
Abstract
Background
Selection pressure exerted by pathogens can influence patterns of genetic diversity in the host. In the immune system especially, numerous genes encode proteins involved in antagonistic interactions with pathogens, paving the way for coevolution that results in increased genetic diversity as a consequence of balancing selection. The complement system is a key component of innate immunity. Many complement proteins interact directly with pathogens, either by recognising pathogen molecules for complement activation, or by serving as targets of pathogen immune evasion mechanisms. Complement genes can therefore be expected to be important targets of pathogen-mediated balancing selection, but analyses of such selection on this part of the immune system have been limited.
Results
Using a population sample of whole-genome resequencing data from wild bank voles (n = 31), we estimated the extent of genetic diversity and tested for signatures of balancing selection in multiple complement genes (n = 44). Complement genes showed higher values of standardised β (a statistic expected to be high under balancing selection) than the genome-wide average of protein coding genes. One complement gene, FCNA, a pattern recognition molecule that interacts directly with pathogens, was found to have a signature of balancing selection, as indicated by the Hudson-Kreitman-Aguadé test (HKA) test. Scans for localised signatures of balancing selection in this gene indicated that the target of balancing selection was found in exonic regions involved in ligand binding.
Conclusion
The present study adds to the growing evidence that balancing selection may be an important evolutionary force on components of the innate immune system. The identified target in the complement system typifies the expectation that balancing selection acts on genes encoding proteins involved in direct interactions with pathogens.
Similar content being viewed by others

Background
The complement system is an evolutionarily ancient branch of innate immunity that occurs in both invertebrates and vertebrates. In mammals, it consists of more than 40 soluble and membrane-bound proteins. It provides manifold utility in immune defence, including recognition, opsonization and killing of pathogens, clearance of apoptotic cells, induction of inflammation, and priming of other innate and adaptive responses [1, 2]. Depending on the initial cue, the complement cascade can be activated via the classical, lectin and alternative pathways. All three pathways converge at complement component 3 (C3) and may ultimately lead to the insertion of a membrane-attack complex (MAC) into the surface of the pathogen to cause its lysis, along with other effects in the host (see Fig. 1 for details).

Overview of the complement cascade. The cue to signal the complement cascade can be varied and helps determine the pathway that is activated. The classical pathway (CP) is primarily triggered when C1Q recognises and binds to the Fc region of antibodies or pentraxins bound to antigens. The lectin pathway (LP) is initiated when lectins of the complement system bind carbohydrates on cell surfaces. The alternative pathway (AP) is active constantly at low levels due to spontaneous hydrolysis of complement component C3, leading to indiscriminate C3b deposition on nearby cell surfaces. This is immediately cleared by regulators of complement activation (RCA) present on host cells, preventing complement activation. Microbes lack RCA proteins and thereby activate the alternative pathway. Thus, only the lectin pathway requires specific recognition of MAMPs. In all cases, C3 convertases are produced once the pathway is triggered which in turn cleaves complement component C5, with all pathways converging at this stage. Ultimately, a membrane-attack complex (MAC) is assembled and inserted on the pathogen surface, and this pore lyses the pathogen cell. In addition, complement activation leads to opsonization and activation of other inflammatory and immune pathways. The figure is for representative purposes and only a single role for complement proteins with multiple functions is presented here. Text in brown indicate genes that were not included in this study. Text in red indicate the gene with signature of balancing selection in the bank vole. Created with BioRender.com.
Immune genes, in particular those encoding proteins that are involved in direct and specific interactions with pathogen ligands, are considered prime candidates for host-pathogen coevolution [3,4,5]. Complement proteins interact with pathogen molecules in multiple ways. First, some complement proteins act as sensors of pathogens by recognising microbe-associated molecular patterns (MAMPs), triggering activation of the complement cascade. Such sensors include the pattern recognition molecules (PRMs) of the lectin pathway (mannose-binding lectins, collectins, and ficolins) and C-reactive protein (CRP). For instance, the PRMs mannose-binding lectins (MBL) recognise carbohydrates on pathogen cell surfaces, activating the lectin pathway [6]. The complement system can also be activated in other ways, including by recognition of antibodies deposited on pathogens (classical pathway) and by “missing self-recognition” (alternative pathway), but these do not involve direct and specific interactions between complement proteins and pathogen ligands (see Fig. 1 for details).
Second, proteins of the complement system are often targets of pathogen immune evasion factors. Common strategies employed by pathogens include direct inhibition of complement proteins and the recruitment or mimicking of endogenous regulators of complement activation (RCA) [7]. For example, species of the Lyme disease spirochaete Borrelia burgdorferi sensu lato complex employ a multi-pronged (and often redundant) approach to prevent destruction by the complement system [8, 9]. Multiple Borrelia proteins belonging to the complement regulator-acquiring surface proteins (CRASPs) recruit RCA protein complement factor H (FH) and prevent complement activation [10]. Borrelia also produce outer surface proteins and complement protein mimics that directly interact with and inhibit complement proteins or MAC formation [11,12,13]. Other pathogens such as Streptococcus pyogenes, Neisseria meningitidis and Staphylococcus aureus use similar strategies to hijack the complement system to their advantage and escape killing [14,15,16,17].
As described above, many of the complement proteins are involved in direct physical and antagonistic interactions with specific pathogen ligands. As such, complement genes can be expected to be involved in coevolution with pathogens [4, 18, 19]. This view is supported by studies showing complement system genes to be under positive selection across species (i.e., selection driving divergence between species), as demonstrated by genome-wide scans as well as analyses of specific genes. For example, a study looking at genome-wide patterns of positive selection across different mammalian species identified genes involved in the complement system to be significantly enriched for targets of positive selection [20]. Similarly, a comprehensive study looking at positive selection on the complement system across primates identified positively selected sites in numerous complement genes, primarily located in protein domains at the interface of pathogen interactions [5].
Besides positive selection across species, host-pathogen interactions like antagonistic coevolution can also lead to balancing selection, resulting in high genetic diversity within species [21]. However, only a handful of studies have investigated balancing selection in specific complement genes. In the case of human gene MBL2, which encodes a PRM, high diversity in its sequence is observed; however, whether this diversity is a consequence of balancing selection is still debatable [22,23,24]. Another study looking at human complement gene C6, which is often a target of immune evasion by pathogens, found evidence of balancing selection across different human populations [25]. However, a comprehensive analysis examining balancing selection across all complement genes has not been attempted in any species.
In this study, we investigate the extent of balancing selection in complement system genes of bank voles (Myodes glareolous) using whole-genome resequencing data. We first identify complement genes with signatures of balancing selection in coding and/or noncoding regions as indicated by BetaScan2 [26]. Second, for genes picked up by BetaScan2 we used a classical neutrality test (the HKA test;[27]) to test for signatures of balancing selection across the whole protein-coding sequence (CDS) [28]. Finally, we look for factors influencing which complement genes have signatures of balancing selection as indicated by BetaScan2. In this case, we hypothesise that complement genes encoding proteins that participate in direct and specific interactions with pathogens, such as those involved in pathogen recognition (i.e., PRMs) or targets of immune evasion, are more likely to be under balancing selection (henceforth collectively referred to as “candidates of balancing selection”) compared to those that are not involved in similar interactions.
Results
We curated complement genes from literature and public databases, of which 44 complement genes were annotated in our bank vole genome assembly that could be used for analyses of nucleotide diversity and balancing selection. A few additional complement genes were found in the assembly but were excluded due to annotation issues, insufficient coverage as compared to the mouse CDS (< 50%) or poor alignment with mouse CDS (Additional File 1: Table S1).
We used whole genome resequencing data from 31 bank voles to provide estimates of genetic diversity and balancing selection. To shortlist genes with signatures of balancing selection, we adopted an outlier analysis for the β estimates provided by BetaScan2 [26]. β is a summary statistic based on allele frequency correlations between neighbouring SNPs, where elevated values are indicative of balancing selection due to genetic hitchhiking and drift of neutral variants. We standardised the β values with locus-specific mutation rate (βstd), which was calculated in 2 kb windows across the whole genome (see Methods for details). The maximum value of βstd in each gene was identified (βstd.max). Four genes (CFHR1, FCNA, ITGAX and HC; Table 1) were outliers for βstd.max, with βstd.max ≥ 95th percentile of a set of control genes (n = 8465; 95th percentile of control βstd.max = 7.03).
We then used the maximum likelihood Hudson-Kreitman-Aguadé test (MLHKA) [27], which tests for selection by comparing polymorphism and divergence to a neutral scenario. We used MLHKA to test for signatures of balancing selection across full coding sequences. We limited the MLHKA tests to the four genes that were outliers of βstd.max. Of these, only FCNA (ficolin A) was significant by MLHKA, with selection increasing the diversity by 2.72x (as indicated by the selection parameter k; Table 1). We thus focused further analyses on this gene.
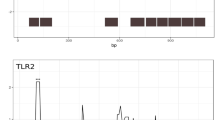
To identify gene regions that were targets of balancing selection in FCNA, we plotted βstd, nucleotide diversity (π), and Tajima’s D along the gene, and calculated linkage disequilibrium (LD) using the normalised LD coefficient D’ (Fig. 2). The parameters βstd, π and Tajima’s D primarily reflect long-term balancing selection and are expected to be elevated under this scenario [29,30,31]. Tajima’s D is based on comparing two different estimates of genetic variation; nucleotide diversity and number of segregating sites. Tajima’s D is sensitive to both selection and demographic processes: D > 0 when there is an excess of intermediate frequency alleles as a result of balancing selection or demographic processes like population contraction; D < 0 when there is an excess of rare alleles, for example, after a selective sweep or population expansion; D = 0 under neutral evolution and constant population size [32]. To visualise the number and frequency of different haplotypes present in the population, we also constructed a haplotype network bordering the most polymorphic region using the whole-genome resequencing data complemented with Sanger sequencing data from an additional 30 bank voles. Under balancing selection, one would expect to see at least two well-separated haplotype groups.

Sliding window analysis of βstd, π, and Tajima’s D, haplotype network, and LD plot for FCNA. Red dashed line indicates the 95th percentile of β std max of control genes. The haplotype network was constructed from the region marked with black square. Gene structure below sliding window plots show protein domains as predicted by Pfam [33]. The LD plot was constructed for the whole gene using D’, with LD blocks shown. D’ refers to the normalised values of the coefficient of LD, with high values indicating strong LD. Gene structure corresponding to the LD plots show introns in light blue and exons in dark blue
FCNA showed high values of π and Tajima’s D almost throughout the entirety of its short length (Fig. 2). Twelve SNPs in FCNA had βstd above the 95th percentile of βstd.max of a set of 8465 control genes, all in the region from exons 6 to 8. It is notable that these exons encode the fibrinogen C domain, which is known to recognise carbohydrates with N-acetylation markers. As a possible consequence of its short length, the whole gene formed a single LD block. A haplotype network based on ~ 700 bp covering a part of the peak in nucleotide diversity showed two well separated haplotype groups. Similar analyses of the other three genes that were outliers for βstd.max are presented in Supplementary Figure S1 (Additional File 2).
We hypothesised that complement genes that are engaged in direct and specific interactions with pathogens, such as those activating the complement cascade by recognising pathogens or are subject to immune evasion, would be more likely to evolve under balancing selection. To test this, we compared βstd.max of complement genes that were candidates of balancing selection (i.e., complement genes encoding PRM/targets of immune evasion; n = 25) and other complement genes (n = 12). There was no difference in βstd.max between these two categories of complement genes (Mann-Whitney U test: p = 0.86; Fig. 3). However, complement genes overall (n = 37) showed elevated βstd.max as compared to control genes (Mann-Whitney U test test: p = 0.014).

Violin plots of βstd.max for non-complement control genes (n = 8465), complement genes that are candidates of balancing selection (i.e., involved in pathogen recognition or are targets of immune evasion; n = 25), and other complement genes (n = 12)
Discussion
In this study, we investigated the extent of balancing selection in complement system genes of bank voles. Based on analyses of whole gene sequences, we found signatures of balancing selection in four complement genes, as indicated by the βstd.max outlier analysis (FCNA, CFHR1, HC, ITGAX). However, only FCNA showed a significant departure from neutrality as assessed by the MLHKA test. FCNA also showed divergent haplotype groups at intermediary frequencies. Taken together, this is consistent with evolution under balancing selection at FCNA. We acknowledge the risk for false positives when testing for signatures of selection in multiple genes [34] and note that the MLHKA test for FCNA does not pass the Bonferroni-adjusted threshold (which would be α = 0.05/37 = 0.0014). However, the fact that both BetaScan2 and the MLHKA test, which are based on partly different information, detected signatures of balancing selection in FCNA indicates that it is not a false positive. Ultimately, tests for signatures of selection should be complemented with functional analyses of alternative alleles [35].
FCNA (ficolin A) in bank voles is orthologous to FCN2 in humans, with both genes encoding a lectin PRM that recognises carbohydrates. Information from human and mice indicate that it forms homotrimers that preferentially recognise N-acetylated carbohydrates such as N-acetylglucosamine (GlcNAc) and N-acetyl galactosamine [36,37,38]. In microbes, GlcNAc is fundamental to forming chitin in fungal cell walls and the peptidoglycan layer of bacterial cell walls, a component abundant in gram-positive bacteria and present to a smaller extent also in gram-negative bacteria. In conjunction with this information, FCNA knockout mice are impaired in their ability to respond to certain pathogens such as the gram-positive bacteria Streptococcus pneumoniae and the fungus Aspergillus fumigatus [39,40,41], highlighting the importance of the protein to the immune response.
Previous studies of balancing selection on complement system genes are few and have been limited to humans. FCNA, is functionally analogous to MBL2, which is contested to be under balancing selection in humans [22,23,24]. However, unlike in humans where there is some evidence for balancing selection on C6 [25], we were unable to corroborate the presence of balancing selection in any of the bank vole genes encoding complement components deposited on pathogens.
We also looked for general differences between different categories of complement genes. Balancing selection due to pathogen-mediated pressure is expected to act on genes that are the focal points of host-pathogen interactions. A previous study of balancing selection in bank voles by Lundberg et al. [19] analysed genes of innate immune signalling pathways. Genes encoding Pattern Recognition Receptors (PRR) – specifically, those recognising microbial cell wall components – displayed higher βmax values than genes involved in downstream signal transduction. Similarly, a study by Cagliani et al. [5] on the complement system of primates found that many genes under positive selection directly interacted with pathogens. Consequently, we hypothesised that certain complement genes (those involved in pathogen recognition or are targets of immune evasion), would show increased signatures of balancing selection. Complement genes overall showed higher βstd.max than control genes. However, in contrast to patterns observed in analyses of PRR signalling pathways [19], complement genes involved in direct interactions with pathogens did not have higher βstd max than other complement genes.
Analysis of LD can be informative in understanding the time since a selection event. When an advantageous allele arises de novo in a population or is favoured from standing variation, it increases in frequency and can often sweep variants at neighbouring linked neutral sites, reducing heterozygosity in these regions. During the early stages of balancing selection, this results in strong LD over large genomic regions, due to insufficient time for recombination to act [31]. However, given sufficient evolutionary time, the frequency of the advantageous allele stabilises at an intermediate level and the regions of LD begin to reduce due to recombination. In our data, we notice that FCNA shows LD, despite signatures of balancing selection indicated by MLHKA and BetaScan2, which are both designed to detect intermediate to ancient selection events [29, 30]. The extremely short length of FCNA (around 3 kb) can potentially explain the lack of recombination observed for this gene.
The targets of balancing selection in immune genes are often expected to occur in coding regions, specifically at sites that are important for interaction with pathogens [3, 5]. In the case of FCNA, the peak in βstd spans three exons coding for the ligand binding region. This exemplifies the expectation that targets of balancing selection within the immune system occur in coding regions that recognise pathogens. However, further analyses are required to attribute the function of polymorphisms found in this gene.
Conclusions
In this study, we demonstrate signatures of balancing selection on FCNA, a complement gene encoding a protein involved in direct interactions with pathogens. Further studies to determine the functional effects of the different FCNA alleles are warranted. The present study is the most comprehensive analysis of balancing selection in the complement system carried out thus far, and adds to the growing evidence that balancing selection is an important evolutionary force not only on genes of the adaptive immune system (e.g. MHC and other genes involved in antigen presentation; [42, 43]), but also on different components of the innate system, such as pattern-recognition receptors, defensins, antiviral restriction factors, and the complement system [19, 44,45,46,47].
Methods
Complement system genes
The primary source for complement system genes was the KEGG [48] pathway “Complement and coagulation cascade” (mmu:04610). Genes pertinent to only the complement system were retained, based on the review by Ricklin et al., [1]. To expand the list to more recently annotated genes, additional information from the Mouse Genome Database (MGD; [49]) in Mouse Genome Informatics (MGI) was included, after verification of function from literature. In total, n = 60 genes were selected. Orthologs of these mouse genes that were annotated in our bank vole genome assembly [19] and covered at least ~ 50% of the mouse coding sequence were retained for analysis. For four genes, annotations were curated manually in Web Apollo [50]. This yielded 44 bank vole complement genes for further analyses.
Whole-genome resequencing
Whole-genome resequencing data from 31 bank voles [19] was used to obtain sequence and polymorphism information. The study sampled adult bank voles collected at Revingehed, a 43 km2 area 20 km east of Lund, in southern Sweden. Samples were collected at six different sites (2–8 samples per site, depending on area of each site). The maximum distance between sites was 7 km, and there are no geographical barriers (e.g. a river) between sites. Paired-end reads (Illumina Hiseq X, 2 × 150 bp) were mapped to the reference genome with BWA [51] with an average coverage of 44x and variants called with freebayes [52]. The raw variants were filtered in a number of steps, including for low quality, coverage and repeat overlap (see [19] for details). Haplotypes were inferred using Beagle version 4.1 [53] and coding sequences were extracted based on annotations. Sequences were edited in Geneious Prime 2020.1.1 (Biomatters) to remove gaps and errors in alignment. The curated sequence data was used to generate basic genetic diversity estimates of complement system genes using the batch mode of DnaSP 6 [54] (Additional File 1: Table S2).
Tests for signatures of selection
We used BetaScan2 [26] to identify localised signatures of balancing selection in coding and non-coding regions. When a SNP is under balancing selection, it leads to correlations in allele frequencies between itself and neighbouring neutral SNPs, and these correlations are summarised as the β statistic, where high values indicate balancing selection. To account for differences in mutation rate across the genome, we estimated the population-scaled mutation rate using Watterson’s θ [55] in windows of 10 kb from the genotype data. For this purpose, we used bedtools 2.29.2 [56] to generate genomic intervals and PopGenome 2.7.5 [57] to calculate θ in each window. To obtain per-base pair values, the estimate in each window was divided by the window size minus the combined length of gaps and annotated repeats within the same window. BetaScan2 was run in 2 kb windows along the genome, with folded allele frequency spectrum and minimum minor allele frequency of 0.15. In each window, β values standardised by θ were calculated (βstd) and the highest βstd in any window along a gene was used (βstd.max). β values for SNPs within 1 kb of an insertion or deletion, as called by DELLY [58], were removed to avoid spurious signals. To identify outlier βstd.max values, the distribution of βstd.max in a set of control genes (n = 8465) was determined, as described in Lundberg et al., [19]. Complement genes with βstd.max values ≥ 95th percentile (βstd.max = 7.03) of the control genes’ βstd.max values were considered for further analyses. GNU Parallel was used to optimise the efficiency of the analysis [59].
To test for signatures of balancing selection reflected in whole coding regions we used the Hudson-Kreitman-Aguadé test (HKA; [60]) on genes that were outliers for βstd.max. The HKA test makes use of interspecific divergence and intraspecific polymorphism data at synonymous sites to detect the presence of balancing selection. A maximum-likelihood approach to the HKA test (MLHKA; [27]) was used. The software uses polymorphism and divergence information to compare models of neutrality and selection between a set of neutrally evolving genes and the candidate gene. For the MLHKA test, a set of 20 effectively neutrally evolving genes were chosen as described in Lundberg et al., [19], with Mus musculus sequences as outgroup. Mouse transcripts orthologous to the neutral genes and the candidate complement genes were identified using TBLASTX. The corresponding mouse and bank vole CDS were then aligned in Geneious Prime for each gene. Alignments were checked manually for errors and indels were removed. Input parameters for each gene such as number of segregating sites (S) and population-scaled mutation rate (θ) were calculated across all bank vole haplotypes using DnaSp. Number of divergent sites was calculated between the mouse ortholog and a randomly chosen bank vole haplotype. The program was run for each candidate gene with chain length of 100,000 for both the neutral and selection models. MLHKA uses the likelihood ratio test (LRT) to indicate significant signature of selection.
Sliding window analysis
For genes with signatures of balancing selection as indicated by the βstd.max outlier analysis, we performed additional analyses across both coding and non-coding regions to identify what part of the gene was the target of selection. First, we performed sliding window analysis of nucleotide diversity and Tajima’s D using PopGenome [57] along the length of each gene, using windows of 2 kb and steps of 500 bp (except FCNA where windows of 500 bp and steps of 50 bp were used due to its short length). High nucleotide diversity and high positive values of Tajima’s D are expected under balancing selection [31, 32].
Sanger sequencing
To ensure the accuracy of the whole-genome resequencing data and spot errors due to mapping and filtering, we performed Sanger sequencing of the gene with signatures of balancing selection based on the MLHKA test, FCNA. Primers targeting regions (~ 700 bp) around the peak of nucleotide diversity were designed to generate amplicons from a separate set of bank vole samples (n = 30). Thermocycling conditions were as follows: 94˚C for 4 min, followed by 37 cycles of denaturation at 94˚C for 30 s, annealing for 30 s at primer-specific Tm (Additional File 1: Table S3), extension at 72˚C for 45 s, and completed with a final extension at 72˚C for 10 min. PCR products were sequenced bidirectionally on a Genetic Analyser 3500 (Applied Biosystems) using BigDye™ terminator (Applied Biosystems). Sequences were aligned and base calls were manually checked and edited in Geneious.
Linkage disequilibrium and haplotype networks
To investigate to what extent localised signatures of selection (identified by BetaScan and sliding window analyses of π and Tajima’s D) were in linkage disequilibrium (LD) with other parts of a gene, we constructed LD plots across the whole gene with the software LDBlockShow [61]. D’ values were used to estimate LD and LD blocks were defined using the method by Gabriel et al., [62]. SNPs with minor allele frequency < 0.15 were filtered out.
To visualise the relationship between alleles at different SNPs in regions with signatures of balancing selection, we constructed a haplotype network from haplotypes inferred from whole-genome resequencing data and Sanger sequences. Haplotype network was constructed in popart [63] using a median joining network [64].
Statistical analyses
Certain characteristics of genes such as their functional category (gene category) could conceivably determine if they are under balancing selection and therefore influence βstd.max. We considered genes as “candidates of balancing selection” if they are: (a) involved in pathogen recognition (complement activation via MAMP recognition), or (b) targets of immune evasion (Additional File 1: Table S2). Targets of immune evasion were based on data in humans and primarily compiled from Lambris et al., [7], Ermert et al., [16] and from a literature survey for other complement genes that were not reported in these references. Human and rodents are infected by many related pathogens. Therefore, it is reasonable to assume that many of the targets of immune evasion identified in human pathogens are also conserved in the bank vole. To check if βstd.max differed between different categories, Mann-Whitney U test was used in R(v4) [65].
Data availability
Genome sequences have been deposited in SRA under BioProject PRJNA335935. The genome assembly has been deposited in GenBank with accession no. MULK00000000.1. Supplementary tables are presented in Additional File 1 and supplementary figure in Additional File 2.
Abbreviations
- MAMPs:
-
Microbe-associated molecular patterns
- PRMs:
-
Pattern recognition molecules
- CDS:
-
Coding sequence
- MLHKA:
-
Maximum likelihood Hudson-Kreitman-Aguadé
- LD:
-
Linkage disequilibrium
- FCNA:
-
Ficolin-A
- RCA:
-
Regulator of complement activation
- MAC:
-
Membrane attack complex
References
Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–97.
Reis ES, Mastellos DC, Hajishengallis G, Lambris JD. New insights into the immune functions of complement. Nat Rev Immunol. 2019;19:503–16.
Sackton TB, Lazzaro BP, Schlenke TA, Evans JD, Hultmark D, Clark AG. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 2007;39:1461–8.
Enard D, Messer PW, Petrov DA. Genome-wide signals of positive selection in human evolution. Genome Res. 2014;24:885–95.
Cagliani R, Forni D, Filippi G, Mozzi A, De Gioia L, Pontremoli C, et al. The mammalian complement system as an epitome of host–pathogen genetic conflicts. Mol Ecol. 2016;25:1324–39.
Degn SE, Thiel S, Jensenius JC. New perspectives on mannan-binding lectin-mediated complement activation. Immunobiology. 2007;212:301–11.
Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008;6:132–42.
Caine JA, Coburn J. Multifunctional and redundant roles of Borrelia burgdorferi outer surface proteins in tissue adhesion, colonization, and complement evasion. Front Immunol. 2016;7:442.
Skare JT, Garcia BL. Complement evasion by Lyme Disease Spirochetes. Trends Microbiol. 2020;28:889–99.
Lin Y-P, Frye AM, Nowak TA, Kraiczy P. New insights into CRASP-Mediated complement evasion in the Lyme Disease Enzootic cycle. Front Cell Infect Microbiol. 2020;10:1.
Pietikäinen J, Meri T, Blom AM, Meri S. Binding of the complement inhibitor C4b-binding protein to Lyme disease Borreliae. Mol Immunol. 2010;47:1299–305.
Hammerschmidt C, Klevenhaus Y, Koenigs A, Hallström T, Fingerle V, Skerka C, et al. BGA66 and BGA71 facilitate complement resistance of Borrelia bavariensis by inhibiting assembly of the membrane attack complex. Mol Microbiol. 2016;99:407–24.
Caine JA, Lin Y-P, Kessler JR, Sato H, Leong JM, Coburn J. Borrelia burgdorferi outer surface protein C (OspC) binds complement component C4b and confers bloodstream survival. Cell Microbiol. 2017;19.
Serruto D, Rappuoli R, Scarselli M, Gros P, van Strijp JAG. Molecular mechanisms of complement evasion: learning from staphylococci and meningococci. Nat Rev Microbiol. 2010;8:393–9.
Zipfel PF, Hallström T, Riesbeck K. Human complement control and complement evasion by pathogenic microbes – tipping the balance. Mol Immunol. 2013;56:152–60.
Ermert D, Ram S, Laabei M. The hijackers guide to escaping complement: Lessons learned from pathogens. Mol Immunol. 2019;114:49–61.
Laabei M, Ermert D. Catch me if you can: Streptococcus pyogenes complement evasion strategies. J Innate Immun. 2019;11:3–12.
Dybdahl MF, Jenkins CE, Nuismer SL. Identifying the molecular basis of host-parasite coevolution: merging Models and Mechanisms. Am Nat. 2014;184:1–13.
Lundberg M, Zhong X, Konrad A, Olsen R-A, Råberg L. Balancing selection in pattern recognition receptor signalling pathways is associated with gene function and pleiotropy in a wild rodent. Mol Ecol. 2020;29:1990–2003.
Kosiol C, Vinař T, Fonseca RR, da, Hubisz MJ, Bustamante CD, Nielsen R, et al. Patterns of positive selection in six mammalian genomes. PLoS Genet. 2008;4:e1000144.
Woolhouse MEJ, Webster JP, Domingo E, Charlesworth B, Levin BR. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat Genet. 2002;32:569–77.
Bernig T, Taylor JG, Foster CB, Staats B, Yeager M, Chanock SJ. Sequence analysis of the mannose-binding lectin (MBL2) gene reveals a high degree of heterozygosity with evidence of selection. Genes Immun. 2004;5:461–76.
Verdu P, Barreiro LB, Patin E, Gessain A, Cassar O, Kidd JR, et al. Evolutionary insights into the high worldwide prevalence of MBL2 deficiency alleles. Hum Mol Genet. 2006;15:2650–8.
Mukherjee S, Sarkar-Roy N, Wagener DK, Majumder PP. Signatures of natural selection are not uniform across genes of innate immune system, but purifying selection is the dominant signature. PNAS. 2009;106:7073–8.
Soejima M, Tachida H, Tsuneoka M, Takenaka O, Kimura H, Koda Y. Nucleotide sequence analyses of human complement 6 (C6) gene Suggest Balancing Selection. Ann Hum Genet. 2005;69:239–52.
Siewert KM, Voight BF. BetaScan2: standardized Statistics to detect balancing selection utilizing Substitution Data. Genome Biol Evol. 2020;12:3873–7.
Wright SI, Charlesworth B. The HKA test revisited: a maximum-likelihood-ratio test of the standard neutral model. Genetics. 2004;168:1071–6.
Charlesworth B, Charlesworth D. Elements of evolutionary genetics. 2010.
Siewert KM, Voight BF. Detecting long-term balancing selection using allele frequency correlation. Mol Biol Evol. 2017;34:2996–3005.
Fijarczyk A, Babik W. Detecting balancing selection in genomes: limits and prospects. Mol Ecol. 2015;24:3529–45.
Charlesworth D. Balancing selection and its Effects on sequences in nearby genome regions. PLoS Genet. 2006;2:e64.
Tajima F. Statistical method for testing the Neutral Mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, et al. Pfam: the protein families database in 2021. Nucleic Acids Res. 2021;49:D412–9.
Thornton KR, Jensen JD. Controlling the false-positive rate in Multilocus Genome Scans for Selection. Genetics. 2007;175:737–50.
Barrett RDH, Hoekstra HE. Molecular spandrels: tests of adaptation at the genetic level. Nat Rev Genet. 2011;12:767–80.
Garlatti V, Belloy N, Martin L, Lacroix M, Matsushita M, Endo Y, et al. Structural insights into the innate immune recognition specificities of L- and H-ficolins. EMBO J. 2007;26:623–33.
Krarup A, Thiel S, Hansen A, Fujita T, Jensenius JC. L-ficolin is a pattern Recognition Molecule Specific for Acetyl Groups*. J Biol Chem. 2004;279:47513–9.
Lynch NJ, Roscher S, Hartung T, Morath S, Matsushita M, Maennel DN, et al. L-Ficolin specifically binds to Lipoteichoic Acid, a cell Wall Constituent of Gram-Positive Bacteria, and activates the lectin pathway of complement 1. J Immunol. 2004;172:1198–202.
Endo Y, Takahashi M, Iwaki D, Ishida Y, Nakazawa N, Kodama T, et al. Mice deficient in Ficolin, a lectin complement pathway Recognition Molecule, are susceptible to Streptococcus pneumoniae infection. J Immunol. 2012;189:5860–6.
Genster N, Takahashi M, Sekine H, Endo Y, Garred P, Fujita T. Lessons learned from mice deficient in lectin complement pathway molecules. Mol Immunol. 2014;61:59–68.
Genster N, Cramer EP, Rosbjerg A, Pilely K, Cowland JB, Garred P. Ficolins promote fungal clearance in vivo and modulate the inflammatory cytokine response in host defense against Aspergillus fumigatus. J Innate Immun. 2016;8:579–88.
Radwan J, Babik W, Kaufman J, Lenz TL, Winternitz J. Advances in the Evolutionary understanding of MHC polymorphism. Trends Genet. 2020;36:298–311.
Forni D, Cagliani R, Tresoldi C, Pozzoli U, Gioia LD, Filippi G, et al. An evolutionary analysis of Antigen Processing and Presentation across different Timescales reveals pervasive selection. PLoS Genet. 2014;10:e1004189.
Hollox EJ, Armour JA. Directional and balancing selection in human beta-defensins. BMC Evol Biol. 2008;8:113.
Cagliani R, Fumagalli M, Riva S, Pozzoli U, Comi GP, Menozzi G, et al. The signature of long-standing balancing selection at the human defensin β-1 promoter. Genome Biol. 2008;9:R143.
Minias P, Vinkler M. Selection balancing at innate immune genes: adaptive polymorphism maintenance in toll-like receptors. Mol Biol Evol. 2022;:msac102.
Cagliani R, Fumagalli M, Biasin M, Piacentini L, Riva S, Pozzoli U, et al. Long-term balancing selection maintains trans-specific polymorphisms in the human TRIM5 gene. Hum Genet. 2010;128:577–88.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–62.
Bult CJ, Blake JA, Smith CL, Kadin JA, Richardson JE. Mouse Genome Database Group. Mouse genome database (MGD) 2019. Nucleic Acids Res. 2019;47:D801–6.
Lee E, Helt GA, Reese JT, Munoz-Torres MC, Childers CP, Buels RM, et al. Web Apollo: a web-based genomic annotation editing platform. Genome Biol. 2013;14:R93.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arXiv:12073907 [q-bio]. 2012.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–97.
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–302.
Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7:256–76.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Pfeifer B, Wittelsbürger U, Ramos-Onsins SE, Lercher MJ. PopGenome: an efficient Swiss Army Knife for Population genomic analyses in R. Mol Biol Evol. 2014;31:1929–36.
Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–9.
Tange O. GNU Parallel. Zenodo. 2022. https://doi.org/10.5281/zenodo.6479152.
Hudson RR, Kreitman M, Aguadé M. A test of Neutral Molecular Evolution based on Nucleotide Data. Genetics. 1987;116:153–9.
Dong S-S, He W-M, Ji J-J, Zhang C, Guo Y, Yang T-L. LDBlockShow: a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief Bioinform. 2020. https://doi.org/10.1093/bib/bbaa227.
Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of Haplotype Blocks in the Human Genome. Science. 2002;296:2225–9.
Leigh JW, Bryant D. popart: full-feature software for haplotype network construction. Methods Ecol Evol. 2015;6:1110–6.
Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48.
R Core Team. R: a Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2020.
Acknowledgements
We would like to thank Science for Life Laboratory, the National Genomics Infrastructure, NGI, Stockholm, and Uppmax for providing assistance in massive parallel sequencing and computational infrastructure.
Funding
This work was supported by grants from the Swedish Research Council (2015–05418 to L.R.), the Crafoord foundation (20150741 to L.R.), the Erik Philip-Sörensen foundation (to L.R.). The funding bodies played no role in the design of the study and collection, analysis, interpretation of data, and in writing the manuscript.
Open access funding provided by Lund University.
Author information
Authors and Affiliations
Contributions
M.N., L.R., M.L. and F.C. conceptualised the study. L.R. conducted fieldwork and labwork. M.L. performed genome assembly and whole-genome resequencing analyses. M.N. performed Sanger sequencing, selection analysis and wrote the first draft of the manuscript. All authors contributed to revising the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Protocols for animal experiments were approved by the Malmö/Lund board for animal experiment ethics (permission M47-14). All methods were in compliance with relevant guidelines and regulations issued by the Swedish Board of Agriculture. All methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org) for the reporting of animal experiments.
Consent for publication
Not applicable.
Competing interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12862_2023_2122_MOESM1_ESM.xlsx
Table S1: List of excluded complement genes. Table S2: Summary of test statistics and categorization for all complement genes. Table S3: Primer details for Sanger sequencing.
12862_2023_2122_MOESM2_ESM.pdf
Supplementary Figure S1: Sliding window analysis of βstd, π, and Tajima’s D, haplotype network, and LD plot for the three genes that were outliers for βstd.max
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nandakumar, M., Lundberg, M., Carlsson, F. et al. Balancing selection on the complement system of a wild rodent. BMC Ecol Evo 23, 21 (2023). https://doi.org/10.1186/s12862-023-02122-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-023-02122-0