
Abstract
Background
The most severe form of human malaria is caused by the protozoan parasite Plasmodium falciparum. This unicellular organism is a member of a subgenus of Plasmodium called the Laverania that infects apes, with P. falciparum being the only member that infects humans. The exceptional virulence of this species to humans can be largely attributed to a family of variant surface antigens placed by the parasites onto the surface of infected red blood cells that mediate adherence to the vascular endothelium. These proteins are encoded by a large, multicopy gene family called var, with each var gene encoding a different form of the protein. By changing which var gene is expressed, parasites avoid immune recognition, a process called antigenic variation that underlies the chronic nature of malaria infections.
Results
Here we show that the common ancestor of the branch of the Laverania lineage that includes the human parasite underwent a remarkable change in the organization and structure of elements linked to the complex transcriptional regulation displayed by the var gene family. Unlike the other members of the Laverania, the clade that gave rise to P. falciparum evolved distinct subsets of var genes distinguishable by different upstream transcriptional regulatory regions that have been associated with different expression profiles and virulence properties. In addition, two uniquely conserved var genes that have been proposed to play a role in coordinating transcriptional switching similarly arose uniquely within this clade. We hypothesize that these changes originated at a time of dramatic climatic change on the African continent that is predicted to have led to significant changes in transmission dynamics, thus selecting for patterns of antigenic variation that enabled lengthier, more chronic infections.
Conclusions
These observations suggest that changes in transmission dynamics selected for significant alterations in the transcriptional regulatory mechanisms that mediate antigenic variation in the parasite lineage that includes P. falciparum. These changes likely underlie the chronic nature of these infections as well as their exceptional virulence.
Similar content being viewed by others


Background
Eukaryotic parasites of the genus Plasmodium infect a broad range of vertebrate species, including birds, reptiles and mammals [1]. There are five species that infect humans and cause malaria, a disease that can be severe, resulting in significant morbidity and mortality within human populations in tropical and subtropical regions of the world [2]. Malaria parasites exhibit multiple morphological states as they transition between their mosquito vectors and their vertebrate hosts, but all symptoms of the disease result from asexual replication of the parasites within the circulating red blood cells (RBCs) of the infected individual. This stage of the infection is associated with a massive increase in parasite numbers and can lead to severe anemia, extensive inflammation and vascular occlusion that can disrupt circulation [3]. The influence of malaria on human evolution is thought to have been substantial, and infection by Plasmodium parasites has been proposed to have played a significant role in shaping the human genome [4].
Of the five parasite species that infect humans, by far the most virulent is P. falciparum. The virulence of this species is due in large part to the cytoadhesive properties of infected RBCs that result from the placement on the RBC surface of parasite encoded variable antigens, included proteins referred to as RIFIN (repetitive interspersed family), STEVOR (subtelomeric variant open reading frame), PfMC-2TM (P. falciparum Maurer’s cleft-2 transmembrane domain proteins) and PfEMP1 (P. falciparum erythrocyte membrane protein 1) [5, 6]. The contribution of RIFINs, STEVORs and PfMC-2TMs to cytoadherence is unclear, however PfEMP1 has been shown to be the major cytoadhesive molecule and is thought to be the primary virulence determinant of P. falciparum [7]. P. falciparum is the sole human-infective member of a subgenus of Plasmodium that infects apes called the Laverania. Unlike malaria species from other evolutionary lineages, the seven characterized species of the Laverania are unique for their expression of EMP1 on the surface of infected RBCs [7] where it binds to molecules on the host endothelial surface and enables the infected cell to adhere to blood vessel walls, thus sequestering the parasites from the peripheral circulation and avoiding destruction in the spleen [8]. However, placement of EMP1 on the RBC surface stimulates the host to generate antibodies against this protein, thus parasites must continuously vary the form of EMP1 they express to avoid clearance by the adaptive immune response. This phenomenon is referred to as antigenic variation, and it is remarkably effective at enabling parasites to maintain chronic infections that can last over a year [3, 9,10,11].
The ability of the Laverania parasites to continuously change the expressed form of EMP1 over the course of an infection is rooted in the expansion of the genes encoding different forms of the protein into large, multicopy gene families called var. A recent comparative study of the genomes of seven Laverania species identified var gene families ranging in size from 28 to 105 copies, with the genes organized into clusters found within the subtelomeric regions near the chromosome ends or in tandem arrays within the internal regions of the chromosomes [12]. Extensive studies in P. falciparum indicate that the genes are transcribed in a mutually exclusion fashion, with all but a single gene maintained in condensed heterochromatin and thus transcriptionally silent [13,14,15]. By switching the single var gene that is actively transcribed, parasites change the form of EMP1 expressed on the infected cell surface and thereby undergo antigenic variation. Whole genome analysis of many P. falciparum isolates also indicates that the var gene family is highly recombinogenic [16, 17], resulting in rapid diversification of the EMP1 coding regions and thus preventing the acquisition of cross-reactive immunity in geographical regions with high transmission rates. The var gene family therefore represents a key attribute of the Laverania that has substantially contributed to its evolutionary success and the increased virulence displayed by P. falciparum compared to other human-infective species of malaria parasites.
The var gene family is present in the genomes of all seven known members of the Laverania subgenus, indicating that it evolved in the common ancestor of these parasites. Interestingly however, in their recent comparative study, Otto and colleagues documented a remarkable change in the organization of protein domain structures within the EMP1 proteins encoded by different clades within the Laverania subgenus [12]. Specifically, they found that four species that constitute a clade which includes P. falciparum have diverged significantly in EMP1 protein structure, suggesting that parasites from this clade cytoadhere to different host endothelial receptors when compared to the more distantly related members of the subgenus. If correct, this marks a significant moment in the evolution of this lineage of pathogens, particularly since this alteration in cytoadhesive properties directly shaped the virulence of the human parasite, P. falciparum. The selective forces that led to this change in EMP1 structure are unknown, however Otto et al. found that an unusual and highly conserved var gene called var2csa appears to be the sole remnant of the more ancient type of EMP1 remaining in P. falciparum. This gene was originally identified as encoding a form of EMP1 that binds to receptors in the placenta [18], but more recently has also been proposed to function as an intermediate in var transcriptional switching [19, 20]. They therefore hypothesized that this ancient gene has been maintained as a single copy in P. falciparum to serve the dual functions of cytoaherence within the placenta as well as regulating var gene transcriptional switching [12]. This study highlighted key steps in the evolution of the var gene family, both in terms of the virulence linked to cytoadherence by EMP1 and the transcriptional regulation that enables antigenic variation.
Here we extend these findings to investigate the evolution of other elements implicated in transcriptional regulation of the var gene family. We found that both the regulatory proteins previously implicated in the control of var gene transcription and a unique regulatory element found within the conserved intron of var genes are present throughout the entire Laverania subgenus, indicating that many aspects that control var gene expression evolved in the common ancestor of this entire group of parasites. In contrast, the divergence of var upstream regulatory regions into distinct subtypes linked to virulence, as well as the evolution of var2csa and a second strain-transcendent var gene called var1csa, occurred relatively recently within the Laverania subgenus, within the clade that includes P. falciparum. These changes all appear to be related to the evolution of a more structured transcriptional regulatory mechanism, potentially enabling parasites to more precisely coordinate the process of antigenic variation. Phylogenetic analysis indicates that this change in the structure of the var gene family coincides with a well-documented alteration in African climatic conditions that likely altered parasite transmission dynamics. Thus, the selective pressures that shaped the evolution of virulence of the sole human-infective member of the Laverania lineage can be traced to two key events, one involving a change in host receptor recognition for cytoadherence and a second that influenced how antigenic variation is controlled.
Results
Proteins implicated in var gene regulation are found encoded in the genomes of all Laverania species
Given the importance of var genes and EMP1 to the virulence of P. falciparum, and the recent identification of significant changes in EMP1 structure over the course of the evolution of the Laverania [12], we were interested in systematically considering the evolution of the mechanisms controlling var gene regulation and antigenic variation in the parasite lineage that gave rise to P. falciparum. While the precise mechanisms that control var gene activation, silencing and mutually exclusive expression are not completely understood, numerous proteins have been implicated in various aspects of var gene regulation, included the histone methyltransferases SET2 [21, 22] and SET10 [23], the demethylase JmJC1 [22, 24], the histone deacetylases SIR2A and SIR2B [25,26,27], the translation factor PTEF [28] and the RECQ helicases RQ1 and WRN [29, 30]. Syntenic orthologues of each protein are found throughout the Laverania, and with the exception of PTEF, all can also be identified in the non-Laverania parasites P. vivax, P. berghei and P. gallinaceum (Table 1), therefore they must have existed in the common ancestor of all Plasmodium species. These proteins represent orthologues of factors known from model organisms to play roles in chromatin assembly, transcriptional regulation or aspects of DNA repair, thus they are likely to have broader roles in parasite nuclear functions beyond their functions in var gene regulation. The histone methyltransferase SET2 and its cognate demethylase JmJC1 are similarly found throughout the Laverania as well as in P. vivax and the bird parasite P. gallinaceum, however they are not encoded in the rodent parasite genomes (Table 1). It was previously suggested that these proteins were specific to primate parasites [22], however the availability of whole genome sequences from additional parasite species has identified syntenic orthologues in parasites of birds, suggesting an origin prior to the root of the Plasmodium genus and more general functions not specific for var gene regulation.
In contrast to the other regulatory proteins associated with var gene regulation, PTEF (Plasmodium translation enhancing factor) appears to be specific to the Laverania (Table 1). BLAST searches of the genomes of non-Laverania parasite species failed to identify genes with significant similarity, and the syntenic genomic positions of P. vivax, P. berghei and P. gallinaceum do not contain open reading frames encoding similar proteins. Thus, the evolution of PTEF appears to be coincident with the appearance of var genes in this branch of the Plasmodium genus. In P. falciparum, PTEF has been described as a protein specifically upregulated during pregnancy where it enhances translation of the EMP1 encoded by the unique var gene called var2csa [28]. var2csa is unusual in that the gene includes a regulatory upstream open reading frame (uORF) that can suppress translation of the EMP1 encoding exons [31, 32]. PTEF was shown to interact with the translating ribosome complex to enable translational reinitiation [28], thereby overcoming the translational repression of the uORF and promoting expression of a form of EMP1 that binds to the proteoglycan chrondroitin sulfate A on the syncytiotrophoblasts of the placenta [33]. var2csa was therefore proposed to be translationally repressed in the absence of a placenta through the uORF, but expression of PTEF when parasites infect pregnant women relieved this repression and enabled parasites to take advantage of this new anatomical site for cytoadhesion. However, var2csa and its regulatory uORF appear to have evolved relatively late in the Laverania (see below), while PTEF is present in all members of the subgenus, indicating that PTEF must have a function in addition to or beyond regulation of var2csa translation. A recent genome-wide assessment of P. falciparum transcripts noted that the number of potential uORFs is remarkably high [34] and given the substantial degree of synteny and sequence homology within the entire subgenus, this extends to the other Lavarania species as well. The role of PTEF in overcoming the repressive effects of uORFs on mRNA translation therefore could include a broader class of mRNAs beyond var2csa, thus explaining its conservation in parasites that do not possess this particular var gene.
The regulatory element found in var introns is conserved throughout the Laverania subgenus
Initial characterization of var genes in P. falciparum identified a conserved, two exon structure that includes an unusual intron that separates the extracellular domain of EMP1 from the intracellular portion that anchors the protein in the RBC membrane [35]. This intron has independent promoter activity, giving rise to long, noncoding RNAs of unknown function [35,36,37], and it has been implicated in the regulation of var gene silencing and mutually exclusive expression [38, 39], although the precise mechanism remains undefined [40]. Close examination of the sequence of var introns identified a distinct strand asymmetry, with each intron easily divided into three well defined regions based on base content, with regions 1 and 3 displaying “mirror images” of one another [41]. The strand asymmetry is easily discernible through the display of G vs C and A vs T content within the separate regions, with region 1 containing high G content and virtually no Cs and region 3 displaying the inverse relationship, with respect to the positive DNA strand (Fig. 1). A bi-directional promoter driving expression of long, noncoding RNAs has been mapped to region 2 [36, 41]. This conserved structure is found within the introns of all var genes in P. falciparum, with the exception of an unusual type called var3 or “type 3” var genes. Type 3 var genes have been identified in some, but not all, strains of P. falciparum, and their function is unknown [42,43,44]. The introns of these genes lack region 2.

Conservation of var intron structure in all seven Laverania species. A Schematic showing the division of var introns into three regions based on base content and strand asymmetry. B Calculation of base content of the positive strand of var introns for each species of the Laverania. Note the prominent G vs C asymmetry in regions 1 and 3 and the conservation of this asymmetry in all seven species
To determine if the regulatory element found in the var introns of P. falciparum is also observed in var genes from parasites throughout the Laverania subgenus, we obtained var intron sequences from annotated var genes from the 6 other Laverania species. Each intron sequence was divided into three regions and the average base content calculated for each region as previously described [41]. The base pair content for each region was displayed as histograms (Fig. 1) and all seven species show virtually identical patterns of strand asymmetry. The unusual base pair structure of var introns originally observed in P. falciparum therefore is also found in var genes throughout the Laverania and likely extends to the origins of var genes themselves. With the exception of the Type 3 var genes mentioned above, the intron structure does not appear to vary based on the chromosomal location or gene type, consistent with what was previously reported for P. falciparum [45], Interestingly, similar to a previous report [46], we were unable to identify Type 3 vars in species other than P. falciparum, although more complete genome assemblies of these species might enable identification of var genes that are currently missing.
Evolution of strain-transcendent var genes in a subset of Laverania species that includes P. falciparum
The var gene family has been extensively studied in P. falciparum, including the vast diversity of individual genes when the complete genome sequences of multiple, independent geographical isolates were compared [44]. Attempts to measure the level of var sequence diversity within populations of naturally circulating parasites have similarly described enormous variation [9, 47] that appears to result from frequent segmental recombination events between genes [48, 49], a phenomenon documented in cultured parasites [50,51,52]. Remarkably, two var genes appear to escape recombination with other members of the var gene family and remain highly conserved in all P. falciparum isolates. The first of these genes, called var1csa, is found near the “right” end of chromosome 5 at the boundary between the subtelomeric region and the core genome. Unlike other members of the var gene family, it is transcribed late in the asexual cycle and appears to not be subjected to mutually exclusive expression [53, 54]. In addition, in all isolates examined to date, the gene carries a premature stop codon in exon 2, leading to its annotation as a pseudogene, and in the 3D7 reference genome the gene is truncated due to a subtelomeric deletion event. To determine if the conservation of var1csa extends beyond P. falciparum, we examined the syntenic region of chromosome 5 in all 7 members of the Laverania subgenus. We found var genes with 92% and 93% sequence identity at this genomic position in P. praefalciparum and P. reichenowi, respectively, but no genes with similar levels of identity in any other species (Fig. 2). BLAST searches of the complete genome sequences from the remaining species to determine if var1csa might exist at an alternative genomic position only identified non-conserved var genes with substantially less sequence identity (Fig. 2), indicating that var1csa is limited to P. praefalciparum, P. reichenowi and P. falciparum. Interestingly, the gene in P. praefalciparum and P. reichenowi also contains a predicted premature stop codon in exon 2. The selective pressure maintaining this unique var gene is unknown but seems unlikely to be related to its protein coding capacity given the unusual transcription pattern displayed by the gene in P. falciparum as well as the premature stop codon.

Conservation of the unusual var gene var1csa in P. reichenowi, P. praefalciparum and P. falciparum. A A phylogenetic tree of the Laverania as described by Otto et al. [12]. B Identification of var1csa in P. falciparum (isolate SN1) and its orthologues in P. reichenowi and P. praefalciparum. The chromosomal region from each species containing the var gene with the highest sequence identity to var1csa from P. falciparum is shown. var genes are shown in blue and members of other multicopy gene families are shown in green. The genes with the highest sequence identity to var1csa in each species are aligned, and the percentage identity is shown below the gene. For P. reichenowi and P. praefalciparum, sequence identity exceeds 90%, and the genes display clear synteny (boxed in blue). For the remainder of the Laverania, the sequence identity is substantially lower and the most similar gene is not located in the syntenic position, indicating these species do not contain true orthologues of var1csa
Similar to var1csa, var2csa is conserved in the genomes of all P. falciparum isolates described to date [16]. The gene is duplicated in some isolates of P. falciparum and was also observed as multiple copies in the sequenced isolate of P. praefalciparum [44, 55], but all isolates of P. falciparum maintain a syntenic copy at the boundary between the subtelomeric region and the core genome at the “left” end of chromosome 12 (Fig. 3A). Also similar to var1csa, var2csa is conserved in P. praefalciparum and P. reichenowi, specifically a conserved copy is located at the syntenic position of the left end of chromosome 12, but no copies of var2csa are found in the other members of the Laverania subgenus. As mentioned above, var2csa is unique amongst var genes in its inclusion of a 360 bp uORF within the 5’ leader of its mRNA. This uORF serves as a translational repressor of the EMP1 encoded by the main open reading frame [31, 32] and is also highly conserved in all three species. This conservation extends to the amino acid sequence of the peptide encoded by the uORF (Fig. 3B). This gene was originally identified for the role of the encoded form of EMP1 in binding to the host cell surface receptor chrondroitin sulfate A in the placenta [18], however more recently it has been suggested to play a role in mediating or coordinating var gene expression switching, potentially as a default gene or node in a hypothetical var gene switching network [19, 20, 22]. In a more recent analysis of the domain structure of EMP1 proteins throughout the Laverania, Otto and colleagues identified a surprising change in structure of the domains that make up EMP1 in the clade that includes P. billcollinsi. P. reichenowi, P. praefalciparum and P. falciparum [12]. However, the EMP1 encoded by var2csa appears to be more closely related to those found in the more distantly related Laverania parasites, leading them to suggest that it might be the sole remnant of an ancient form of the protein that remains within the genomes of these parasites [12]. They further suggested that the selective pressure that has prevented this particular gene from recombining with other members of the family could be related to its putative role in transcriptional regulation rather than in the cytoadherence properties of the encoded protein. If correct, this suggests that the function of var2csa in regulating var gene transcription evolved in the common ancestor of P. reichenowi, P. praefalciparum and P. falciparum, and implicates the uORF in this function.

Conservation of the var gene var2csa in P. reichenowi, P. praefalciparum and P. falciparum. A Schematic showing the syntenic region of chromosome 12 in P. reichenowi, P. praefalciparum and three isolates of P. falciparum (SN01, 7G8 and 3D7). The positions of the single copy genes fikk12 and acs7 are shown and represent the border between the core genome and the highly variable subtelomeric domain. The orthologues of var2csa are aligned and boxed in blue. B Alignment of the amino acid sequence encoded by the uORF of var2csa from P. praefalciparum, P. reichenowi and three isolates of P. falciparum (SN01, 7G8 and 3D7)
Divergence of var gene regulatory regions into different types
With the initial completion and assembly of the P. falciparum reference genome [56], it became evident that var genes are typically found in one of three genomic positions: in subtelomeric regions transcribed away from the telomere, in subtelomeric regions transcribed toward the telomere, or in tandem arrays found within the interior of the chromosomes. The significance of this arrangement was not immediately apparent since all var genes, regardless of their position in the genome, appear to be subject to similar transcriptional regulation, including epigenetically regulated activation and silencing, as well as mutually exclusive expression. However, more detailed analysis of the upstream regulatory regions of all members of the family found that they could be organized into three basic types, called UpsA, B or C, based on DNA sequence similarity [45, 57], and that these types correlated closely with the genes’ position within the chromosomes. Two additional types, called UpsD and E, are associated with var1csa and var2csa, respectively. The importance of Ups type was suggested by studies of clinical samples obtained from individuals with either severe or asymptomatic disease. Parasites isolated from children with severe disease were most often found to express UpsA or B genes [58,59,60], while UpsC genes were not specifically associated with disease condition. Studies with cultured parasites found that UpsA and B genes also displayed higher switching rates than observed for UpsC genes, which are more stable once activated [61, 62]. These studies suggest that the upstream transcriptional regulatory regions have evolved into different types to influence how expression of the gene family progresses over the course of an infection.
Given the potential importance of the organization of the upstream regions for regulating var gene expression in P. falciparum, we investigated when this organization evolved in the Laverania lineage and whether a similar organization exists in other members of the subgenus. Similar to the previous analyses that identified the UpsA/B/C/D and E types [45, 57], we constructed maximum-likelihood phylogenetic trees to identify subgroups of upstream regulatory domains based on sequence similarities. Using this method, we readily detected the three primary types (UpsA/B/C) in P. falciparum, and similarly found them to generally correlate with genomic position, thus replicating the previously published analyses [45, 57] (Additional file 1, Fig. 1A). Applying the same methodology to the var gene upstream regions from P. praefalciparum and P. reichenowi, we were similarly able to detect clearly differentiated upstream types, yielding phylogenetic trees with separated branches that again loosely correlated with genomic position (Additional File 1, Fig. 1B, C) suggesting that in these species the var gene family is organized into Ups types similar to P. falciparum. For the other four Laverania species, fewer full-length upstream var sequences were available, however similar analysis yielded trees with less well defined branches and with the upstream regulatory regions from the different chromosomal locations distributed throughout the trees (Additional file 1, Fig. 1D–G), suggesting that these regulatory regions are either not organized into separate Ups types, or that the organization is less strictly defined.
To more closely investigate the origins of the Ups promoter type organization, we combined the var upstream regulatory sequences from all seven Laverania species and constructed a single maximum-likelihood phylogenetic tree (Fig. 4). Of particular interest are the branches that represent the UpsA and B sequences. Both of these var regulatory types segregate into groups that are relatively distant from the other members of the var gene family. More importantly, these branches only include sequences from P. falciparum, P. praefalciparum and P. reichenowi, with no sequences from the other four species represented within these branches. This indicates that the UpsA and B promoter types evolved at the root of the branch of the Laverania that includes P. praefalciparum, P. reichenowi and P. falciparum, coinciding with the evolution of var1csa and var2csa into distinct, strain and species transcendent genes. UpsC sequences are similarly found within a distinct branch that includes P. falciparum, P. praefalciparum and P. reichenowi, but this group also includes two sequences from P. billcollinsi, indicating that the UpsC type evolved somewhat earlier than either UpsA or B.

Maximum-likelihood phylogenetic tree of 0.5–1.5 kb upstream regulatory regions of var genes from seven Laverania species. The annotation number for each gene (from Plasmodb.org) is shown and the colour of the text signifies the species, as shown in the lower right panel. Sequences of the UpsB type are shaded in pink, UpsA in light blue, UpsC in yellow, var2csa in gray and var1csa in dark blue. Five genes with atypical upstream sequences from P. falciparum that segregate outside of the UpsA/B/C/D/E groups are marked with an asterisk. The evolutionary history was inferred using the Maximum Likelihood method and Jukes–Cantor model [63], with bootstrap values for 1000 replicates shown for various nodes. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. Evolutionary analyses were conducted in MEGA X [64]
Interestingly, while the highly conserved aspects of var2csa, including the presence of the uORF in the 5’ UTR of the transcript, its location in the subtelomeric region of chromosome 12 and the structure of the encoded form of EMP1, all originated in the common ancestor of P. falciparum, P. praefalciparum and P. reichenowi (see Fig. 3), the upstream region of var2csa, also called UpsE, invariable clusters with var genes from the more distant Laverania species (Fig. 4). This indicates that var2csa evolved from a type of var gene that is no longer found in P. falciparum, thus explaining why it departs from the UpsA/B/C organization. This is consistent with the conclusion of Otto and colleagues, based on the structure of the encoded protein, that var2csa is the sole remaining example in P. falciparum of an ancient var gene [12]. We similarly found that the upstream region of var1csa (called UpsD) also segregates with genes from the more distant Laverania species, suggesting that it too might be derived from an ancient var gene type that is no longer found in P. falciparum. In addition, the Ups regions from five P. falciparum var genes (Pf3D7_0809100, Pf3D7_1240300, Pf3D7_0617400, Pf3D7_0712900, Pf3D7_080600) segregate within branches apart from the UpsA/B/C organization that includes the rest of the P. falciparum var gene family. Four of these Ups regions were previously noted by Lavstsen et al. to have atypical sequences [45], and they are all located at the boundaries between var gene containing chromosomal segments and the core genome, within the interior regions of the chromosomes. Genes at such positions are likely to be more constrained in their ability to undergo recombination with other family members due to the adjacent unique, highly conserved sequences, thus preventing them from diverging as rapidly. This is consistent with the clustering of these Ups regions with sequences from the more distantly related P. reichenowi and P. praefalciparum rather than with other P. falciparum var genes.
Discussion
Both the evolution of the UpsA and B var upstream regulatory domain subtypes and the development of the species-transcendent, conserved genes var1csa and var2csa arose in the common ancestor of P. praefalciparum, P. falciparum and P. reichenowi as well as P. lomamiensis [65], a parasite of bonobos with insufficient genome sequence data to be included in the analysis described here (Fig. 5A). Our analysis suggests that a strong selective pressure on parasites at this particular moment in the evolutionary history of the Laverania led to the evolution of var1csa, var2csa and the UpsA/B/C organization of the var gene family. Further, this selection pressure appears to have been sufficiently powerful and sustained to maintain these changes as the subsequent parasite species diverged, despite the hyper-recombinogenic nature of the var gene family and the plasticity of the subtelomeric regions of the genome, two characteristics that would be predicted to rapidly disrupt this organization in the absence of strong selection. Thus, the selection pressure that led to this change in var gene organization likely represents an exceptionally strong influence that shaped the genome of the most virulent of the human malaria parasite species.

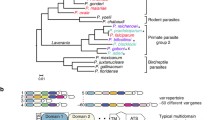
Phylogenetic trees marking key moments in the evolution of the var gene family. A The evolutionary relationships of the Laverania are shown, with each parasite species and its ape host denoted. var genes, the regulatory intronic element and the putative regulatory protein PTEF are thought to have originated in the common ancestor of the entire subgenus (shown by green asterisk). A significant shift in the structure of the var encoded protein EMP1 and the UpsC upstream regulatory type occurred in the common ancestor of the bottom five species (shown by red asterisk) while the evolution of the conserved genes var1csa and var2csa, as well as the UpsA and B types, occurred prior to the branch that includes the human parasite P. falciparum (shown by purple asterisk). B The codivergence model of parasite speciation is displayed showing the branch of the Laverania that evolved var1csa, var2csa and the UpsA/B/C organization. The phylogenetic tree shows the evolution of gorillas, humans, chimpanzees and bonobos from a common ancestor ~ 8–9 million years ago. This model presumes that parasites species diverged along with their hosts, resulting in P. praefalciparum, P. reichenowi and P. lomamiensis infecting gorillas, chimpanzees and bonobos, respectively. Humans are hypothesized to have lost their original parasites, then become reinfected through a recent gorilla-to-human transmission event, resulting in P. falciparum. These ape species (and their parasites) initially diverged during the late Miocene, a period of major climatic change in Africa marked by aridification and a regional shift from rainforests to grasslands and savannah
The functional significance of the development of the var1csa/var2csa/UspA/B/C organization of the var gene family is unknown but is likely to involve how the family is transcriptionally regulated. Mathematical modelling has previously proposed that coordination of var gene transcriptional switching might be achieved through central organizing var genes that serve as nodes within a switching network [66], and additional work has implicated var2csa in this function [19, 20]. If this model holds, var1csa might similarly contribute to coordinating var gene switching patterns, thereby providing an explanation for its universal conservation despite a timing of transcription inconsistent with the production of EMP1 and its annotation as a pseudogene. The divergence of var upstream transcriptional regulatory domains into UpsA/B/C/D/E types could provide additional structure to the switching network, thereby organizing the family into a hierarchy of genes with different switching rates. This would influence the probability of specific genes being activated at different time points of an infection or in the presence of different degrees of host immunity, as has been observed [59, 60, 67], and would contribute to a coordinated order of gene activation. More sophisticated coordination of var gene switching has been proposed as a way for parasite populations that can number in the billions of individuals to limit var gene expression to a single or very small number of var genes at any given time [66]. In contrast, uncoordinated, random var gene switching by individual parasites within such large circulating populations would result in much more rapid immune exposure of the entire var repertoire. Therefore, we hypothesize that the evolution of var1csa, var2csa and the UpsA/B/C/D/E organization of the var gene family enabled parasite populations to maintain more lengthy infections, although why this trait would have been under such strong selection pressure specifically in the common ancestor of this clade of the Laverania is not immediately apparent.
To better understand what might have given rise to the evolution of a more coordinated mechanism of var gene transcriptional regulation, we considered the current models for the speciation of the Laverania. Using estimates of mutation rates and Bayesian whole-genome estimates, Otto and colleagues dated the origins of the Laverania to 0.7–1.2 million years ago, and the common ancestor of P. reichenowi/P. praefalciparum/P. falciparum to 140–230 thousand years ago [12]. Others have also dated speciation events within the Laverania to timepoints in the relatively recent past [68]. In contrast, Hahn and colleagues proposed a much older origin of this parasite clade. Their analysis indicates that the species within this branch of the Laverania arose through codivergence with their ape hosts, beginning with the ancestors of gorillas and chimpanzees approximately 8–9 million years ago (Fig. 5B) [65, 69]. This model suggests that humans lost their initial Laverania parasites after diverging from chimpanzees, and subsequently acquired P. falciparum through a relatively recent gorilla-to-human transmission event [70]. The latter model is particularly intriguing since it places the common ancestor of this parasite clade within the late Miocene period, a time of substantial climatic and environmental change on the African continent that is known to have contributed to ape speciation [71]. Specifically, the continent experienced progressive cooling and aridification, resulting in the contraction of continent-wide rainforests and a drastic expansion of grasslands and savannah (reviewed in [72]). This change in environmental conditions would have created large geographical ranges with seasonal transmission, including lengthy time periods in which malaria transmission is greatly reduced or eliminated, for example dry seasons when the mosquito vector can be absent for months. In order to sustain transmission under such conditions, parasites would have been required to extend individual infections to span such extended time intervals when transmission is absent. This is precisely what more coordinated var gene transcriptional switching patterns have been proposed to do [66], and provides a compelling hypothesis for the selection pressure that underlies the origin and maintenance of var1csa/var2csa/UpsA/B/C/D/E organization of the gene family. While the current geographical distribution of both host and parasite species have been documented [65, 69], the selective pressures contributing to these distributions have not been identified, and may or may not reflect the selective pressures in place when the var1csa/var2csa/UpsA/B/C/D/E organization evolved. Similarly, the chronicity of infections with species other than P. falciparum have not been rigorously investigated. Thus, this hypothesis remains speculative.
In conclusion, the strategy of avoiding splenic clearance through cytoadherence, a phenomenon that lies at the heart of the exceptional virulence of P. falciparum in humans, appears to have evolved in the common ancestor of the entire Laverania subgenus. Along with the var gene family and the cytoadhesive protein EMP1, the basic elements for transcriptional regulation of this family, including the proteins implicated in the epigenetic regulation of transcriptional activation, silencing and mutually exclusive expression, were present in the common ancestor of all Laverania. This also includes the regulatory element found in var introns and its associated noncoding RNAs. Thus, strong cytoadhesion and tightly regulated antigenic variation based on mutually exclusive var gene expression are universal characteristics of these parasites and represent an ancient, conserved evolutionary adaptation of the Laverania. However, not all aspects of var gene regulation and cytoadhesion through EMP1 expression have remained unchanged and at least two significant events have helped shape the evolution of the most virulent of the malaria species that infect humans (Fig. 5A). In addition to changes in var transcriptional regulation, Otto and colleagues previously documented a significant alteration in EMP1 structure and speculated that this could indicate a change in the host cell surface receptors used for cytoadherence [12]. How these changes in cytoadhesive properties affected interactions with their hosts is not known, however they have been incorporated into a highly successful human pathogen that continues to cause substantial morbidity and mortality throughout the developing world. Understanding the key evolutionary events that shaped this subgenus of parasites provides valuable insights into the pathogenesis and persistence of malaria caused by P. falciparum.
Methods
var Intron structure analysis
Analysis of var intron base pair content was performed as described by Calderwood et al. [41]. Briefly, 10 var genes were randomly selected for each species in the Laverania clade from the genome sequence database at EupathDB.org. Each gene was uploaded as an individual file into the sequence analysis program SnapGene (www.snapgene.com) and the intron was manually divided into each region based on base content as described by Calderwood et al. [41]. The percentage of each base on the positive strand was calculated per region, averaged, and graphed using Prism (www.graphpad.com).
Presence of regulatory proteins, var1csa, and var2csa in the Laverania Clade
The histone methyltransferases SET2 [21, 22] and SET10 [23], the demethylase JmJC1 [22, 24], the histone deacetylases SIR2A and SIR2B [25,26,27], the translation factor PTEF [28] and the RECQ helicases RQ1 and WRN [29, 30] were chosen based on their proposed roles in the regulation of var gene expression. The amino acid sequences and the genomic position of each gene from the 3D7 reference genome of P. falciparum were obtained from the EupathDb database (www.plasmodb.org) and analysed using the Basic Local Alignment Search Tool (BLAST; ncbi.nlm.gov). The top hit corresponded to the orthologous gene in each species, and the syntenic chromosomal location for each gene was verified. With the exception of PTEF in P. blacklocki (incomplete sequence assembly), every gene resided in the syntenic location based on the 3D7 genome. For var1csa and var2csa, the same procedure was performed.
Upstream var regulatory region analysis
Collecting sequences: Analysis of var upstream sequences was performed as described by Kraemer and Smith [57]. The var genes from each species were compiled by downloading 1.5 kb upstream of each “PfEMP1” or “EMP1” labeled gene on PlasmoDB. The number of compiled sequences were compared with the number of genes reported by Otto et al. [12], and in all cases, the initial number of genes we collected exceeded the number previously reported. Each gene was reevaluated based on the following criteria: Genes with upstream sequences below 500 bp or that had a truncated exon 1 were removed. Similarly, genes with greater than 2 introns or that contained ambiguous nucleotides labeled as “N” in the upstream sequences were discarded unless the ambiguous nucleotides could be deleted from the 5’ end of the sequence without going below 500 bp. Genes on unassembled chromosomes were noted. A full description of the sequences from each species that were included in the analyses is provided in Additional file 2, and a description of the number of genes found for each species analyzed in this study compared to that described by Otto et al. [12] is provided in Additional file 1, Table 1.
Determining Chromosomal Location and Orientation: Chromosomal location and orientation for each var gene was determined as described by Lavstsen et al. [45] and Kraemer et al. [57]. Subtelomeric regions included sequences within 100 kb of either chromosome end while sequences between the two subtelomeric regions were annotated as being within a chromosomal internal region. The orientation of subtelomeric genes was determined based on direction of transcription, either toward or away from the adjacent telomere. For genes located on unassembled chromosomes, chromosomal location could sometimes be inferred by locating the nearest conserved, single copy gene, then determining the chromosome position of its orthologous gene within the 3D7 reference genome sequence.
Constructing maximum-likelihood phylogenetic trees: The collected sequences for the var upstream regions from each species were aligned via Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) [73] and the resulting FASTA sequences were analyzed using MEGA-X [64], a molecular evolutionary genetics analysis tool (https://www.megasoftware.net/). The ML tree was generated with the Jukes-Cantor Model (63) and 1000 bootstrap replicates. The ML Heuristic Method used was Nearest-Neighbor-Interchange (NNI) and BioNJ.
Availability of data and materials
The datasets analysed during the current study are available in the Eupathdb database (Plasmodb.org). In addition to the datasets available through the Eupathdb database, additional genome sequence data were obtained through the databases originally described by Otto et al., Nature Microbiology, 2018. These sequences were submitted to EBI, project ID PRJEB13584 (secondary study accession: ERP015144). These can also be accessed via ftp://ftp.sanger.ac.uk/pub/project/pathogens/Plasmodium/Laverania/.
References
Galen SC, Borner J, Martinsen ES, Schaer J, Austin CC, West CJ, Perkins SL. The polyphyly of Plasmodium: comprehensive phylogenetic analyses of the malaria parasites (order Haemosporida) reveal widespread taxonomic conflict. R Soc Open Sci. 2018;5: 171780.
WHO. 2018. World malaria report 2018. Organization WH,
Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–9.
Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77:171–92.
Templeton TJ. The varieties of gene amplification, diversification and hypervariability in the human malaria parasite, Plasmodium falciparum. Mol Biochem Parasitol. 2009;166:109–16.
Wahlgren M, Goel S, Akhouri RR. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat Rev Microbiol. 2017;15:479–91.
Pasternak ND, Dzikowski R. PfEMP1: an antigen that plays a key role in the pathogenicity and immune evasion of the malaria parasite Plasmodium falciparum. Int J Biochem Cell Biol. 2009;41:1463–6.
Deitsch KW, Dzikowski R. Variant gene expression and antigenic variation by malaria parasites. Annu Rev Microbiol. 2017;71:625–41.
Chen DS, Barry AE, Leliwa-Sytek A, Smith TA, Peterson I, Brown SM, Migot-Nabias F, Deloron P, Kortok MM, Marsh K, Daily JP, Ndiaye D, Sarr O, Mboup S, Day KP. A molecular epidemiological study of var gene diversity to characterize the reservoir of Plasmodium falciparum in humans in Africa. PLoS ONE. 2011;6: e16629.
Childs LM, Buckee CO. Dissecting the determinants of malaria chronicity: why within-host models struggle to reproduce infection dynamics. J R Soc Interface. 2015;12:20141379.
Sama W, Killeen G, Smith T. Estimating the duration of Plasmodium falciparum infection from trials of indoor residual spraying. Am J Trop Med Hyg. 2004;70:625–34.
Otto TD, Gilabert A, Crellen T, Bohme U, Arnathau C, Sanders M, Oyola SO, Okouga AP, Boundenga L, Willaume E, Ngoubangoye B, Moukodoum ND, Paupy C, Durand P, Rougeron V, Ollomo B, Renaud F, Newbold C, Berriman M, Prugnolle F. Genomes of all known members of a Plasmodium subgenus reveal paths to virulent human malaria. Nat Microbiol. 2018;3:687–97.
Chookajorn T, Dzikowski R, Frank M, Li F, Jiwani AZ, Hartl DL, Deitsch KW. Epigenetic memory at malaria virulence genes. Proc Natl Acad Sci U S A. 2007;104:899–902.
Flueck C, Bartfai R, Volz J, Niederwieser I, Salcedo-Amaya AM, Alako BT, Ehlgen F, Ralph SA, Cowman AF, Bozdech Z, Stunnenberg HG, Voss TS. Plasmodium falciparum heterochromatin protein 1 marks genomic loci linked to phenotypic variation of exported virulence factors. PLoS Pathog. 2009;5: e1000569.
Perez-Toledo K, Rojas-Meza AP, Mancio-Silva L, Hernandez-Cuevas NA, Delgadillo DM, Vargas M, Martinez-Calvillo S, Scherf A, Hernandez-Rivas R. Plasmodium falciparum heterochromatin protein 1 binds to tri-methylated histone 3 lysine 9 and is linked to mutually exclusive expression of var genes. Nucleic Acids Res. 2009;37:2596–606.
Otto TD, Bohme U, Sanders M, Reid A, Bruske EI, Duffy CW, Bull PC, Pearson RD, Abdi A, Dimonte S, Stewart LB, Campino S, Kekre M, Hamilton WL, Claessens A, Volkman SK, Ndiaye D, Amambua-Ngwa A, Diakite M, Fairhurst RM, Conway DJ, Franck M, Newbold CI, Berriman M. Long read assemblies of geographically dispersed Plasmodium falciparum isolates reveal highly structured subtelomeres. Wellcome Open Res. 2018;3:52.
Pilosof S, He Q, Tiedje KE, Ruybal-Pesantez S, Day KP, Pascual M. Competition for hosts modulates vast antigenic diversity to generate persistent strain structure in Plasmodium falciparum. PLoS Biol. 2019;17: e3000336.
Salanti A, Staalsoe T, Lavstsen T, Jensen ATR, Sowa MPK, Arnot DE, Hviid L, Theander TG. Selective upregulation of a single distinctly structured var gene in chondroitin sulphate A-adhering Plasmodium falciparum involved in pregnancy-associated malaria. Mol Microbiol. 2003;49:179–91.
Ukaegbu UE, Zhang X, Heinberg AR, Wele M, Chen Q, Deitsch KW. A unique virulence gene occupies a principal position in immune evasion by the malaria parasite Plasmodium falciparum. PLoS Genet. 2015;11: e1005234.
Mok BW, Ribacke U, Rasti N, Kironde F, Chen Q, Nilsson P, Wahlgren M. Default pathway of var2csa switching and translational repression in Plasmodium falciparum. PLoS ONE. 2008;3: e1982.
Jiang L, Mu J, Zhang Q, Ni T, Srinivasan P, Rayavara K, Yang W, Turner L, Lavstsen T, Theander TG, Peng W, Wei G, Jing Q, Wakabayashi Y, Bansal A, Luo Y, Ribeiro JM, Scherf A, Aravind L, Zhu J, Zhao K, Miller LH. PfSETvs methylation of histone H3K36 represses virulence genes in Plasmodium falciparum. Nature. 2013;499:223–7.
Ukaegbu UE, Kishore SP, Kwiatkowski DL, Pandarinath C, Dahan-Pasternak N, Dzikowski R, Deitsch KW. Recruitment of PfSET2 by RNA polymerase II to variant antigen encoding loci contributes to antigenic variation in P. falciparum. PLoS Pathog. 2014;10: e1003854.
Volz JC, Bartfai R, Petter M, Langer C, Josling GA, Tsuboi T, Schwach F, Baum J, Rayner JC, Stunnenberg HG, Duffy MF, Cowman AF. PfSET10, a Plasmodium falciparum methyltransferase, maintains the active var gene in a poised state during parasite division. Cell Host Microbe. 2012;11:7–18.
Kishore SP, Stiller JW, Deitsch KW. Horizontal gene transfer of epigenetic machinery and evolution of parasitism in the malaria parasite Plasmodium falciparum and other apicomplexans. BMC Evol Biol. 2013;13:37.
Duraisingh MT, Voss TS, Marty AJ, Duffy MF, Good RT, Thompson JK, Freitas-Junior LH, Scherf A, Crabb BS, Cowman AF. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium faiciparum. Cell. 2005;121:13–24.
Freitas-Junior LH, Hernandez-Rivas R, Ralph SA, Montiel-Condado D, Ruvalcaba-Salazar OK, Rojas-Meza AP, Mancio-Silva L, Leal-Silvestre RJ, Gontijo AM, Shorte S, Scherf A. Telomeric heterochromatin propagation and histone acetylation control mutually exclusive expression of antigenic variation genes in malaria parasites. Cell. 2005;121:25–36.
Tonkin CJ, Carret CK, Duraisingh MT, Voss TS, Ralph SA, Hommel M, Duffy MF, Silva LM, Scherf A, Ivens A, Speed TP, Beeson JG, Cowman AF. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 2009;7:e84.
Chan S, Frasch A, Mandava CS, Ch’ng JH, Quintana MDP, Vesterlund M, Ghorbal M, Joannin N, Franzen O, Lopez-Rubio JJ, Barbieri S, Lanzavecchia A, Sanyal S, Wahlgren M. Regulation of PfEMP1-VAR2CSA translation by a Plasmodium translation-enhancing factor. Nat Microbiol. 2017;2:17068.
Li Z, Yin S, Sun M, Cheng X, Wei J, Gilbert N, Miao J, Cui L, Huang Z, Dai X, Jiang L. DNA helicase RecQ1 regulates mutually exclusive expression of virulence genes in Plasmodium falciparum via heterochromatin alteration. Proc Natl Acad Sci U S A. 2019;116:3177–82.
Claessens A, Harris LM, Stanojcic S, Chappell L, Stanton A, Kuk N, Veneziano-Broccia P, Sterkers Y, Rayner JC, Merrick CJ. RecQ helicases in the malaria parasite Plasmodium falciparum affect genome stability, gene expression patterns and DNA replication dynamics. PLoS Genet. 2018;14: e1007490.
Amulic B, Salanti A, Lavstsen T, Nielsen MA, Deitsch KW. An upstream open reading frame controls translation of var2csa, a gene implicated in placental malaria. PLoS Pathog. 2009;5: e1000256.
Bancells C, Deitsch KW. A molecular switch in the efficiency of translation reinitiation controls expression of var2csa, a gene implicated in pregnancy-associated malaria. Mol Microbiol. 2013. https://doi.org/10.1111/mmi.12379.
Salanti A, Dahlback M, Turner L, Nielsen MA, Barfod L, Magistrado P, Jensen AT, Lavstsen T, Ofori MF, Marsh K, Hviid L, Theander TG. Evidence for the involvement of VAR2CSA in pregnancy-associated malaria. J Exp Med. 2004;200:1197–203.
Kaur C, Kumar M, Patankar S. Messenger RNAs with large numbers of upstream open reading frames are translated via leaky scanning and reinitiation in the asexual stages of Plasmodium falciparum. Parasitology. 2020;147:1100–13.
Su X, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JV, Peterson DS, Ravetch JV, Wellems TE. A large and diverse gene family (var) encodes 200–350 kD proteins implicated in the antigenic variation and cytoadherence of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82:89–100.
Epp C, Li F, Howitt CA, Chookajorn T, Deitsch KW. Chromatin associated sense and antisense noncoding RNAs are transcribed from the var gene family of virulence genes of the malaria parasite Plasmodium falciparum. RNA. 2009;15:116–27.
Amit-Avraham I, Pozner G, Eshar S, Fastman Y, Kolevzon N, Yavin E, Dzikowski R. Antisense long noncoding RNAs regulate var gene activation in the malaria parasite Plasmodium falciparum. Proc Natl Acad Sci U S A. 2015;112:E982–91.
Deitsch KW, Calderwood MS, Wellems TE. Malaria. Cooperative silencing elements in var genes. Nature. 2001;412:875–6.
Dzikowski R, Li F, Amulic B, Eisberg A, Frank M, Patel S, Wellems TE, Deitsch KW. Mechanisms underlying mutually exclusive expression of virulence genes by malaria parasites. EMBO Rep. 2007;8:959–65.
Bryant JM, Regnault C, Scheidig-Benatar C, Baumgarten S, Guizetti J, Scherf A. CRISPR/Cas9 genome editing reveals that the intron is not essential for var2csa gene activation or silencing in Plasmodium falciparum. MBio. 2017. https://doi.org/10.1128/mBio.00729-17.
Calderwood MS, Gannoun-Zaki L, Wellems TE, Deitsch KW. Plasmodium falciparum var genes are regulated by two regions with separate promoters, one upstream of the coding region and a second within the intron. J Biol Chem. 2003;278:34125–32.
Rask TS, Hansen DA, Theander TG, Gorm PA, Lavstsen T. Plasmodium falciparum erythrocyte membrane protein 1 diversity in seven genomes—divide and conquer. PLoS Comput Biol. 2010. https://doi.org/10.1371/journal.pcbi.1000933.
Wang CW, Lavstsen T, Bengtsson DC, Magistrado PA, Berger SS, Marquard AM, Alifrangis M, Lusingu JP, Theander TG, Turner L. Evidence for in vitro and in vivo expression of the conserved VAR3 (type 3) plasmodium falciparum erythrocyte membrane protein 1. Malar J. 2012;11:129.
Otto T, Assefa S, Böhme U, Sanders M, Kwiatkowski D, Berriman M, Newbold D. Evolutionary analysis of the most polymorphic gene family in falciparum malaria. Wellcome Open Res. 2019;4:193.
Lavstsen T, Salanti A, Jensen ATR, Arnot DE, Theander TG. Sub-grouping of Plasmodium falciparum 3D7 var genes based on sequence analysis of coding and non-coding regions. Malar J. 2003;2:27.
Trimnell AR, Kraemer SM, Mukherjee S, Phippard DJ, Janes JH, Flamoe E, Su XZ, Awadalla P, Smith JD. Global genetic diversity and evolution of var genes associated with placental and severe childhood malaria. Mol Biochem Parasitol. 2006;148:169–80.
Barry AE, Leliwa-Sytek A, Tavul L, Imrie H, Migot-Nabias F, Brown SM, McVean GA, Day KP. Population genomics of the immune evasion (var) genes of Plasmodium falciparum. PLoS Pathog. 2007;3:e34.
Kraemer SM, Kyes SA, Aggarwal G, Springer AL, Nelson SO, Christodoulou Z, Smith LM, Wang W, Levin E, Newbold CI, Myler PJ, Smith JD. Patterns of gene recombination shape var gene repertoires in Plasmodium falciparum: comparisons of geographically diverse isolates. BMC Genomics. 2007;8:45.
Zilversmit MM, Chase EK, Chen DS, Awadalla P, Day KP, McVean G. Hypervariable antigen genes in malaria have ancient roots. BMC Evol Biol. 2013;13:110.
Bopp SE, Manary MJ, Bright AT, Johnston GL, Dharia NV, Luna FL, McCormack S, Plouffe D, McNamara CW, Walker JR, Fidock DA, Denchi EL, Winzeler EA. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 2013;9: e1003293.
Claessens A, Hamilton WL, Kekre M, Otto TD, Faizullabhoy A, Rayner JC, Kwiatkowski D. Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of Var genes during mitosis. PLoS Genet. 2014;10: e1004812.
Zhang X, Alexander N, Leonardi I, Mason C, Kirkman LA, Deitsch KW. Rapid antigen diversification through mitotic recombination in the human malaria parasite Plasmodium falciparum. PLoS Biol. 2019;17: e3000271.
Kyes SA, Christodoulou Z, Raza A, Horrocks P, Pinches R, Rowe JA, Newbold CI. A well-conserved Plasmodium falciparum var gene shows an unusual stage-specific transcript pattern. Mol Microbiol. 2003;48:1339–48.
Winter G, Chen QJ, Flick K, Kremsner P, Fernandez V, Wahlgren M. The 3D7var5.2 (var(COMMON)) type var gene family is commonly expressed in non-placental Plasmodium falciparum malaria. Mol Biochem Parasitol. 2003;127:179–91.
Sander AF, Salanti A, Lavstsen T, Nielsen MA, Magistrado P, Lusingu J, Ndam NT, Arnot DE. Multiple var2csa-type PfEMP1 genes located at different chromosomal loci occur in many Plasmodium falciparum isolates. PLoS ONE. 2009;4: e6667.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511.
Kraemer SM, Smith JD. Evidence for the importance of genetic structuring to the structural and functional specialization of the Plasmodium falciparum var gene family. Mol Microbiol. 2003;50:1527–38.
Jensen ATR, Magistrado P, Sharp S, Joergensen L, Lavstsen T, Chiucciuini A, Salanti A, Vestergaard LS, Lusingu JP, Hermsen R, Sauerwein R, Christensen J, Nielsen MA, Hviid L, Sutherland C, Staalsoe T, Theander TG. Plasmodium falciparum associated with severe childhood malaria preferentially expresses PfEMP1 encoded by group A var genes. J Exp Med. 2004;199:1179–90.
Lavstsen T, Magistrado P, Hermsen CC, Salanti A, Jensen AT, Sauerwein R, Hviid L, Theander TG, Staalsoe T. Expression of Plasmodium falciparum erythrocyte membrane protein 1 in experimentally infected humans. Malar J. 2005;4:21.
Rottmann M, Lavstsen T, Mugasa JP, Kaestli M, Jensen AT, Muller D, Theander T, Beck HP. Differential expression of var gene groups is associated with morbidity caused by Plasmodium falciparum infection in Tanzanian children. Infect Immun. 2006;74:3904–11.
Frank M, Dzikowski R, Amulic B, Deitsch K. Variable switching rates of malaria virulence genes are associated with chromosomal position. Mol Microbiol. 2007;64:1486–98.
Noble R, Christodoulou Z, Kyes S, Pinches R, Newbold CI, Recker M. The antigenic switching network of Plasmodium falciparum and its implications for the immuno-epidemiology of malaria. Elife (Cambridge). 2013;2: e01074.
Jukes TH, Cantor CR. Evolution of protein molecules. In: Munro H, editor. Mammalian protein metabolism. New York: Academic Press; 1969. p. 21–132.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9.
Liu W, Sherrill-Mix S, Learn GH, Scully EJ, Li Y, Avitto AN, Loy DE, Lauder AP, Sundararaman SA, Plenderleith LJ, Ndjango JN, Georgiev AV, Ahuka-Mundeke S, Peeters M, Bertolani P, Dupain J, Garai C, Hart JA, Hart TB, Shaw GM, Sharp PM, Hahn BH. Wild bonobos host geographically restricted malaria parasites including a putative new Laverania species. Nat Commun. 2017;8:1635.
Recker M, Buckee CO, Serazin A, Kyes S, Pinches R, Christodoulou Z, Springer AL, Gupta S, Newbold CI. Antigenic variation in Plasmodium falciparum malaria involves a highly structured switching pattern. PLoS Pathog. 2011;7: e1001306.
Jensen AT, Magistrado P, Sharp S, Joergensen L, Lavstsen T, Chiucchiuini A, Salanti A, Vestergaard LS, Lusingu JP, Hermsen R, Sauerwein R, Christensen J, Nielsen MA, Hviid L, Sutherland C, Staalsoe T, Theander TG. Plasmodium falciparum associated with severe childhood malaria preferentially expresses PfEMP1 encoded by group A var genes. J Exp Med. 2004;199:1179–90.
Volkman SK, Barry AE, Lyons EJ, Nielsen KM, Thomas SM, Choi M, Thakore SS, Day KP, Wirth DF, Hartl DL. Recent origin of Plasmodium falciparum from a single progenitor. Science. 2001;293:482–4.
Sharp PM, Plenderleith LJ, Hahn BH. Ape origins of human malaria. Annu Rev Microbiol. 2020;74:39–63.
Rayner JC, Liu W, Peeters M, Sharp PM, Hahn BH. A plethora of Plasmodium species in wild apes: a source of human infection? Trends Parasitol. 2011;27:222–9.
Joordens JCA, Feibel CS, Vonhof HB, Schulp AS, Kroon D. Relevance of the eastern African coastal forest for early hominin biogeography. J Hum Evol. 2019;131:176–202.
Couvreur TLP, Dauby G, Blach-Overgaard A, Deblauwe V, Dessein S, Droissart V, Hardy OJ, Harris DJ, Janssens SB, Ley AC, Mackinder BA, Sonke B, Sosef MSM, Stevart T, Svenning JC, Wieringa JJ, Faye A, Missoup AD, Tolley KA, Nicolas V, Ntie S, Fluteau F, Robin C, Guillocheau F, Barboni D, Sepulchre P. Tectonics, climate and the diversification of the tropical African terrestrial flora and fauna. Biol Rev Camb Philos Soc. 2020. https://doi.org/10.1111/brv.12644.
Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, Lopez R. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636–41.
Acknowledgements
The authors would like to acknowledge the support and encouragement of the Student Research Program of the Bronx High School of Science to RH.
Funding
This project was funded by National Institutes of Health grants AI52390 and AI99327 to KWD. The Department of Microbiology and Immunology at Weill Medical College of Cornell University acknowledges the support of the William Randolph Hearst Foundation.
Author information
Authors and Affiliations
Contributions
MRG and KWD designed the study. MRG and RH obtained the sequences and performed the analyses. MRG, RH and KWD wrote and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
: Maximum-likelihood phylogenetic trees of the 0.5–1.5 kb upstream regulatory regions of var genes from all seven Laverania species and Collected Ups sequences from all species compared with the number of genes reported by Otto et al., 2018.
Additional file 2
: Description of individual sequences analyzed. This file provides a description of each sequence analyzed, organized according to species. Specific characteristics for each sequence are provided.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gross, M.R., Hsu, R. & Deitsch, K.W. Evolution of transcriptional control of antigenic variation and virulence in human and ape malaria parasites. BMC Ecol Evo 21, 139 (2021). https://doi.org/10.1186/s12862-021-01872-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-021-01872-z