2018;19:1295–307.
Ceballos FC, Joshi PK, Clark DW, Ramsay M, Wilson JF. Runs of homozygosity: Windows into population history and trait architecture. Nat Rev Genet. 2018;19:220–34.
Khan A, Patel K, Shukla H, Viswanathan A, van der Valk T, Borthakur U, et al. Genomic evidence for inbreeding depression and purging of deleterious genetic variation in Indian tigers. Proc Natl Acad Sci U S A. 2021;118(49):e2023018118.
Sánchez-Barreiro F, De Cahsan B, Westbury MV, Sun X, Margaryan A, Fontsere C, et al. Historic sampling of a vanishing beast: population structure and diversity in the black rhinoceros. Mol Biol Evol. 2023;40(9):msad180.
Larison B, Kaelin CB, Harrigan R, Henegar C, Rubenstein DI, Kamath P, et al. Population structure, inbreeding and stripe pattern abnormalities in plains zebras. Mol Ecol. 2021;30:379–90.
Stoffel MA, Johnston SE, Pilkington JG, Pemberton JM. Mutation load decreases with haplotype age in wild Soay sheep. Evol Lett. 2021;5:187–95.
Gonçalves L, Teixeira M, Rodrigues A, Mendes N, Matos C, Pereira C, et al. Molecular detection of bartonella species and haemoplasmas in wild African buffalo (Syncerus caffer) in Mozambique, Africa. Parasitol Open. 2018;4:E15.
Jamieson IG. Founder effects, inbreeding, and loss of genetic diversity in four avian reintroduction programs. Conserv Biol. 2010;25:115–23.
van Hooft P, Dougherty ER, Getz WM, Greyling BJ, Zwaan BJ, Bastos ADS. Genetic responsiveness of African buffalo to environmental stressors: a role for epigenetics in balancing autosomal and sex chromosome interactions? PLoS ONE. 2018;13(2):e0191481.
Rey O, Eizaguirre C, Angers B, Baltazar-Soares M, Sagonas K, Prunier JG, Blanchet S. Linking epigenetics and biological conservation: towards a conservation epigenetics perspective. Funct Ecol. 2020;34:414–27.
Agreco. National census of wildlife in Mozambique: final report. Mozambique, Ministerio da Agricultura; 2008.
Acknowledgements
The authors are grateful to Prof. Mauro M. Colombo, Dr. Tiziano Cirillo (BioForMoz project Manager) and Dr. Elisa Taviani (BioForMoz Scientific Coordinator, DISTAV, University of Genova, Italy) as well as to Dr. Lucilia Chuquela, Director of the National History Museum of Maputo (NHM-UEM) - Eduardo Mondlane University, to Dr. Daniela de Abreu Dir. Adj. for Research of the Natural History Museum (NHM-UEM) and to the whole staff. We are also grateful to Suzana Macuvele for her assistance in the laboratory work. We would like to thank the Mozambique Wildlife Alliance for providing us the samples from Gilè National Park and Coutada 9. We are especially thankful to Dr. Marc Stalman, Director of Science of the Gorongosa National Park, and to the staff of the Marromeu National Reserve, Gilè National Park, and Coutada 9 for their cooperation and support.
Funding
This research was supported by the projects “Training in Biodiversity and Biotechnology for sustainable development” (AID 11096) and BioForMoz “Support for Environmental Research” (AID 12089) managed by the Department of Biomedical Sciences, University of Sassari, Italy, and funded by the Italian Agency for Development Cooperation, Italy.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
P.C. and S.S. conceived the research. P.C. and M.D.C. drafted the manuscript. S.S. handled relations with Mozambican institutions, contributed to procuring funds and coordinated sample collection. C.M.B., L.C.B.G.N., F.C.M. and J.S.A. collected samples and S.S. performed DNA extraction. P.F., A.M. and N.O. performed library preparation and provided sequencing support. P.C., M.D.C., P.F., F.P. and G.S. performed the analyses. All authors contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the research and training committee of the projects AID 11096 “Training in Biodiversity and Biotechnology for sustainable development” and BioForMoz AID 12089 “Support for Environmental Research”, funded by the Italian Development Cooperation Agency, in collaboration with the Natural History Museum of Maputo, Biotechnology Center of the E. Mondlane University of Maputo, Mozambique, the Department of Biology and Biotechnologies “Charles Darwin” Sapienza University of Rome and CNR-IRET, Italy. The research protocol of this study has been reviewed and approved by the Scientific Board of the Biotechnology Centre, Eduardo Mondlane University, Maputo, Mozambique: CB-UEM/COMETH 007 /2017. It complies with the OIE-World Organisation for Animal Health, requirements regarding animal experimentation, care, and welfare. The blood samples used in this study are stored at the BioBank of the Natural History Museum of Maputo (NHM-UEM) and at the Biotechnology Centre of the Eduardo Mondlane University (CB-UEM), Maputo, Mozambique. Blood samples were collected during past veterinary operations within the Gorongosa National Park (research permit: PNG/DSCi/C156/2019) and from animals immobilized under the mandate granted by the National Administration of Conservation Areas (ANAC) to the vet team of the Mozambique Wildlife Alliance (MWA) in accordance with Mozambican legislation.
Consent for publication
Not applicable.
Competing interests
The author(s) declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Colangelo, P., Di Civita, M., Bento, C.M. et al. Genome-wide diversity, population structure and signatures of inbreeding in the African buffalo in Mozambique. BMC Ecol Evo 24, 29 (2024). https://doi.org/10.1186/s12862-024-02209-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-024-02209-2