
Abstract
Background
Using molecular data generated by high throughput next generation sequencing (NGS) platforms to infer phylogeny is becoming common as costs go down and the ability to capture loci from across the genome goes up. While there is a general consensus that greater numbers of independent loci should result in more robust phylogenetic estimates, few studies have compared phylogenies resulting from smaller datasets for commonly used genetic markers with the large datasets captured using NGS. Here, we determine how a 5-locus Sanger dataset compares with a 377-locus anchored genomics dataset for understanding the evolutionary history of the pseudoxyrhophiine snake radiation centered in Madagascar. The Pseudoxyrhophiinae comprise ~86 % of Madagascar’s serpent diversity, yet they are poorly known with respect to ecology, behavior, and systematics. Using the 377-locus NGS dataset and the summary statistics species-tree methods STAR and MP-EST, we estimated a well-supported species tree that provides new insights concerning intergeneric relationships for the pseudoxyrhophiines. We also compared how these and other methods performed with respect to estimating tree topology using datasets with varying numbers of loci.
Methods
Using Sanger sequencing and an anchored phylogenomics approach, we sequenced datasets comprised of 5 and 377 loci, respectively, for 23 pseudoxyrhophiine taxa. For each dataset, we estimated phylogenies using both gene-tree (concatenation) and species-tree (STAR, MP-EST) approaches. We determined the similarity of resulting tree topologies from the different datasets using Robinson-Foulds distances. In addition, we examined how subsets of these data performed compared to the complete Sanger and anchored datasets for phylogenetic accuracy using the same tree inference methodologies, as well as the program *BEAST to determine if a full coalescent model for species tree estimation could generate robust results with fewer loci compared to the summary statistics species tree approaches. We also examined the individual gene trees in comparison to the 377-locus species tree using the program MetaTree.
Results
Using the full anchored dataset under a variety of methods gave us the same, well-supported phylogeny for pseudoxyrhophiines. The African pseudoxyrhophiine Duberria is the sister taxon to the Malagasy pseudoxyrhophiines genera, providing evidence for a monophyletic radiation in Madagascar. In addition, within Madagascar, the two major clades inferred correspond largely to the aglyphous and opisthoglyphous genera, suggesting that feeding specializations associated with tooth venom delivery may have played a major role in the early diversification of this radiation. The comparison of tree topologies from the concatenated and species-tree methods using different datasets indicated the 5-locus dataset cannot beused to infer a correct phylogeny for the pseudoxyrhophiines under any method tested here and that summary statistics methods require 50 or more loci to consistently recover the species-tree inferred using the complete anchored dataset. However, as few as 15 loci may infer the correct topology when using the full coalescent species tree method *BEAST. MetaTree analyses of each gene tree from the Sanger and anchored datasets found that none of the individual gene trees matched the 377-locus species tree, and that no gene trees were identical with respect to topology.
Conclusions
Our results suggest that ≥50 loci may be necessary to confidently infer phylogenies when using summaryspecies-tree methods, but that the coalescent-based method *BEAST consistently recovers the same topology using only 15 loci. These results reinforce that datasets with small numbers of markers may result in misleading topologies, and further, that the method of inference used to generate a phylogeny also has a major influence on the number of loci necessary to infer robust species trees.
Similar content being viewed by others


Background
Phylogenetic studies are undergoing a massive jump in the scale of the molecular datasets used to estimate phylogenies, due to the ease of collecting hundreds of loci sampled throughout the genomes of non-model taxa (e.g., [1, 2]). Increasing the number of loci is expected to have a positive effect on phylogenetic estimation [3]. Both simulation and empirical studies [4–8] demonstrate strong correlation between the number of independent loci and phylogenetic accuracy, though the exact number of markers required to resolve relationships varies depending on the informativeness of the markers, the method of inference used, the number of taxa, and the time-scale being examined. Typically though, the ability to generate hundreds or thousands of loci that provide the appropriate amount of variation at differing time scales has been a challenge, especially across many non-model taxa. Recently, two different next-generation sequencing (NGS) protocols have produced large datasets composed of generally longer loci (in contrast to shorter length reads from restriction site-associated markers) useful for estimating phylogenies at varying temporal scales—the ultra-conserved element procedure of Faircloth et al. [9] and the anchored phylogenomics approach of Lemmon et al. [1]. These methods differ in the specific regions targeted and the numbers of loci produced, yet both produce orthologous markers across multiple, non-model taxa with substantial genetic variation for inferring phylogenies at both shallow and deep-time scales [1, 2, 7, 9–16].
While generating DNA datasets that cover the genome has become easier, estimating species-tree phylogenies with these data remains problematic [17]. Multi-species coalescent methods that jointly estimate gene trees and species trees, such as *BEAST [18], have proved robust for species-trees estimation. Although these full-coalescent methods can be highly accurate when using relatively few loci (e.g., [7]), they may not be suitable for the large numbers of loci produced using NGS techniques due to computational time and a lack of convergence as the number of taxa or loci increases [14, 19].
Alternatively, methods that use summarized information from user-provided gene trees to quickly estimate species trees, such as MP-EST [20] and STAR [21], are promising for analyzing NGS datasets. These methods can accommodate many taxa and loci and have the statistically desirable properties of being accurate when used with large numbers of loci and low levels of missing data [19, 20]. However, species-tree methods that depend on summarized gene-tree uncertainty may suffer when markers are short and uninformative [22, 23] or when incomplete-lineage sorting is not the main cause of gene-tree discordance [24–26]. While the concern of uninformative markers can be circumvented by using high quality datasets (i.e., longer loci with more informative sites), gene-tree discordance due to mutational variance or migration between species is a problem that still exists when estimating species trees even under a full-coalescent model such as *BEAST [24, 26, 27]. Despite these potential challenges, summary statistics approaches remain a viable option for NGS dataset analyses and several recent empirical studies with these types of data have used MP-EST and STAR to estimate well-resolved species trees that confirm previously hypothesized taxonomy as well as discover novel relationships (e.g., [13, 15, 28].
Here, we use an anchored phylogenomics dataset to construct a generic-level species tree for the Malagasy pseudoxyrhophiines and simultaneously explore how different datasets, with respect to locus number, influence phylogenetic inference. The subfamily Pseudoxyrhophiinae is part of the family Lamprophiidae, a mainly African radiation of snakes [29–31]. Pseudoxyrhophiines are among the most poorly studied of Colubroids, with little known with respect to ecology and reproduction (e.g., [32–34]), as well as basic morphology (e.g., hemipenial structure; [35]). This is unfortunate since pseudoxyrhophiines are unique among the world’s snake fauna as being the only island snake lineage where the majority of diversification takes place in situ on the island rather than through dispersal from the mainland [36], potentially making pseudoxyrhophiines an excellent model system for determining what factors promote ecological and morphological diversification within a closed system.
Of the currently recognized 89 species of pseudoxyrhophiine, 80 are endemic to Madagascar (excepting possible introductions to the Comoros; [37]), with the remaining taxa distributed in mainland Africa (five spp.), the Comoros islands (three spp.), and Socotra (one sp.). Previous studies have indicated the African and Socotran species are the sister lineage(s) to a monophyletic radiation of Malagasy/Comoros taxa [38, 39] but this has not been supported by the most recent phylogenetic estimates for the group [30, 31]. Prior molecular phylogenetic studies have included up to 54 of the recognized species of Pseudoxyrhophiinae for a single gene [40] and no study has used more than 10 loci to determine relationships among the genera, with the majority of taxa having only 1–6 loci available [30]. These studies also used concatenated gene-tree methodologies, rather than species-tree approaches, which are more likely to be misleading when using small numbers of loci [6]. Although under certain circumstances concatenation results in identical topologies when compared to a species trees [6, 41, 42], empirical studies have demonstrated that concatenation may overestimate branch lengths, causing inaccuracies in downstream phylogenetic analyses (e.g., [43]).
Using a NGS dataset comprised of 377 loci covering 77 % of the genera of Pseudoxyrhophiinae, we first estimate species trees using the full dataset and the summary statistics approaches STAR and MP-EST, and subsets of the loci using the multi-species coalescent method *BEAST. This is the first species tree to be produced for the subfamily, clarifying the intergeneric relationships of these snakes, and allowing us to more robustly examine the monophyly of the Malagasy genera, specifically with respect to the African mainland genus Duberria. Recent studies have found Duberria within a clade also containing the Malagasy pseudoxyrhophiine genus Compsophis, with this clade then sister taxon to the remaining Malagasy pseudoxyrhophiines [30, 31]. That relationship suggests that Malagasy genera are not a single monophyletic radiation, which is a key assumption for future studies examining diversification of these snakes in Madagascar [44]. Second, using varying numbers of loci with different tree-inference methodologies, we compare the tree topologies estimated from the full NGS dataset (377 loci) to smaller subsets of the NGS data (3–200 loci), as well as to a typical, Sanger-sequenced dataset comprised of five loci that are commonly used to infer squamate phylogenies. From these empirical results we determine if the numbers of loci and methods used influence phylogenetic inference for pseudoxyrhophiines.
Methods
Taxa sampled
We sampled 25 taxa: 23 pseudoxyrhophiines across 21 species, in 17 different genera and included two psammophiines as outgroups, Mimophis mahfalensis, and Rhamphiophis rubropunctatus. With the exception of the outgroup Rhamphiophis rubropunctatus and the pseudoxyrhophiine Parastenophis betsileanus, the same individuals were used for the Sanger and Anchored NGS methods (Additional file 1).
Ethics statement
All sample collection complied with the policies and guidelines of relevant institutions, including the Ministries des Eaux et Forêts, Madagascar National Parks, the Université d’Antananarivo, Departement de Biologie Animale, the University of Michigan Museum of Zoology, and the American Museum of Natural History.
DNA extraction and Sanger sequencing
DNA was extracted using a Quiagen® DNEasy kit, following the tissue protocol. Sanger sequencing was used to sequence five loci that have been previously utilized to infer squamate phylogenies [30, 43, 45]. This dataset consisted of two protein-coding mitochondrial genes (COI, 623 bp; CytB, 550 bp), two nuclear protein-coding genes (Cmos 562 bp; Rag2, 645 bp), and one nuclear intron (Nav intron 5, 610 bp) (PCR and sequencing details in Additional file 2). Sanger sequencing was performed at the American Museum of Natural History on an ABI 3730 sequencer. Sequences were edited and aligned using the Geneious alignment algorithm in Geneious® v.6.1.4 and checked by eye to ensure that protein coding loci did not contain stop codons.
Anchored phylogenomics locus selection and probe design
We used the following approach to identify a set of loci that could be obtained efficiently in snakes using Anchored Hybrid Enrichment. First, in order to improve gene-tree resolution, we increased the size of each target region described in [1] to approximately 1350 bp by including flanking regions that contained sufficient sequence conservation between Homo and Anolis. Because some of the original anchor regions were near each other, some of these loci were joined to form a single locus. Loci that performed poorly in [1] were removed. The resulting locus set contained 394 loci comprising a total of 468,296 bp target region (referred to as the version 2 vertebrate loci; genomic coordinates available on Dryad doi:10.5061/dryad.kp400). In order to improve the capture efficiency for snakes, we also obtained homologous sequences from the Python molurus genome (NCBI accession AEQU000000000) and 15× genomic reads obtained for the brown reed snake Calamaria pavimentata (Illumina PE100bp; specimen obtained from California Academy of Sciences, accession CAS235364). After aligning the Anolis, Python, and Calamaria sequences using MAFFT [46], alignments were trimmed to produce the final probe region alignments (alignments available on Dryad doi:10.5061/dryad.kp400), and probes were tiled at approximately 1.5X tiling density per species (probe specification available on Dryad doi:10.5061/dryad.kp400).
The NGS dataset was generated by the Center for Anchored Phylogenetics [47] using the anchored hybrid enrichment methodology described by [1]. We used a Covaris E220 Focused-ultrasonicator to fragment each genomic DNA sample to a fragment size of ~150–350. A Beckman-Coulter Biomek FXp liquid-handling robot was then used to prepare indexed Illumina libraries following protocol modified from Meyer and Kircher [48] (with SPRIselect size-selection after blunt-end repair using a 0.9x ratio of bead to sample volume). A single pool containing all of the libraries was then enrichment for the target using an Agilent Custom SureSelect kit (Agilent Technologies) that contained the probes described above. The enriched library pool was then sequenced on 1 PE150 Illumina HiSeq2000 lane by the Translational Science Laboratory in the College of Medicine at Florida State University.
NGS data assembly
Paired reads were merged following [49]. Briefly, for each degree of overlap for each read we computed the probability of obtaining the observed number of matches by chance, and selected degree of overlap that produced the lowest probability, with a p-value less than 10−10 required to merge reads. When reads were merged, mismatches are reconciled using base-specific quality scores, which were combined to form the new quality scores for the merged read (see [49] for details). Reads failing to meet the probability criterion were kept separate but still used in the assembly. Between 50 and 75 % of the sequenced library fragments had an insert size between 150 bp and 300 bp.
The reads for each sample were assembled into contigs using a reference assembly approach to map reads to the Calamaria probe regions and a de-novo assembly approach to extend the assembly into the flanks (java scripts available upon request from A. Lemmon). The reference assembler uses a library of spaced 20-mers derived from the sites conserved across squamates. A preliminary match resulted if at least 17 of 20 matches existed between the positions in a read and the corresponding positions in one of the spaced. Reads obtaining a preliminary match were then compared to the Calamaria, with greater than 55 matches out of 100 was considered a significant match. Approximate alignment positions of mapped reads were estimated using the position of the spaced 20-mers, and all 60-mers existing in the read were stored in a hash table used by the de-novo assembler. The de-novo assembler identifies exact matches between a read and one of the 60-mers found in the hash table. Simultaneously using the two levels of assembly described above, the read files were traversed repeatedly until an entire pass through the reads produced no additional mapped reads.
Mapped reads were then clustered for each locus into clusters using 60-mer pairs observed in the reads mapped to that locus. In short, a list of all 60mers found in the mapped reads was compiled, the 60-mers were clustered if found together in at least two reads. The 60-mer clusters were then used to separate the reads into clusters for contig estimation. Relative alignment positions of reads within each cluster were then refined in order to increase the agreement across the reads. Up to one gap was also inserted per read if needed to improve the alignment. Note that given sufficient coverage and an absence of contamination, each single-copy locus should produce a single assembly cluster. Low coverage (leading to a break in the assembly), contamination, and gene duplication, can all lead to an increased number of assembly clusters. A whole genome duplication, for example would increase the number of clusters to two per locus.
Consensus bases were called from assembly clusters as follows. For each site an unambiguous base was called if the bases present were identical or if the polymorphism of that site could be explained as sequencing error, assuming a binomial probability model with the probability of error equal to 0.1 and alpha equal to 0.05. If the polymorphism could not be explained as sequencing error, the ambiguous base was called what corresponded to all of the observed bases at that site (e.g. ‘R’ was used if ‘A’ and ‘G’ were observed). Called bases were soft-masked (made lowercase) for sites with coverage lower than 5. A summary of the assembly results is presented in the additional files (Additional file 3).
In order to filter out possible low-level contaminants, consensus sequences derived from very low coverage assembly clusters (<10 reads) were removed from further analysis. After filtering, consensus sequences were grouped by locus (across individuals) in order to produce sets of homologs. Orthology was then determined for each locus using a distance-based approach. First, a pairwise distance measure was computed for pairs of homologs by computing the percent of 20-mers observed in the two sequences that were found in both sequences. The list of 20-mers was constructed from consecutive 20-mers as well as spaced 20-mers (every third base) in order to allow increased levels of sequence divergence. Finally, we clustered the sequences using the distance matrix and a Neighbor-Joining algorithm that allowed at most one sequence per species to be assigned to a cluster. Only clusters containing at least 50 % of the species were utilized downstream.
Following orthology assessment, sequences in each orthologous set were aligned using MAFFT v7.023b [46]. The flags --genafpair and --maxiterate 1000 were utilized. The alignments for each locus was masked/trimmed in three steps. First, each alignment site was identified as “good” if the most common character observed was present in >50 % of the sequences. Second, 20 bp regions of each sequence that contained <10 good sites were masked. Third, sites with fewer than 4 unmasked bases were removed from the alignment.
Phylogenetic analyses
We estimated species trees using the summary statistics methods STAR [21] and MP-EST [20]. The STAR method estimates the species tree from a distance matrix constructed from the average ranks of gene-coalescence events from gene trees, while MP-EST estimates the species tree from gene trees by maximizing a pseudo-likelihood function of the triplets (rooted 3-taxon statements) of the species tree. Both of these methods require user-provided gene trees for all loci and allow for some missing data. We generated maximum-likelihood gene trees for all loci (NGS and Sanger-sequenced loci) using RAxML v.7.0.4 [50] under the GTRGAMMA model. For each locus, we performed 100 bootstrap replicates, which permits error estimates to be generated on the species trees; this entire process was streamlined using a Perl-scripted pipeline developed by FTB and A. Narechania [51]. We used the Species Tree Analysis Web Server [52] to run all STAR and MP-EST analyses. The STAR and MP-EST programs were run using several different subsets of loci. For the Sanger dataset, we used the complete dataset of 5 loci and subsets of 4 loci (removing CytB), 4 loci (removing COI), and 3 (nucDNA only) loci. For the NGS dataset, we randomly subsampled the entire 377-locus dataset into 200, 100, 50, 25, 10, 5, 4, and 3-locus datasets; these subsets were run five times using different sets of random loci to determine, on average, how discordant the resulting trees were compared to the 377-locus species trees. The topological comparisons between all trees were performed using Robinson-Foulds (RF) distances in the R statistics platform [53] using the package phangorn [54]. The RF distance is a metric that determines the number of bipartitions that differ between trees to indicate the amount of topological discordance between two trees [55]. To show relative differences between the trees using the RF distances, we used the percentage of the maximum RF distance, which was calculated using 2(n - 2), where n is the number of taxa, and n - 2 represents the maximum number of inner branches for a rooted tree [56]. The % RF distance between trees is the ratio of the RF distance divided by the maximum RF distance. We also used the coefficient of variation for each locus-set to determine in the MP-EST and STAR analyses at what point increasing the number of loci reached diminishing returns, where additional loci no longer resulted in an improvement to the species tree; this is indicated by the lowest coefficient of variation value for each locus set. We also compared the NGS and Sanger species trees using the same numbers of loci (5, 4, and 3) to examine how similar the resulting trees are topologically when using the same numbers of loci, again using RF distances.
We next used RAxML to generate concatenated trees using the complete 377-locus NGS dataset and the complete 5-locus Sanger dataset; the Sanger dataset was partitioned by locus and codon position for the protein coding genes (the mtDNA genes were considered a single partition).. These results were compared with the explicit species-tree approaches using % RF distances. To determine how locus number affects bootstrap-support values for the species trees, a mean bootstrap value was taken across the entire tree for each of the NGS subset species trees (200, 100, 50, 25, 10, 5, 4, and 3-locus datasets) and then averaged across the five replicates for each of those subset species trees.
Although the complete 377-locus NGS dataset is too large for the full-coalescent species-tree model implemented in *BEAST [14], we ran an additional series of species trees with subsets of 15 loci to determine if this method could estimate the same topology as the full 377-locus dataset using MP-EST and STAR. A recent study of chickadee phylogenetics [7] found that among several coalescent-based species-tree inference methods, *BEAST was the most robust and consistently converged on the same tree estimated from the authors’ full dataset (40 loci) when using 15 loci. We took five random subsets of 15 NGS loci and estimated five species trees using *BEAST [18] in BEAST v1.8 [57]. In addition, we also ran *BEAST on the 5-locus Sanger dataset to determine how well it performed using a dataset similar to those typically used to infer squamate phylogenies. We used jModelTest v2.1.6 [58] with the Bayesian Information Criteria to choose the most appropriate model of sequence evolution for each locus in the *BEAST analyses, as *BEAST allows for multiple models (details available on Dryad doi:10.5061/dryad.kp400). As in our STAR and MP-EST analyses Mimophis mahfalensis and Ramphiophis rubropuntatus were used as outgroups. The *BEAST analyses were run for 2 × 108 generations each using a log-normal relaxed clock model, a Yule-process speciation prior, and were sampled every 10000 generations. Tracer v1.4 [59] was used to assess stationarity for each of the runs and determine burnin. A summary table of the various methods used with the different datasets and loci is included as an additional file (Additional file 4).
To visualize congruence between resulting gene trees and the species trees, we used the program MetaTree [60]. MetaTree builds a “tree-of-trees” that shows the relationships between alternative phylogenies. This program takes user-inputted phylogenies with fixed sets of taxa to construct a tree that clusters similar topologies together, allowing the user to examine a set of trees and determine how similar topologies are to one another; here we use this program to examine whether gene trees from both the Sanger and NGS datasets are similar to the species tree topology. For our MetaTree analyses, we compared the gene trees from the Sanger dataset to the 377-locus species tree. Since Micropisthodon and Rhamphiophis were not available for all of the Sanger loci (Appendix 1), they were not included and pruned from the species tree for that comparison. For the NGS MetaTree analysis, we used subsets of 50 gene trees (enhancing visual clarity of the results) from the NGS dataset (using the 344 loci with all 25 taxa/individuals) to compare to the 377-locus species tree.
Results
Sequencing
For the Sanger dataset, sequences were aligned in Geneious® using the Geneious alignment algorithm and were checked by eye to assure no gaps in protein-coding genes. The five loci ranged from a minimum of 1.9% (Cmos) to a maximum of 20 % (CytB) uncorrected pairwise divergence across taxa (Rag2 = 2.2 %; Nav intron 5 = 8 %; COI = 17.8 %), with a mean of 10 % uncorrected pairwise divergence for all loci. For all taxa, parsimony informative sites (PI) = 26 % of sites (594/2990 total bp; from 26 PI Cmos–242 PI CytB) and for ingroup taxa only, PI = 25 % of sites (568/2990 total bp; from 20 PI Cmos–235 PI CytB). All but one locus had at least 24 of the 25 taxa (details available in additional files; Additional file 1).
For the NGS dataset, 387 loci were successfully sequenced for an average of 549,304 bp per sample before trimming. A total of 377 loci remained after filtering and orthology assessment. The resulting dataset after aligning in Mafft and trimming/masking as described above contained 513,877 alignment sites. Loci ranged in length from 424–2156 bp, with a mean locus length of 1374 bp. The matrix had very little missing data (1.5 %). All loci had at least one representative from each genus; 91.2 % of loci (344/377 loci) included all 25 terminal taxa, and 99.7 % of loci (376/377 loci) included 24 of the 25 taxa, with a single locus missing two taxa. The uncorrected pairwise divergence for loci ranged from 0.5 − 11.8 % divergent, with a mean of 3.1 % divergence across the matrix. For all taxa across all loci, PI = 8 % of sites (40147/517276 bp total; maximum = 225/1464 bp, minimum = 16/1527 bp) and for ingroup taxa, PI = 6 % of sites (30290/517276 bp total; maximum = 175/1464 bp, minimum = 12/1527 bp); the mean PI per locus = 106 bp for all taxa and 80 bp for ingroup taxa.
Phylogenetic estimates
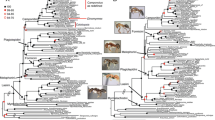
The STAR and MP-EST species trees inferred for Pseudoxyrhophiinae using the complete 377-locus NGS dataset were identical (RF distance = 0 %; Fig. 1). This is our preferred tree for all subsequent topological comparisons and we consider it the species tree for the pseudoxyrhophiines throughout the remainder of the manuscript, as it was estimated using species-tree methods, included the most data and was topologically identical across a variety of methods. The concatenated 377-locus tree and all five replicates using 15 random NGS loci in *BEAST were also identical to the pseudoxyrhophiine topology of the 377-locus species tree (RF distance = 0 %), while the 5-locus Sanger *BEAST tree differed slightly from the full NGS dataset species tree (RF distance =10 %; Table 1). All *BEAST runs were found to reach stationarity by 2 × 108 generations, with no trends in the trace plots suggesting that the MCMC had not converged; we discarded the first 25 % of generations as burnin. Effective sample size values were high (>200) for almost all parameters. The mean RF distances for the NGS datasets of 3–200 loci varied from 1 − 21 % (RF distances ranged from 0 − 33 % for each individual species tree estimated under each subset of loci) when compared to the 377-locus species tree. Species trees inferred using greater numbers of loci produced more similar topologies with respect to the 377-locus species tree; as locus number increased, mean RF distances generally decreased and mean bootstrap support values generally increased (Fig. 2). At ≥50 loci, the placement of a single taxon, Liopholidophis (Figs. 1 and 2) is responsible for all discordance among the NGS subset trees and the 377-locus species tree and the coefficients of variation calculated for MP-EST and STAR indicated that after 50 loci, increasing locus number did improve the topology with respect to matching the 377-locus species tree (all RF values and supporting data available on Dryad doi:10.5061/dryad.kp400). The species trees and concatenated trees from the Sanger datasets had RF distances similar to those from the NGS subsets of <50 loci when compared to the 377-locus species tree, with none being identical to the species tree (Table 1).

Species tree for the pseudoxyrhophiines using STAR and MP-EST (topologies identical) based on the 377-locus NGS data set. Bootstrap values are 100 for all nodes except those labeled with the STAR value first, followed by the MP-EST value. Photos CJR & SR

NGS dataset mean Robinson-Foulds (RF) % distances for each species tree (5 replicates with random loci subsets) with respect to the number of NGS loci used (3, 4, 5, 10, 25, 50, 100, 200) compared to the 377-locus species tree and the mean bootstrap values for each species tree (5 replicates with random loci subsets) with respect to the number of loci used (3, 4, 5, 10, 25, 50, 100, 200, 377)
Mean bootstrap values generally increased for the NGS species-tree analyses as the number of loci increased (Fig. 2). The bootstrap values for the 377-locus trees were high for both methods of species-tree inference, with the placement of only two taxa supported <0.95 (Liopholidophis, Micropisthodon; Fig. 1). The runs from *BEAST generally gave high posterior probability support for the same nodes, with the same two taxa (Liopholidophis, Micropisthodon) having lower posterior probability values (<0.95). Interestingly, the concatenated 377-locus tree had slightly higher support values, with only one of the nodes having a value <1.0 (Micropisthodon + Ithycyphus, bootstrap values = 0.39 concatenated tree/0.52 species tree). This is in contrast to the bootstrap values for the concatenated 5-locus Sanger tree, which had a lower mean bootstrap value of 0.69 across the tree versus 0.97 for the concatenated 377-locus tree.
For the MetaTree comparison of the Sanger dataset to the 377-locus species tree, each Sanger gene has a unique topology and none of the Sanger gene trees were closely clustered with the 377-locus species tree, with the two mtDNA gene trees (COI, CytB) and Rag2 gene tree being the most discordant (Fig. 3). The branches for the gene trees are also relatively long, indicating high degrees of conflict between the topologies and have few shared splits between trees [60]. For the NGS gene trees compared to the 377-locus species tree, few of the gene trees were similar to the full species tree and no gene trees were identical to the species tree; no gene tree topologies were identical to each other as well. We present these results to illustrate the large range of discordance between the species tree and the various gene trees with both the Sanger and NGS data; for brevity, one MetaTree set of 50 loci is shown in comparison with the species tree (Fig. 3; full MetaTree available on Dryad doi:10.5061/dryad.kp400).

Metatree visualization showing similarity of the species tree topology (as shown in Fig. 1) with that of the 5 Sanger loci gene trees (left) and 50 NGS loci gene trees (right). The species tree is labeled for each. Sanger loci gene trees are indicated by locus name, NGS loci gene trees are indicated by number. In both data sets no gene trees were identical to the species tree
Within pseudoxyrhophiines, the three genera with multiple species were monophyletic in all analyses. Several of the resulting clades have been recovered by other studies (e.g., Langaha + (Micropisthodon + Ithycyphus); [39]. Our species tree (Fig. 1) also provides well-supported placement for some enigmatic taxa such as Elapotinus picteti (formerly Exallodontophis albignaci, see [61]), which was found as the sister taxon to a clade including Liophidium, Liopholidophis, Pseudoxyrhopus, Heteroliodon. We also find strong support for a single monophyletic Malagasy clade of pseudoxyrhophiines with respect to the African taxon Duberria. We discuss the taxonomy of the group in detail below.
Availability of supporting data
The datasets supporting the results of this article and the information pertaining to the anchored loci probes are available in the Dryad repository (doi:10.5061/dryad.kp400).
Discussion
Phylogenetic inference and dataset size
The number of loci required to accurately infer phylogenies has generally been addressed in a theoretical framework, but how this translates for empirical studies has not been extensively examined. Here, using datasets with varying numbers of loci and multiple phylogenetic methods, we estimate a well-supported species tree for pseudoxyrhophiine snake genera that suggests a monophyletic radiation of these snakes on Madagascar. Identical tree topologies were inferred using datasets of both 15 and ≥50 loci, but the number of loci necessary for accurate inference depends on the species-tree method.
When using the summary statistics methods STAR and MP-EST, large numbers of loci (≥50) are required to consistently produce phylogenies equal to those using all 377 loci and that the differences between topologies generally decreases as more loci are added to the analyses (Fig. 2). These results are comparable to another recent empirical study which found that increasing locus number improved topological estimates, but that ultimately the number of requisite loci plateaued [7]. There is a threshold where increasing the number of loci no longer provides greater resolution; for our dataset this is ~50 loci for both STAR and MP-EST (Fig. 2) with bootstrap-support values increasing similarly (Fig. 3). While STAR and MP-EST generate strongly supported trees for pseudoxyrhophiines, which is likely due in part to the high quality loci in our dataset, these methods may fail when gene trees are estimated from short, uninformative loci [22]. Additionally, despite the overall increase of support values as loci number increases, the position of some taxa remain poorly supported regardless of the number of loci included (Liopholidophis, Micropisthodon; Fig. 1). In particular, the instability of one taxon (Liopholidophis) is responsible for all topological discordance at 50 or more loci with respect to the subset NGS trees and the 377-locus tree. This suggests that some taxa may remain problematic regardless of how many loci are included. Similarly, a recent study of major snake lineages using 333 anchored phylogenomics loci and species-tree methodologies found low support values within lamprophiids, the family to which pseudoxyrhophiines belong [15]. Previous work indicates that including more individuals per species increases phylogenetic accuracy [25, 62–64] and future studies with multiple individuals per species and full species sampling per genus will determine whether more complete sampling provides better resolution for the weakly supported taxa in the pseudoxyrhophiine phylogeny.
In contrast to the summary statistics methods requiring ≥50 loci, we found that with only 15 loci, *BEAST consistently inferred the same pseudoxyrhophiine topology as the 377-locus trees from STAR and MP-EST. This suggests that for smaller datasets (e.g., <50 loci), full-coalescent methods may outperform other types of species-tree analyses requiring more loci, though the generality of this trend should be investigated. While the matching topologies of the 15-locus *BEAST analyses and all other methods using the complete dataset make us confident in the results from *BEAST here, we acknowledge that full coalescent analyses such as *BEAST should ideally include multiple individuals per terminal taxon, as this greatly improves all aspects of tree estimation [18]. Estimating population sizes in particular requires at least two individuals per species [18], as well as greater numbers of loci when compared to accurately estimating only the tree topology [7]. The results presented here may not be typical when including population-level sampling, poorly defined species, or very large numbers of taxa, since these scenarios may result in shorter branch lengths and represent recent time-scales, which likely require larger numbers of loci for good resolution and support [8]. Furthermore with a dataset that includes hundreds of species, coalescent-based methods such as *BEAST are unsuitable due to computational time and failure of parameters to converge [14, 19].
Unlike the 15-locus NGS subsets used in *BEAST, the 5-locus Sanger dataset was unable to recover the same topology as the 377-locus tree, despite these particular loci having higher average numbers of parsimony informative sites compared to the NGS loci (26 % of Sanger loci sites versus 8 % of NGS loci sites). When using STAR and MP-EST, neither the Sanger datasets nor NGS subsets of 3–5 loci resulted in species trees congruent with the 377-locus tree (Fig. 2; Table 1). Interestingly, the most congruent tree using 3–5 loci was produced using the complete 5-Sanger locus dataset in *BEAST (RF distance = 10 %; Table 1), further demonstrating that for small datasets, a full-coalescent model approach may yield the most accurate topology. Full-coalescent-model approaches may be robust to low numbers of loci for topological inference when a high proportion of gene trees match the species tree [18, 65]. However, our results indicate that datasets of ≤5 loci within any of the species-tree frameworks do not generate trees that fully correspond to the 377-locus tree for pseudoxyrhophiines (Table 1; Fig. 2). Additionally, neither of the two mtDNA gene trees (COI, CytB) closely matches the species tree, further underscoring that mtDNA gene trees, at least in this case, do not suitably represent the species tree (Fig. 3). While many studies use mtDNA gene trees as a proxy for the species tree, our results suggest that relying entirely on a single locus, whether mitochondrial or nuclear, is a potentially risky practice. It is likely that estimating credible species trees using small numbers of loci with conflicting gene-tree histories is impossible. Simulations have shown that when the majority of gene trees are highly discordant with the species tree, as many as 120 loci are necessary to recover the species tree in a coalescent-based framework [65]. Similar to our Sanger dataset results, an empirical study on lizards [66] using varying numbers of loci found that when estimating trees with four loci, *BEAST was unable to correctly infer the species tree, even when only the most informative loci were included. Therefore if a reduced set of loci are discordant it is expected that numerous additional markers are required to generate a credible species tree.
We find no topological difference for the pseudoxyrhophiines between the 377-locus concatenated tree and the 377-locus species tree (RF distance = 0 %). It is expected that when incomplete lineage sorting is low, which is likely when population sizes at the time of speciation are small and the time between speciation events is long, concatenation may accurately infer species relationships [14, 52, 67–70], especially at deep time scales [22]. However, incomplete lineage sorting can still be pervasive at deeper time scales and coalescent-based species-tree methods are able to account for this [27]. Furthermore, concatenation may overestimate branch-lengths [43] and result in positively misleading support values [65]. Finally, species-tree methods may be better suited to modeling the phylogenetic signal of very large datasets compared to concatenation (reviewed in [17]).
Pseudoxyrhophiine generic relationships
The 377-locus species tree for the pseudoxyrhophiines presented here is well supported (Fig. 1); although several (for the most part rare) genera missing from this analysis (three Malagasy genera-Lycodryas, Brygophis, Pararhadinaea; one African genus-Amplorhinus; one Socotran genus-Ditypophis), taxonomic relationships are largely similar to those found in prior studies with respect to affinities between genera based both on morphology as well as molecular datasets, as is much of the overall structure of the tree [30, 39, 71–76]. We found two major clades of pseudoxyrhophiines: one which includes the genera Alluaudina, Compsophis, Dromicodryas, Thamnosophis, Elapotinus, Liophidium, Liopholidophis, Pseudoxyrhopus, and Heteroliodon and a second that includes Leioheterodon, Parastenophis, Phisalixella, Madagascarophis, Langaha, Micropisthodon, and Ithycyphus.
The major subdivision of this Malagasy snake radiation, in two approximately equally diverse clades is intriguing, in that this largely corresponds to a division of the aglyphous genera (with non-grooved teeth, nearly equal in size) and opisthoglyphous genera (with enlarged grooved teeth, at the rear of the maxillae). Historically, this tooth character was considered important for classifying snakes (e.g., [77]), and was used to organize the most recent monographic treatment of the Malagasy snakes [78]. All genera in the Alluaudina, Compsophis, Dromicodryas, Thamnosophis, Elapotinus, Liophidium, Liopholidophis, Pseudoxyrhopus, and Heteroliodon clade are aglyphous, with the exception of Alluaudina and Compsophis. Similarly, all genera in the Leioheterodon, Parastenophis, Phisalixella, Madagascarophis, Langaha, Micropisthodon, and Ithycyphus clade are opisthoglyphous with the exception of Leioheterodon and Micropisthodon. It is worth noting that Leioheterodon have enlarged rear teeth, and are known to be capable of mild envenomation [79]; and that more generally, the dentition of most of the rarer Malagasy snakes have not been recently re-evaluated. Nevertheless, our results presented here do suggest that the evolution in dentition, envenomation, and thus feeding strategies, may have influenced the early diversification of the pseudoxyrhophiine radiation, though the impact of these traits on the evolution of these groups requires further testing.
Inclusion of the continental African pseudoxyrhophiine genus Duberria allows us to address the biogeography of pseudoxyrhophiines in Africa and Madagascar. Many prior studies have supported a single dispersal event from continental Africa to Madagascar for the pseudoxyrhophiines, with the African taxa Amplorhinus and Duberria and the Socotran Ditypophis as sister taxa to the Malagasy species [38, 39, 73, 80]. This suggests the Malagasy species are monophyletic, with subsequent dispersal to the Comoros islands [81, 82]. However, not all previously used datasets/tree-methodologies, even within the same publications, support this scenario, and there is also some evidence for Ditypophis and/or Duberria nested within Malagasy taxa [30, 39, 80]. Since we do not include Amplorhinus or Ditypophis here, we cannot directly comment on their position or biogeographic origins, but our results strongly suggest that the Malagasy genera form a monophyletic group with respect to Duberria. While we do not show all of the summary-statistics species trees generated from the NGS subsets of loci, we note that 81/82 of those species trees place Duberria as the basal taxon to a monophyletic Malagasy Pseudoxyrhophiinae. Future datasets including Amplorhinus and Ditypophis will allow further exploration regarding the biogeography of both continental African and Malagasy pseudoxyrhophiines. Within the aglyphous clade, the genera Elapotinus, Heteroliodon, Liopholidophis, Liophidium, Pseudoxyrhopus have been previously referred to as the “Pseudoxyrhopus group” (minus Liopholidophis in the initial description; [71]) and were originally united based on hinged teeth and tooth replacement pattern [71]. The exact placement of Liopholidophis within this clade remains somewhat unclear as it is poorly supported in our species tree (bootstrap values = 66/65 %; Fig. 1), but importantly all of the discordant trees do place Liopholidophis within this clade. Previous molecular-based studies have indicated that Liopholidophis may be the sister taxon to Liophidium [74] but most studies [30, 39, 72, 73, 76] have found the same relationship presented here, with Liopholidophis being the sister taxon to Heteroliodon + Pseudoxyrhopus, and Liophidium in turn being the sister taxon to this inclusive clade. Although none of these prior studies have included all five genera in the same analysis, our results show the same relationships reported in previous studies [30, 39, 72–74, 76]. Additional sampling within these genera will help determine if these genera are all monophyletic or if Heteroliodon renders Pseudoxyrhopus paraphyletic, as has been previously suggested [40].
The remaining aglyphous genera include two sister taxon pairs; Thamnophis + Dromicodryas, and Alluaudina + Compsophis. Thamnosophis (formerly Bibilava, see [74, 83]) and Dromicodryas are both diurnal and active terrestrial predators, sharing external morphological similarities [78]. A sister-taxon relationship between these genera has been indicated by prior studies [30, 74, 76], and our results continue to support this relationship, as well as the recognition of Thamnosophis as distinct from Liopholidophis (see [84, 85]).
Alluaudina and Compsophis have been shown to share morphological characters with respect to hemipenes [35] and several molecular studies have found Alluaudina and Compsophis as sister taxa [39, 73] as we do here (Fig. 1). However, at least one recent molecular study [30] indicates that Compsophis is sister to Ditypophis and part of the clade that includes the mainland African/Socotran taxa (Amplorhinus, Duberria, Ditypophis). We find very high support for Compsophis being the sister taxon to Alluaudina (Fig. 1), rather than being placed in a clade with Duberria, similar to other studies where Ditypophis, Duberria, and/or Amplorhinus fall outside of the Malagasy pseudoxyrhophiines [39, 73].
Within the mostly opisthoglyphic clade of pseudoxyrhophiines, the currently monotypic Parastenophis was formerly a subgenus within the genus Stenophis (now considered paraphyletic, see [75]), which also included the currently recognized species in the genera Phisalixella and Lycodryas. However, as was suggested in a taxonomic revision of Stenophis [75] and noted in other molecular studies [73], it appears that Parastenophis, a nocturnal tree-snake, is the sister taxon to the large, diurnal and terrestrial Malagasy hognose snakes, Leioheterodon. The highly disparate morphology, ecology, and behavior between these sister genera [75, 79, 86], is intriguing and deserves further investigation. Although Lycodryas was not included in the present study, previous work [75], as well as our own unpublished results, indicate that Lycodryas is likely the sister taxon to Phisalixella. Both of these genera share very similar morphologies, and are nocturnal tree-snakes, that until recently were also included in the genus Stenophis [75]. As previously suggested [75] and indicated in our species tree here (Fig. 1), the sister taxon to Phisalixella (plus Lycodryas) is the similarly nocturnal and elliptical-pupiled Madagascarophis. Madagascarophis is a broadly distributed group of snakes in Madagascar that is also partly arboreal, but lacks the lateral body flattening and slender necks of Phisalixella and Lycodryas.
The remaining clade within the opisthoglyphic pseudoxyrhophiines contains Langaha, Micropisthodon, Ithycyphus, which are mostly diurnal and arboreal snakes (Fig. 1). These relationships have been supported by previous molecular studies [30, 39, 76]. Although the support for Micropisthodon + Ithycyphus is the lowest for any node in the species tree (bootstrap value = 52 %), it was consistently inferred in the species-tree analyses even when using subsets of the NGS dataset and was also found with high support values in the aforementioned studies. The behavior and ecology of Micropisthodon and Langaha are not yet well described, and the evolution and function of the unique head ornamentation in Langaha still not clearly understood [87, 88].
Conclusions
The phylogeny presented here is a first step in using large-scale phylogenomic data to determine relationships for snakes of Madagascar. This phylogeny, produced using both summary statistics and full-coalescent models for species-tree estimation, is well supported and shows a monophyletic clade of Malagasy pseudoxyrhophiines. Within this radiation of snakes Madagascar, we also find strong support for two major subclades which largely correspond to the aglyphous and opisthoglyphous genera, and thus different forms of tooth venom delivery. Our results suggest that while hundreds of loci are not always essential for accurate topological inference, the method used for tree estimation may affect the resulting phylogeny and that for smaller datasets (<50 loci), full-coalescent models of tree inference are likely more accurate than summary statistics methods. Because full-coalescent model-based methods are unable to computationally handle large amounts of data, these summary statistics provide essentially the only current option for generating species trees using hundreds of loci obtained from hundreds of taxa. Importantly, our results indicate that summary statistics methods, given enough loci (~ ≥50), are able to estimate a robust species tree comparable with a full-coalescent model method. However, whether these results are applicable across different study systems is not known and it is unclear if other datasets will give similar results with respect to the numbers of loci necessary for inferring tree topologies with the methodologies used here. Using the maximum number of loci available may be the best strategy for resolving trees, though this does not account for time and expense for developing probes and sequencing these loci. Considering the alternative, where the investigator uses the minimum number of loci due to time or financial constraints, it would be difficult to know a priori which loci in combination yield trees with the most resolution and highest node support. Future NGS studies that include a far more complete sampling of species, and even populations for broadly distributed pseudoxyrhophiine taxa, will inevitably improve our ability to estimate relationships among species, and thus provide the groundwork to examine biogeography and diversification processes that generated this spectacular insular snake diversity.
References
Lemmon AR, Emme SA, Lemmon EM. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst Biol. 2012;61:727–44.
Lemmon EM, Lemmon AR. High-throughput genomic data in systematics and phylogenetics. Annu Rev Ecol Evol Syst. 2013;44:99–121.
Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. J Mol Evol. 1983;19:153–70.
Corl A, Ellegren H. Sampling strategies for species trees: the effects on phylogenetic inference of thenumber of genes, number of individuals, and whether loci are mitochondrial, sex-linked, or autosomal. Mol Phylogenet Evol 2013;67:358–66.
McCormack JE, Harvey MG, Faircloth BC, Crawford NG, Glenn TC, Brumfield RT. A phylogeny of birds based on over 1,500 loci collected by target enrichment and high-throughput sequencing. PLoS One. 2013;8:e54848.
Rokas A, Williams BL, King N, Carroll SB. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature. 2003;425:798–804.
Harris RB, Carling MD, Lovette IJ. The influence of sampling design on species tree inference: a new relationship for the New World chickadees (Aves: Poecile). Evolution (N Y). 2014;68:501–13.
Leaché AD, Rannala B. The accuracy of species tree estimation under simulation: a comparison of methods. Syst Biol. 2011;60:126–37.
Faircloth BC, McCormack JE, Crawford NG, Harvey MG, Brumfield RT, Glenn TC. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst Biol. 2012;61:717–26.
McCormack JE, Faircloth BC, Crawford NG, Gowaty PA, Brumfield RT, Glenn TC. Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species-tree analysis. Genome Res. 2012;22:746–54.
Crawford NG, Faircloth BC, McCormack JE, Brumfield RT, Winker K, Glenn TC. More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs. Biol Lett. 2012;8:783–6.
Smith BT, Harvey MG, Faircloth BC, Glenn TC, Brumfield RT. Target capture and massively parallel sequencing of ultraconserved elements for comparative studies at shallow evolutionary time scales. Syst Biol. 2014;63:83–95.
Leaché AD, Wagner P, Linkem CW, Böhme W, Papenfuss TJ, Chong RA, et al. A hybrid phylogenetic-phylogenomic approach for species tree estimation in African Agama lizards with applications to biogeography, character evolution, and diversification. Mol Phylogenet Evol. 2014;79:215–30.
O’Neill EM, Schwartz R, Bullock CT, Williams JS, Shaffer HB, Aguilar-Miguel X, et al. Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum) species complex. Mol Ecol. 2013;22:111–29.
Pyron RA, Hendry CR, Chou VM, Lemmon EM, Lemmon AR, Burbrink FT. Effectiveness of phylogenomic data and coalescent species-tree methods for resolving difficult nodes in the phylogeny of advanced snakes (Serpentes: Caenophidia). Mol Phylogenet Evol. 2014;81:221–31.
Brandley MC, Bragg JG, Singhal S, Chapple DG, Jennings CK, Lemmon AR, et al. Evaluating the performance of anchored hybrid enrichment at the tips of the tree of life: a phylogenetic analysis of Australian Eugongylus group scincid lizards. BMC Evol Biol. 2015;15:62.
Liu L, Xi Z, Wu S, Davis CC, Edwards SV. Estimating phylogenetic trees from genome-scale data. Ann N Y Acad Sci 2015; doi:10.1111/nyas.12747
Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010;27:570–80.
Bayzid MS, Warnow T. Estimating optimal species trees from incomplete gene trees under deep coalescence. J Comput Biol. 2012;19:591–605.
Liu L, Yu L, Edwards SV. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol Biol. 2010;10:302.
Liu L, Yu L, Pearl DK, Edwards SV. Estimating species phylogenies using coalescence times among sequences. Syst Biol. 2009;58:468–77.
Gatesy J, Springer MS. Phylogenetic analysis at deep timescales: unreliable gene trees, bypassed hidden support, and the coalescence/concatalescence conundrum. Mol Phylogenet Evol. 2014;80:231–66.
Lanier HC, Huang H, Knowles LL. How low can you go? The effects of mutation rate on the accuracy of species-tree estimation. Mol Phylogenet Evol. 2014;70:112–9.
Leaché AD, Harris RB, Rannala B, Yang Z. The influence of gene flow on species tree estimation: a simulation study. Syst Biol. 2014;63:17–30.
Patel S, Kimball RT, Braun EL. Error in phylogenetic estimation for bushes in the Tree of Life. J Phylogenetics Evol Biol. 2013;1:110.
Knowles LL, Lanier HC, Klimov PB, He Q. Full modeling versus summarizing gene-tree uncertainty: method choice and species-tree accuracy. Mol Phylogenet Evol. 2012;65:501–9.
Lanier HC, Knowles LL. Applying species-tree analyses to deep phylogenetic histories: challenges and potential suggested from a survey of empirical phylogenetic studies. Mol Phylogenet Evol. 2014;83:191–199.
Ilves KL, López-Fernández H. A targeted next-generation sequencing toolkit for exon-based cichlid phylogenomics. Mol Ecol Resour. 2014;14:802–11.
Vidal N, Delmas A-S, David P, Cruaud C, Couloux A, Hedges SB. The phylogeny and classification of caenophidian snakes inferred from seven nuclear protein-coding genes. C R Biol. 2007;330:182–7.
Pyron RA, Burbrink FT, Wiens JJ. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol Biol. 2013;13:93.
Pyron RA, Burbrink FT, Colli GR, de Oca ANM, Vitt LJ, Kuczynski CA, et al. The phylogeny of advanced snakes (Colubroidea), with discovery of a new subfamily and comparison of support methods for likelihood trees. Mol Phylogenet Evol. 2011;58:329–42.
Andreone F, Raxworthy CJ. The colubrid snake Brygophis coulangesi (Domergue 1988) rediscovered in north-eastern Madagascar. Trop Zool. 1998;11:249–57.
Raxworthy CJ, Nussbaum RA. A review of the Madagascan snake genera Pseudoxyrhopus, Pararhadinaea, and Heteroliodon (Squamata: Colubridae). 1994.
Cadle JE, de Freitas S, Penny ND, Adams PA, Gonzales VA, Castañeda GG, et al. Sexual dimorphism and reproductive biology in the Malagasy snake genus Liopholidophis (Lamprophiidae: Pseudoxyrhophiinae). Proc Calif Acad Sci. 2009;60:461.
Ziegler T, Vences M, Glaw F, Böhme W. Genital morphology and systematics of Geodipsas Boulenger, 1896 (Reptilia: Serpentes: Colubridae), with description of a new genus. Rev Suisse Zool. 1997;104:95–114.
Pyron RA, Burbrink FT. Ecological and evolutionary determinants of species richness and phylogenetic diversity for island snakes. Glob Ecol Biogeogr. 2014;23:848–56.
Meirte D. New Records of Leioheterdon madagascariensis from the Comoros. J Herpetol Assoc Africa. 1993;42:21–3.
Kelly CMR, Barker NP, Villet MH, Broadley DG. Phylogeny, biogeography and classification of the snake superfamily Elapoidea: a rapid radiation in the late Eocene. Cladistics. 2009;25:38–63.
Nagy ZT, Joger U, Wink M, Glaw F, Vences M. Multiple colonization of Madagascar and Socotra by colubrid snakes: evidence from nuclear and mitochondrial gene phylogenies. Proc R Soc B Biol Sci. 2003;270:2613–21.
Nagy ZT, Sonet G, Glaw F, Vences M. First large-scale DNA barcoding assessment of reptiles in the biodiversity hotspot of Madagascar, based on newly designed COI primers. PLoS One. 2012;7:e34506.
Cranston KA, Hurwitz B, Ware D, Stein L, Wing RA. Species trees from highly incongruent gene trees in rice. Syst Biol. 2009;58:489–500.
Salas-Leiva DE, Meerow AW, Calonje M, Griffith MP, Francisco-Ortega J, Nakamura K, et al. Phylogeny of the cycads based on multiple single-copy nuclear genes: congruence of concatenated parsimony, likelihood and species tree inference methods. Ann Bot. 2013;112:1263–78.
Ruane S, Bryson RW, Pyron RA, Burbrink FT. Coalescent species delimitation in milksnakes (genus Lampropeltis) and impacts on phylogenetic comparative analyses. Syst Biol. 2014;63:231–50.
Vences M, Wollenberg KC, Vieites DR, Lees DC. Madagascar as a model region of species diversification. Trends Ecol Evol. 2009;24:456–65.
Utiger U, Helfenberger N, Schätti B, Schmidt C, Ruf M, Ziswiler V. Molecular Systematics and Phylogeny of Old and New World Ratsnakes, Elaphe Auct., and Related Genera (Reptilia, Squamata, Colubridae). Russian Journal of Herpetology 2013;9:105–124.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Center for Anchored Phylogenomics [www.anchoredphylogeny.com]. Accessed Oct. 5, 2015
Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010;2010:pdb.prot5448.
Rokyta DR, Lemmon AR, Margres MJ, Aronow K. The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genomics. 2012;13:312.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
The Burbrink Lab Software [http://csivc.csi.cuny.edu/Frank.Burbrink/files/Software.html]. Accessed Oct. 5, 2015
Shaw TI, Ruan Z, Glenn TC, Liu L. STRAW: Species TRee Analysis Web server. Nucleic Acids Res. 2013;41(Web Server issue):W238–41.
R Core Team. R: A language and environment for statistical computing. 2014.
Schliep KP. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–3.
Robinson DF, Foulds LR. Comparison of phylogenetic trees. Math Biosci. 1981;53:131–47.
Kuhner MK, Yamato J. Practical performance of tree comparison metrics. Syst Biol. 2015;64:205–14.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–73.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772.
Rambaut A, Drummond AJ. Tracer v1. 5. 2009: http://tree.bio.ed.ac.uk/software/tracer/. Accessed Oct. 5, 2015
Nye TMW. Trees of trees: an approach to comparing multiple alternative phylogenies. Syst Biol. 2008;57:785–94.
Kucharzewski C, Raselimanana AP, Wang C, Glaw F. A taxonomic mystery for more than 150 years: Identity, systematic position and Malagasy origin of the snake Elapotinus picteti Jan, 1862, and synonymy of Exallodontophis Cadle, 1999 (Serpentes: Lamprophiidae). Zootaxa. 2014;3852:179–202.
McCormack JE, Huang H, Knowles LL. Maximum likelihood estimates of species trees: how accuracy of phylogenetic inference depends upon the divergence history and sampling design. Syst Biol. 2009;58:501–8.
Maddison WP, Knowles LL. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 2006;55:21–30.
Huang H, He Q, Kubatko LS, Knowles LL. Sources of error inherent in species-tree estimation: impact of mutational and coalescent effects on accuracy and implications for choosing among different methods. Syst Biol. 2010;59:573–83.
Edwards SV, Liu L, Pearl DK. High-resolution species trees without concatenation. Proc Natl Acad Sci U S A. 2007;104:5936–41.
Camargo A, Avila LJ, Morando M, Sites JW. Accuracy and precision of species trees: effects of locus, individual, and base pair sampling on inference of species trees in lizards of the Liolaemus darwinii group (Squamata, Liolaemidae). Syst Biol. 2012;61:272–88.
Townsend TM, Mulcahy DG, Noonan BP, Sites JW, Kuczynski CA, Wiens JJ, et al. Phylogeny of iguanian lizards inferred from 29 nuclear loci, and a comparison of concatenated and species-tree approaches for an ancient, rapid radiation. Mol Phylogenet Evol. 2011;61:363–80.
Wielstra B, Arntzen JW, van der Gaag KJ, Pabijan M, Babik W. Data concatenation, bayesian concordance and coalescent-based analyses of the species tree for the rapid radiation of triturus newts. PLoS One. 2014;9:e111011.
McVay JD, Carstens BC. Phylogenetic model choice: justifying a species tree or concatenation analysis. J Phylogen Evolution Biol 1:114. doi:10.4172/2329-9002.1000114
Lambert SM, Reeder TW, Wiens JJ. When Do Species-Tree and Concatenated Estimates Disagree? An Empirical Analysis with Higher-Level Scincid Lizard Phylogeny. Mol Phylogenet Evol 2014;82:146–155.
Cadle JE. The dentition, systematics, and phylogeny of Pseudoxyrhopus and related genera from Madagascar (Serpentes: Colubridae), with descriptions of a new species and a new genus. Bull Museum Comp Zool. 1999;155:381–443.
Franzen M, Raselimanana A, Nagy Z, D’Cruze N, Glaw F, Vences M, et al. A new black-bellied snake (Pseudoxyrhophiinae: Liophidium) from western Madagascar, with notes on the genus Pararhadinaea. Amphibia Reptilia. 2009;30:173–83.
Vidal N, Branch WR, Pauwels OSG, Hedges SB, Broadley DG, Wink M, et al. Dissecting the major African snake radiation: a molecular phylogeny of the Lamprophiidae Fitzinger (Serpentes, Caenophidia). Zootaxa. 2008;1945:51–66.
Glaw F, Nagy ZT, Franzen M, Vences M. Molecular phylogeny and systematics of the pseudoxyrhophiine snake genus Liopholidophis (Reptilia, Colubridae): evolution of its exceptional sexual dimorphism and descriptions of new taxa. Zool Scr. 2007;36:291–300.
Nagy ZT, Glaw F, Vences M. Systematics of the snake genera Stenophis and Lycodryas from Madagascar and the Comoros. Zool Scr. 2010;39:426–35.
Vieites DR, Ratsoavina FM, Randrianiaina R-D, Nagy ZT, Glaw F, Vences M. A rhapsody of colours from Madagascar: discovery of a remarkable new snake of the genus Liophidium and its phylogenetic relationships. Salamandra. 2010;46:1–10.
Boulenger GA. Catalogue of the snakes in the British museum (Natural History), Volume 2. London: British Museum London; 1894.
Guibé J. Les serpents de madagascar. Mémoires l’Institut Sci Madagascar (sér A, Biol Anim. 1958;12:189–260.
Glaw F, Vences M. Amphibians and Reptiles of Madagascar. 3rd ed. Verlag: Cologne, Germany; 2007.
Lawson R, Slowinski JB, Crother BI, Burbrink FT. Phylogeny of the Colubroidea (Serpentes): new evidence from mitochondrial and nuclear genes. Mol Phylogenet Evol. 2005;37:581–601.
Glaw F, Nagy ZT, Köhler J, Franzen M, Vences M. Phylogenetic relationships of a new species of pseudoxyrhophiine snake (Reptilia: Lamprophiidae: Thamnosophis) suggest a biogeographical link between western and northern Madagascar. Org Divers Evol. 2009;9:13–22.
Hawlitschek O, Nagy ZT, Glaw F. Island evolution and systematic revision of Comoran snakes: why and when subspecies still make sense. PLoS One. 2012;7:e42970.
Cadle JE, Ineich I. Nomenclatural status of the Malagasy snake genus Bibilava Glaw, Nagy, Franzen, and Vences, 2007: resurrection of Thamnosophis Jan and designation of a lectotype for Leptophis lateralis Duméril, Bibron, and Duméril (Serpentes: Colubridae). Herpetol Rev. 2008;39:285.
Mocquard F. Description de quelques reptiles et d’un batracien nouveaux de la collection du Muséum. Bull du Muséum Natl d’Histoire Nat Paris 1904:10;301–309.
Cadle JE. Snakes of the genus Liopholidophis (Colubridae) from eastern Madagascar: New species, revisionary notes, and an estimate of phylogeny. Bull Museum Comp Zool. 1996;154:369–464.
Mori A, Randriamboavonjy TM. Field observation of maternal attendance of eggs in a madagascan snake, Leioheterodon madagascariensis. Curr Herpetol. 2010;29:91–5.
Krysko KL. Feeding behaviour of the Madagascar leaf‐nosed snake, Langaha madagascariensis (Serpentes: Colubridae: Pseudoxyrhophiinae), with an alternative hypothesis for its bizarre head structure. 2005.
Tingle JL. Field observations on the behavioral ecology of the Madagascan Leaf-Nosed Snake, Langaha madagascariensis. Herpetol Conserv Biol. 2012;7:442–8.
Acknowledgements
Field studies in Madagascar were made possible due to the assistance of the Ministries des Eaux et Forêts, Madagascar National Parks, and the Université d’Antananarivo, Departement de Biologie Animale. Research support was provided by the National Science Foundation DEB 1257610 (CJR) and DEB 1257926 (FTB), the American Museum of Natural History Gerstner Scholars Program (Gerstner Family Foundation), and the Richard Guilder Graduate School. Thanks to R. Lawson†, the American Museum of Natural History Ambrose Monell Cryo Collection (J. Feinstein), and the University of Michigan Museum of Zoology (G. Schneider, R. Nussbaum) for tissue loans. Thanks to X. Chen for aiding in extract preparation, E. Myers for comments that substantially improved this manuscript, T. Nye for help in running MetaTree, and A. Kuhn for computational aid.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SR produced the Sanger dataset, conducted the analyses, contributed to the conception of the study, and drafted the manuscript. CJR contributed to the conception of the study, helped to draft the manuscript, and contributed to the sample collection. ARL & LML produced the anchored phylogenomics dataset. FTB contributed to the conception of the study and helped to draft the manuscript. All authors contributed to the editing of the manuscript and approved the final version.
Additional files
Additional file 1:
Samples used in analyses and which loci were successfully sequenced (NGS=Next generation sequencing; Cmos, COI, CytB, Rag2, and NAV intron 5=Sanger sequencing). (XLSX 11 kb)
Additional file 2:
PCR and Sequencing Protocols. All PCR reactions were 10 μL reactions consisting of 5 μL of GoTaq® Green Master Mix, 3 μL of H2O, 0.5 μL each of forward and reverse primers at a 10 μM concentration, and 1 μL of DNA extract. Samples were incubated at 96 °C for 15 min initially, 96 °C for 45 s, followed by 45 s at the appropriate temperature for the primer pair (see below), with a 72 °C extension period for 1 min. This procedure, minus the initial 15 min incubation period, was repeated for 35 cycles. Reactions were cleaned with 2 μL of ExoSap-IT® following the Exosap-IT® protocol. Sequencing reactions used the same primers as for the PCR reactions. We used the BigDye® Terminator v3.1 Cycle Sequencing Kit; each sequencing reaction consisted of 0.2 μL of ABI BigDye® Terminator Ready Reaction Mix, 1.5 μL of ABI BigDye® 5X Sequencing Buffer, 4.3 μL of H2O, 1 μL of the cleaned PCR reaction template, and 0.5 μL of the 10 μM primer for each direction. Sequencing reactions were incubated initially at 96 °C for 1 min, followed by 96 °C for 10 s, 50 °C for 5 s, and 60 °C for 1 min 15 s and repeated for 15 cycles, minus the initial 1 min incubation period; the reaction was then incubated at 96 °C for 10 s, 50 °C for 5 s, and 60 °C for 1 min 30 s and repeated for 6 cycles; the reaction was then incubated at 96 °C for 10 s, 50 °C for 5 s, and 60 °C for 2 min and repeated for 5 cycles. Sequencing reactions were cleaned prior to sequencing using ethanol precipitation. (PDF 155 kb)
Additional file 3:
Summary of the Anchored Phylogenomics assembly results. (XLSX 17 kb)
Additional file 4:
Tree estimation methods, with the dataset type (Sanger and NGS) and the number(s) of loci used with each method. (PDF 4 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ruane, S., Raxworthy, C.J., Lemmon, A.R. et al. Comparing species tree estimation with large anchored phylogenomic and small Sanger-sequenced molecular datasets: an empirical study on Malagasy pseudoxyrhophiine snakes. BMC Evol Biol 15, 221 (2015). https://doi.org/10.1186/s12862-015-0503-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-015-0503-1