
Abstract
Background
Caribbean anole lizards (Dactyloidae) have frequently been used as models to study questions regarding biogeography and adaptive radiations, but the evolutionary history of Central American anoles (particularly those of the genus Norops) has not been well studied. Previous work has hypothesized a north-to-south dispersal pattern of Central American Norops, but no studies have examined dispersal within any Norops lineages. Here we test two major hypotheses for the dispersal of the N. humilis/quaggulus complex (defined herein, forming a subset within Savage and Guyer’s N. humilis group).
Results
Specimens of the N. humilis group were collected in Central America, from eastern Mexico to the Canal Zone of Panama. Major nodes were dated for comparison to the geologic history of Central America, and ancestral ranges were estimated for the N. humilis/quaggulus complex to test hypothesized dispersal patterns. These lineages displayed a northward dispersal pattern. We also demonstrate that the N. humilis/quaggulus complex consists of a series of highly differentiated mitochondrial lineages, with more conserved nuclear evolution. The paraphyly of the N. humilis species group is confirmed. A spatial analysis of molecular variance suggests that current populations are genetically distinct from one another, with limited mitochondrial gene flow occurring among sites.
Conclusions
The observed south-to-north colonization route within the Norops humilis/quaggulus complex represents the first evidence of a Norops lineage colonizing in a south-to-north pattern, (opposite to the previously held hypothesis for mainland Norops). One previously described taxon (N. quaggulus) was nested within N. humilis, demonstrating the paraphyly of this species; while our analyses also reject the monophyly of the Norops humilis species group (sensu Savage and Guyer), with N. tropidonotus, N. uniformis, and N. marsupialis being distantly related to/highly divergent from the N. humilis/quaggulus complex. Our work sheds light on mainland anole biogeography and past dispersal events, providing a pattern to test against other groups of mainland anoles.
Similar content being viewed by others


Background
Central America is an important region for understanding historical biogeography and intercontinental dispersal in the Western Hemisphere. Mesoamerica has served as a pathway of dispersal for many taxa, some of which originated in North America and dispersed south (e.g. Pitvipers [1], but see [2]; Bolitoglossine salamanders [3]) and others that moved north following a southern origin (e.g. hylid frogs [4]; toads [5]). Lower Central America (LCA) displays extraordinary high levels of species diversity in many taxonomic groups including insects [6], fish [7, 8] and herpetofauna [9, 10]. Many phylogeographic studies in LCA have been conducted on a diverse assemblage of organisms, including trees [11, 12], mammals [13–15], fish [16, 17], and amphibians [3, 18, 19]. In reptiles, there have been some work on squamate biogeography [1, 20–24], but the majority of these studies have centered on snakes, with no published multi-locus phylogeographic studies on lizards in Central America (Hasbún et al. [25] uses a single mtDNA marker to investigate biodiversity in a species of Ctenosaura). Given the rich history of scientific work on lizards in the Caribbean, especially anoles (family Dactyloidae [26–29]), the lack of biogeographic work on mainland lizards is somewhat surprising.
Anoles, present throughout Central America [10, 30], are an ideal group to remedy this deficiency. Several species groups in the genus Norops are widespread, and present excellent opportunities to study the colonization of Mesoamerica. Among these taxa, we selected the Norops humilis species group as a model for refining biogeographic hypotheses for Central American lizards in general, and Central American anoles in particular. The N. humilis species group (described below) ranges from Mexico to Panama, although the timing of diversification within this group has not been determined. Our first objective was to estimate the date of origin for the group, which may in part provide support for one of two hypotheses. Hypothesis 1 (H1; see Fig. 1) predicts a north-to-south dispersal pattern, while Hypothesis 2 (H2; see Fig. 1) predicts a south-to-north dispersal pattern. Our second objective was to test biogeographic patterns within this group, as interpreted from the above hypotheses. The N. humilis species group [31] is an ideal group for examining these hypothesized patterns as well as their timing. As originally defined, the group includes N. compressicauda, N. humilis, N. notopholis, N. tropidonotus, and N. uniformis. To these we add the newly described N. wampuensis [32] and N. quaggulus [33], plus N. marsupialis, a former subspecies of N. humilis that has recently been treated as a species [34]. Several studies suggest the N. humilis species group may be paraphyletic [35–37]. Therefore, our third objective was to confirm the polyphyly of the N. humilis species group. We also examined the N. humilis/quaggulus complex, which we define to include N. humilis plus N. quaggulus, a sister species described based on putatively unique hemipenial morphology and evidence of reciprocal monophyly based on mitochondrial genes [33]. This complex also has a wide distribution (Panama to Honduras). Our analyses provide the opportunity to further evaluate the relationship between N. humilis and N. quaggulus, as well as to test the specific status of N. marsupialis.

Representation of two alternative dispersal hypotheses for the N. humilis/quaggulus complex investigated in this study. Solid black lines demark the rough geologic boundaries of major tectonic blocks as they correspond to present-day Central America. The dotted line marks the subdivision of the Chortis block into highland and lowland regions. The phylogenetic representation of the first dispersal hypothesis (H1) is indicated by the dotted lines; the alternative (H2) is shown with solid lines
Methods
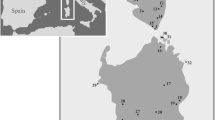
Samples from several of the species in the Norops humilis species group (N. humilis/quaggulus, N. marsupialis, N. tropidonotus and N. uniformis) were collected throughout Central America or acquired from tissue loans (Fig. 2, Additional file 1: Appendix S1). For the N. humilis/quaggulus complex, 147 specimens were sampled throughout Honduras, Nicaragua, Costa Rica, and Panama. DNA was extracted from liver or muscle tissues using Qiagen DNeasy kits (Qiagen, USA). PCR was conducted following protocol and using lizard-specific primers from Macey et al. [38] for mtDNA and Nicholson [35] for nucDNA. Purified PCR reactions were sent to Michigan State University’s Research and Technology Support Facility for sequencing of the following gene regions: NADH-ubiquinone oxidoreductase chain 2 (ND2), tRNATrp, tRNAAla, tRNAAsn, tRNACys, tRNATyr, origin of light strand replication, and partial CO1 from mitochondrial DNA. A subset of these samples representing each major lineage detected by the mitochondrial analyses was selected for nuclear DNA analysis using a nuclear internal transcribed spacer unit (ITS-1). A nuclear marker was included because many studies have shown the limitations of mtDNA to reflect levels of gene flow or the extent of reproductive isolation among populations (e.g. [39, 40]; see [41] for a review). Analyses based solely on mtDNA can also provide results that are in conflict with the nuclear genome [42, 43].

Geographic distribution of Norops humilis species group lineages used in this study. This map of the Norops humilis species group denotes the six main lineages in the N. humilis/quaggulus complex as hypothesized by the phylogenetic reconstructions from the Maximum Likelihood and Bayesian analyses. Note that two localities contain multiple lineages of N. humilis. An additional site for N. tropidonotus from Veracruz, Mexico, is not included in this map. Abbreviations: PAN = Panama, CR = Costa Rica, NICA = Nicaragua, HON = Honduras, ES = El Salvador, GUA = Guatemala, BEL = Belize and MEX = Mexico
Within the N. humilis species group, a total of 1451 aligned bp of mitochondrial data were collected for 192 individuals and 1522 aligned bp of the nuclear gene region ITS-1 was collected for 48 individuals. All newly acquired data were combined with published sequences for 65 additional Norops species (Additional file 2: Appendix S2) in order to investigate the monophyly of the N. humilis species group and of the N. humilis/quaggulus complex. Sequences were edited using Sequencher 4.9 (GeneCodes Corp., Ann Arbor, MI, USA) and aligned initially using MUSCLE in MEGA 5.2.2 [44], then adjusted manually.
The relatively continuous geographic distribution of the N. humilis/quaggulus complex combined with a lack of distinct phenotypic differences made a priori separation of sampling localities into populations somewhat arbitrary. In order to describe the genetic structure and identify the best maximally differentiated number of populations within the N. humilis/quaggulus complex, a spatial analysis of molecular variance (SAMOVA 1.0) was used to group 15 localities selected in this study (each with n ≥ 4 specimens; Additional file 3: Appendix S3) into a number of user-defined clusters (K). For each cluster, the proportion of total genetic variance (high F CT index) due to differences between populations [45] was estimated and evaluated to select the optimal number of genetic groups. A simulated annealing process for each cluster (K = 2 to 14) was repeated 1023 times for each of 100 sets of initial conditions to ensure that the final population groups were not affected by the initial configuration. Significance of the F SC index was used to obtain the suggested number of genetic groupings for the localities selected [46]. This analysis was based on sequences for the mitochondrial region only, since ITS-1 data were only obtained for a limited number of specimens, and were not sufficient for population genetic analysis.
Phylogenetic estimations were conducted under Bayesian analytical methods. PARTITIONFINDER [47] was used to select models of evolution as well as to examine the suitability of partitioning each dataset (mitochondrial, nuclear, and concatenated). In all cases each gene (including tRNAs) was entered as a potential partition. Protein coding genes were further partitioned by codon position for the PartitionFinder analysis. Branch lengths were unlinked, all models of evolution available in MRBAYES 3.2.2 [48] were tested, and a BIC information criterion and greedy algorithm were used. The PARTITIONFINDER analysis recommended a HKY + I + Γ model of evolution for the mtDNA segment, and GTR + Γ for the nuclear data with no partitioning recommended within either region. For the combined dataset, two partitions were recommended (mt and nuc) with GTR + I + Γ as the selected model of evolution for both partitions.
Bayesian analyses for each dataset (as above) were conducted using MRBAYES and BEAST 1.7.5 [49]. The phylogenetic hypotheses using MRBAYES 3.2.2 was developed using 20 million generations, sampling every 1000 generations for two independent runs using four Markov chains with node support evaluated via posterior probabilities (BAPP). We evaluated stationarity of variables by examining our output via TRACER v1.5 [50]. The first 20 % of trees were discarded as burnin and a majority rule consensus tree was generated to summarize the post-burnin results. With the phylogeny constructed using BEAST, we estimated divergence dates, employing a lognormal relaxed clock and a calibration rate of 0.65 % per million years for mtDNA [38] with a Yule Process speciation prior. This rate has been used for our selected mitochondrial gene region for reptile and amphibian groups [51, 52] including anoles (e.g. [53, 54]; mean of a prior distribution, SD = 0.0025 for ucld.mean parameter). The calibration rate was only applied to the mtDNA analysis, as there are no calibration rates available for the nuclear ITS-1 gene. In addition to Bayesian analyses, ML analyses were conducted on each dataset using MEGA 5.2.2 with node support evaluated via bootstrap analyses (MLBS) based on 2000 replications. In addition to the constructed topologies, pairwise genetic distance (uncorrected-p) was estimated between all individuals included in the phylogenetic analyses using both the mitochondrial and concatenated datasets. These distances were compared among all major lineages (as indicated by phylogenetic analysis) of N. humilis species group members, as well as within each species or lineage using MEGA 5.2.2.
Phylogeographic hypotheses were tested using likelihood-based inference in LAGRANGE [55, 56]. The analysis was conducted using the phylogeny constructed with BEAST for mtDNA only, as divergence dating was not available for the nuclear (ITS) tree. Dispersal events between regions were examined and ancestral ranges were estimated within the N. humilis/quaggulus complex to evaluate the support for each of the hypotheses presented above (Fig. 1). The geographic regions coded were (1) the Caribbean versant of the Chorotega Block, (2) the Pacific versant of the Chorotega Block, (3) the lowlands of the Chortis Block and (4) Highlands of the Chortis Block. These areas represent the entire range of the N. humilis/quaggulus complex and were selected based on major geographic barriers. For the analysis, dispersal was constrained to be possible only between adjacent regions because the anoles studied herein are both small and ground dwelling, which would make rapid dispersal to very distant and non-adjacent regions unlikely. If the complex originated in the northern part of the range (i.e. Honduras or northern Nicaragua) with younger clades in the south, it would indicate a north-to-south dispersal, while a southern origin would support a south-to-north colonization route.
Results
All phylogenetic hypotheses demonstrate the paraphyly of the N. humilis species complex (Figs. 3, 4, 5 and 6). Each phylogenetic analysis is discussed in detail below in regard to the N. humilis/quaggulus complex. Six main lineages were present when using the concatenated dataset (mt + nucDNA) for both Bayesian (Fig. 3) and ML (not shown) phylogenetic analyses. One lineage (lineage 6) corresponded to samples currently classified as N. quaggulus: a clade nested within the rest of the N. humilis/quaggulus complex, rendering the current species designation of N. humilis paraphyletic.

Bayesian phylogenetic hypothesis from both mitochondrial and nuclear genes combined. Bayesian posterior probabilities are located at all major nodes

Bayesian reconstruction of Norops using the mitochondrial genes. Genes used were ND2, tRNATrp, tRNAAla, tRNAAsn, tRNACys, tRNATry, origin of light strand replication, and partial CO1, including multiple samples of N. humilis species group. The numbers at nodes are posterior probabilities followed by bootstrap values for nodes that were congruent between the analyses (i.e. posterior/bootstrap)

Bayesian reconstruction of Norops using the nuclear gene ITS-1. This tree includes multiple samples of N. humilis species group members. The numbers at nodes are posterior probabilities followed by bootstrap values for nodes that were congruent between the analyses (i.e. posterior/bootstrap)

Divergence dating of the N. humilis group in relation to other Norops. Tree was created in BEAST 1.7.5 using a calibration rate from Macey et al. [38]. Nodes with <0.95 posterior probabilities have their values listed on the tree. Where present, all grey bars on the nodes correspond to the 95 % confidence interval of the date
The trees from our mitochondrial analyses were similar in topology to those from the combined data with the only difference being a lineage of the N. humilis/quaggulus complex restricted to the Pacific versant of Costa Rica and Nicaragua (N. humilis lineage 3, Figs. 2, 3, 4, 5 and 6). In the Bayesian analysis of mtDNA, this lineage formed two separate clades: one was comprised of samples from Monteverde, Costa Rica and the other was comprised of all additional nearby samples from Guanacaste, Costa Rica plus specimens from southern Nicaragua (Fig. 4). These samples formed a single lineage for the concatenated dataset using both Bayesian (Fig. 3) and ML as well as for the mtDNA dataset using ML. We also constructed a phylogenetic tree on the mtDNA dataset with BEAST. Results of the BEAST analysis provided a topology in agreement with the tree constructed using MRBAYES for all major lineages (Fig. 6). When considering 0.65 % per lineage per million years for the mtDNA data, the origin of the N. humilis/quaggulus complex was estimated to be 17.2 Myr BP (95 % CI = 14.2-20.6 Myr BP). The stem age for each of the distinct mitochondrial lineages of the N. humilis/quaggulus complex were estimated to have originated between 10.3 and 3.2 Myr BP (Fig. 6).
When using only the nuclear ITS-1 dataset, lineages were less divergent than either mitochondrial dataset described above. The Bayesian analyses differed slightly from the mtDNA and combined analyses by presenting only four main clades of the N. humilis/quaggulus complex (Fig. 5). Primary differences included the presence of a conjoined Panamanian/eastern Costa Rican clade (lineages 1 and 2), a single Pacific Costa Rican clade (monophyletic lineage 3), one clade (lineage 4) of a single Caribbean Costa Rican locality (la Lola, Limon Province) and the remaining Caribbean Costa Rican samples (lineages 5 and 6) grouping together in an unresolved clade. The ML analysis of ITS-1 reduced the N. humilis/quaggulus complex to three main clades: Eastern Costa Rica/Panama (lineages 1 and 2), Pacific Costa Rica plus one Caribbean site (lineages 3 and 4), and the remaining Caribbean Costa Rican samples (lineages 5 and 6; figure not shown). However, the clades in the ML analysis lack significant support, possibly due to the conservative nature of ITS-1, which failed to yield significant variation among closely related lineages.
When we calculated pairwise genetic distances using the concatenated dataset, genetic distance (uncorrected p) was very high between the N. humilis/quaggulus complex and other members of the N. humilis species group: 18-20 % from N. tropidonotus, 20–25 % from N. uniformis, and 21–26 % from N. marsupialis. In addition, within the N. humilis/quaggulus complex, genetic distances (uncorrected p, Table 1) ranged up to 10 % among all N. humilis/quaggulus complex lineages. Distances within lineage 6, shown as N. quaggulus, ranged up to 3 %, and were up to 9 % within each of the other lineages, shown as N. humilis 1–5 (>2 % for all clades except N. humilis lineage 3).
SAMOVA was used to designate the optimal number of distinct genetic clusters within the N. humilis/quaggulus complex. Grouping of the 15 sample localities into 12 clusters by SAMOVA yielded an F SC value (0.03) that approached zero (Additional file 3: Appendix S3). A lack of shared haplotypes among the localities used in the analysis indicated limited gene flow, at least for the genes in question. K = 12 was chosen as the most likely number of clusters, because it maximized the variation among clusters (F CT = 0.86) while minimizing the variation among localities within clusters (F SC ). While F CT increased for K = 13 and K = 14, the difference was slight (F CT = 0.86 and 0.87 respectively). Rodríguez-Robles et al. [46] stated that F CT should peak where F SC = 0, so we use F SC as the determining factor in our analysis, given the low magnitude of increases in F CT created by additional partitioning. The variance among clusters (85.5 %) accounted for the majority of genetic diversity within the N. humilis/quaggulus complex (F CT = 0.86), 0.4 % by the variation among localities within these clusters (F SC = 0.03), and 14.1 % by the variation among each of the localities (F ST = 0.86). The 12 populations identified in the N. humilis/quaggulus complex by the SAMOVA analysis was double the number of major lineages we designate in our phylogenetic analyses (n = 6), because the population analysis identified additional substructure that was not recovered in our phylogenetic hypothesis.
The LAGRANGE analysis using the tree recovered from BEAST yielded a log likelihood of –lnL = 22.4 at the root node with dispersal and extinction probabilities of 0.01 and 7.1e-10 respectively. The ancestral range estimation indicates a south-to-north distribution for the N. humilis/quaggulus complex (Fig. 7). The Panamanian lineage (N. humilis 1) is sister to the rest of the N. humilis/quaggulus complex. A clade containing all Honduras and Caribbean Nicaragua specimens was younger than any clades restricted to Costa Rican or Panamanian samples in both ML and Bayesian analyses. Coupled with our ancestral range estimation, the topology suggests that the N. humilis/quaggulus complex originated in the south before dispersing northwards.

Ancestral range estimation of the Norops humilis/quaggulus complex created using LAGRANGE. The probabilities for the present scenario are indicated at each node unless the scenario has a probability of 1, with all probabilities reflected on each internal branch. Probabilities correspond to the branches stemming from a node, rather than the node itself, they are not reflective of the confidence in the topology. Geographic regions are coded as follows: Pacific versant of the Chorotega Block (A), Caribbean versant of the Chorotega Block (B), lowlands Chortis Block (C) and highlands Chortis Block (D). Branches indicating simultaneous occupation of regions are black (AB), orange (BC), yellow (BD), green (CD), or gray (three or more regions). PLEASE NOTE the colors on this figure pertain to geographical regions, whereas in all other figures correspond to lineages. The tree used for this analysis was created in BEAST v1.7.5 and only contains mitochondrial data since divergence dating was not possible for ITS-1. All nodes included in this tree are strongly supported (posterior probabilities > 0.95)
Discussion and conclusions
To address our first objective, we estimated the age of the Norops humilis/quaggulus species complex and evaluated two potential dispersal patterns for this group. The BEAST analysis yielded a mean crown group age of 17.2 Myr BP (range = 14.2–20.6) for the clade. We report this date cautiously because it was calculated from mitochondrial data only. To evaluate the second objective, we used the ancestral range estimation to evaluate dispersal patterns. Using the LAGRANGE output, we infer that the common ancestor of the N. humilis/quaggulus complex originated in Panama before dispersing west to Costa Rica, and then north into Nicaragua and Honduras. The south-to-north pattern and origin in Panama suggests isolation of the ancestor of the N. humilis/quaggulus complex in the Talamancan region of extreme LCA. The divergence between Pacific and Caribbean N. humilis in northern Costa Rica (Chorotega Block) was estimated to be 6.6 Myr BP (range = 4.12–6.94), which corresponds to the estimate of 5.4 Myr BP for the rise of the lower Central American highlands [57]. This uplift represents a potential vicariant event responsible for the separation of the Pacific (lineage 3) and Caribbean lineages (N. humilis lineages 4 and 5 + N. quaggulus). However, the Tilarán range in northern Costa Rica is estimated to have originated around 2 Myr BP [57], after the present split between Caribbean and Pacific lineages. Sister species are often found on opposite sides of the Central American Highlands [20, 21], and while Caribbean lineages of N. humilis form a paraphyletic group, the fact that one clade is separated by the continental divide is unsurprising. The ancestral area estimation suggests that early ancestors of some of the lineages were present on both sides of the continental divide in Costa Rica and Panama, corresponding to dates prior to the rise of the Central American Highlands.
As in many other taxa ([58] and sources within), LCA has served as a region of diversification for the Norops humilis/quaggulus complex (once again, distinct from the N. humilis species group). The northward distribution observed here is similar to that found in eleutherodactyline (genera Craugastor [19]; Pristimantis [59]) and hylid frogs (Dendropsophus [60]) that originated in South America and dispersed to Central America prior to the most recent completion of the Isthmus of Panama (3–4 Myr BP). This is also similar to findings in other groups of squamates [2], although it remains to be seen if this pattern of dispersal is shared by other lizard species.
Our third objective was to examine polyphyly described by others [35, 36] of the N. humilis species group. The lack of support for a monophyletic N. humilis species group is consistent with results from Nicholson [35] and Poe [36], further illustrating the extent of convergence in morphological characters of mainland anoles. All analyses agreed in assigning several species of the N. humilis species group into separate areas of the tree. Norops marsupialis, N. tropidonotus, and N. uniformis were placed in areas of the tree distant to a monophyletic N. humilis/quaggulus complex. Initially N. marsupialis was included in this study as a member of the N. humilis/quaggulus complex. Taylor [61] described N. marsupialis as a subspecies of N. humilis in 1956, and it was not elevated to specific status until 2015 [34]. All of our analyses (Bayesian and ML for each gene region and combined) found support for the specific status of N. marsupialis, and agreed to its placement as sister to a clade containing N. aquaticus and N. woodi (approx. 30 Myr divergent from the N. humilis/quaggulus complex, Fig. 6). Therefore, we confirm N. marsupialis as a distinct species morphologically similar to, but evolutionary distant from N. humilis/quaggulus, in concordance with with Köhler et al. [34]. Morphological convergence within anoles has long been reported, particularly for Caribbean species [27, 62–66] and is further demonstrated by our analysis of the N. humilis species group. Inclusion of the other three members of the group (N. compressicauda, N. notopholis and N. wampuensis) into the molecular phylogeny of Norops may yield further insight on their placement in the phylogeny, but their inclusion is not likely to significantly alter the results obtained here. Additional work within N. tropidonotus may also be necessary to examine the evolution that has occurred in that species, as it occupies a broad geographic range (Köhler 2008 [30]). Our data suggest that N. tropidonotus contains at least three deep mitochondrial divergences, and could potentially represent multiple cryptic lineages. While N. quaggulus is nested within N. humilis, we do not recommend synonymizing the two species until further work is done to investigate this complex. The deep divergences seen among the N. humilis clades may correspond to cryptic species, and if further work confirms the presence of two or more species, the name N. quaggulus would have priority as the senior synonym.
The SAMOVA results indicate that limited mitochondrial gene flow is occurring among localities, suggesting 12 distinct genetic groupings among our sample localities. Therefore, we conclude that genetic differentiation within the N. humilis/quaggulus complex is significant enough to conclude that (1) population fragmentation has occurred and (2) the complex does not represent a single panmictic population. The isolation, coupled with the high genetic variance, supports all major lineages identified in the Bayesian analyses as being distinct from one another, as well as further subdivision within most lineages. While we do not suggest that the 12 groups correspond to separate species, the presence of at least six well supported, divergent clades within the N. humilis/quaggulus complex demonstrates that deep mitochondrial divergence has occurred within this group, although nuclear evolution appears to be more conserved. This result is similar to several studies on Caribbean anoles, which also display considerable mitochondrial differentiation, with much less diversity in the nuclear DNA [67–70]. This pronounced phylogeographic substructuring may be explained in part, by low vagility in lizards [9]. Deep mitochondrial divergences coupled with conserved nuclear evolution as seen here, may have at least two implications 1) ITS is more slowly evolving than the mitochondrial genome of anoles, which we consider to be a likely scenario given that nuclear DNA is generally regarded as experiencing slower rates of evolution [71–73] and 2). The mitochondrial-nuclear relationships observed here are characteristic of male-biased dispersal [74, 75], indicating that female N. humilis are more philopatric than males as seen in Caribbean anoles [76, 77].
The novel biogeographic pattern for Central American anoles revealed here illustrates a need for further work on mainland Norops. What remains to be tested is whether the south-to-north dispersal route seen in the N. humilis/quaggulus complex is repeated in other Norops groups. In addition, it is important to clarify where the Central American Norops lineage originated, how it dispersed throughout the mainland, and when these events took place. Investigating the phylogeography of other widespread anoles may be highly informative towards understanding other distribution patterns of the Central American herpetofauna. There are several widespread Norops species and species complexes that vary in their ecological roles with corresponding morphological features. These are grouped into designations called ecomodes, which are distinct from ecomorphs, to accommodate mainland anoles (see [35]). Such an assortment of ecologically diverse anoles may provide good models for testing the biogeographic hypotheses discussed here. Further studies on mainland Norops species are needed to test if the cryptic diversity suggested here is present in other widespread species complexes within the genus.
References
Crother BI, Campbell JA, Hillis DM. Phylogeny and historical biogeography of the palm-pitvipers, genus Bothriechis: biochemical and morphological evidence. In: Campbell JA, Brodie Jr ED, editors. Biology of the Pitviper. Tyler, TX: Selva; 1992. p. 1–20.
Castoe TA, Daza JM, Smith EN, Sasa MM, Kuck U, Campbell JA, et al. Comparative phylogeography of pitvipers suggests a consensus of ancient Middle American highland biogeography. J Biogeogr. 2009;36:88–103.
Elmer KR, Bonett RM, Wake DB, Lougheed SC. Early Miocene origin and cryptic diversification of South American salamanders. BMC Evol Biol. 2013;13:59.
Wiens JJ, Graham CH, Moen DS, Smith SA, Reeder TW. Evolutionary and ecological causes of the latitudinal diversity gradient in hylid frogs: Treefrog trees unearth the roots of high tropical diversity. Am Nat. 2006;168:579–96.
Pramuk JB, Robertson T, Sites Jr JW, Noonan BP. Around the world in 10 million years: biogeography of the nearly cosmopolitan true toads (Anura: Bufonidae). Glob Ecol Biogeogr. 2008;17:72–83.
Morrone JJ. Biogeographic area and transition zones of Latin America and the Caribbean islands based on panbiogeographic and cladistic analyses of the entomofauna. Annu Rev Entomol. 2006;51:467–94.
Bussing WA. Geographic distribution of the San Juan ichthyofauna of Central America with remarks on its origin and ecology. In: Thorson TB, editor. Investigations of the Ichthyofauna of Nicaraguan Lakes. Lincoln, NE: University of Nebraska; 1976. p. 157–75.
Smith SA, Bermingham E. The biogeography of lower Mesoamerican freshwater fishes. J Biogeogr. 2005;32:1835–54.
Savage JM. The enigma of the Central American herpetofauna: dispersals of vicariance? Ann Mo Bot Gard. 1982;69:464–547.
Savage JM. The amphibians and reptiles of Costa Rica: a herpetofaunal between two continents, between two seas. Chicago, IL: University of Chicago Press; 2002.
Cavers S, Navarro C, Lowe J. Chloroplast DNA phylogeography reveals colonization history of a Neotropical tree, Cedrela odorata L., in Mesoamerica. Mol Ecol. 2003;12:1451–60.
Novick RN, Dick C, Lemes M, Navarro C, Caccone A, Bermingham E. Genetic structure of Mesoamerican populations of big-leaf mahogany (Swuetenia macrophylla) inferred from microsatellite analysis. Mol Ecol. 2003;12:2885–94.
Cropp S, Boinski S. The Central American squirrel monkey (Saimiri oesterdii): introduced hybrid or endemic species? Mol Phylogenet Evol. 2000;16:350–65.
Harris D, Rogers DS, Sullivan J. Phylogeography of Peromyscus furvus (Rodentia; Muridae) based on cytochrome b sequence data. Mol Ecol. 2000;9:2129–35.
Villalobos F. Tree squirrels: A key to understand the historic biogeography of Mesoamerica? Mammalian Biol. 2013;78:258–66.
Bermingham E, Martin AP. Comparative mtDNA phylogeography of Neotropical freshwater fishes: testing shared history to infer the evolutionary landscape of lower Central America. Mol Ecol. 1998;7:499–517.
Martin AP, Bermingham E. Regional endemism and cryptic species revealed by molecular and morphological analysis of a widespread species of Neotropical catfish. Proc R Soc Lond B. 2000;264:1135–41.
García-Paris M, Good DA, Parra-Olea G, Wake DB. Biodiversity of Costa Rican salamanders: implications of high levels of genetic differentiation and phylogeographic structure for species formation. Proc Natl Acad Sci U S A. 2000;97:1640–7.
Crawford AJ, Smith EN. Cenozoic biogeography and evolution in direct-developing frogs of Central America (Leptodactylidae: Eleutherodactylus) as inferred from a phylogenetic analysis of nuclear and mitochondrial genes. Mol Phylogenet Evol. 2005;35:536–55.
Zamudio KR, Greene HW. Phylogeography of the bushmaster (Lachesis muta: Viperidae): implications for Neotropical biogeography, systematics, and conservation. Biol J Linn Soc Lond. 1997;62:421–42.
Parkinson CL, Zamudio KR, Greene HW. Phylogeography of the pitviper clade Agkistrodon: historical ecology, species status, and conservation of cantils. Mol Ecol. 2000;9:411–20.
Daza JM, Smith EN, Páez VP, Parkinson CL. Complex evolution in the neotropics: the origin and diversification of the widespread genus Leptodeira (Serpentes: Colubridae). Mol Phylogenet Evol. 2009;53:653–67.
Daza JM, Castoe TA, Parkinson CL. Using regional comparative phylogeographic data from snake lineages to infer historical processes in Middle America. Ecography (Cop). 2010;33:343–54.
Colston TJ, Grazziotin FG, Shepard DB, Vitt LJ, Colli GR, Henderson RW, et al. Molecular systematics and historical biogeography of tree boas (Corallus spp.). Mol Phylogenet Evol. 2013;66:953–59.
Hasbún CR, Gómez A, Köhler G, Lunt DH. Mitochondrial DNA phylogeography of the Mesoamerican spiny-tailed lizards (Ctenosaura quinquecarinata complex): historical biogeography, species status and conservation. Mol Ecol. 2005;14:3095–107.
Williams EE. The ecology of colonization as seen in the zoogeography of anoline lizards on small islands. Q Rev Biol. 1969;44:345–89.
Schoener TW. Size patterns in West Indian Anolis lizards. II. Correlations with the sizes of particular sympatric species-displacement and convergence. Am Nat. 1970;104:155–74.
Roughgarden J. Evolution of niche width. Am Nat. 1972;106:683–718.
Losos JB, Jackman TR, Larson A, de Queiroz K, Rodriguez-Schettino L. Contingency and determinism in replicated adaptive radiations of island lizards. Science. 1998;5539:2115–8.
Köhler G. Reptiles of Central America. 2nd ed. Herpeton: Offenbach, Germany; 2008.
Savage JM, Guyer C. Infrageneric classification and species composition of the anole genera, Anolis, Ctenonotus, Dactyloa, Norops, and Semiurus (Sauria: Iguanidae). Amphib-Reptil. 1989;10:105–16.
McCranie JR, Köhler G. A new species of anole from eastern Honduras related to Norops tropidonotus (Reptilia: Squamata: Polychrotidae). Senckenberg Biol. 2001;81:227–34.
Köhler G, McCranie JR, Nicholson KE, Kreutz J. Geographic variation in hemipenial morphology in Norops humilis (Peters 1863), and the systematic status of Norops quaggulus (Cope 1885) (Reptilia, Squamata, Polychrotidae). Senckenberg Biol. 2003;82:213–22.
Köhler JJ, Poe S, Ryan MJ, Köhler G. Anolis marsupialis Taylor 1956, a valid species from southern Pacific Costa Rica (Reptilia, Squamata, Dactyloidae). Zootaxa. 2015;3915:111–22.
Nicholson KE. Phylogenetic analysis and a test of the current infrageneric classification of Norops (Beta Anolis). Herpetol Monogr. 2002;16:93–110.
Poe S. Phylogeny of anoles. Herpetol Monogr. 2004;18:37–89.
Nicholson KE, Crother BI, Guyer C, Savage JM. It is time for a new classification of anoles (Squamata: Dactyloidae). Zootaxa. 2012;3477:1–108.
Macey JR, Schulte II JA, Anajeva NB, Larson A, Rastegar-Pouyani N, Shammakov SM, et al. Phylogenetic relationships among agamid lizards of the Laudakia caucasia species group: testing hypotheses of biogeographic fragmentation and an area cladogram for the Iranian Plateau. Mol Phylogenet Evol. 1998;10:118–31.
Funk DJ, Omland KE. Species level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Evol Syst. 2003;34:397–423.
Chan KMA, Levin SA. Leaky prezygotic isolation and porous genomes: rapid introgression of maternally inherited DNA. Evolution. 2005;59:720–29.
Toews DPL, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol. 2012;21:3907–30.
Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol Ecol. 2004;13:729–44.
Rice WR. Nothing in genetics makes sense except in the light of genomic conflict. Annu Rev Ecol Evol Syst. 2013;44:217–37.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11:2571–81.
Rodríguez-Robles JA, Jezkova T, Leal M. Climatic stability and genetic divergence in the tropical insular lizard Anolis krugi, the Puerto Rican ‘Lagartijo Jardinero de la Montaña’. Mol Ecol. 2010;19:1860–76.
Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29:1695–701.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:1–4.
Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214.
Rambaut A, Drummond AJ. 2009 Tracer v1.5. Available at: http://tree.bio.ed.ac.uk/software/.
Weisrock DW, Macey JR, Ugurtas IH, Larson A, Papenfuss TJ. Molecular phylogenetics and historical biogeography among salamandrids of the ‘true’ salamander clade: rapid branching of numerous highly divergent lineages in Mertensiella luschani associated with the rise of Anatolia. Mol Phylogenet Evol. 2001;18:434–48.
Townsend TM, Mulcahy DG, Noonan BP, Sites Jr JW, Kuczynski CA, Wiens JJ, et al. Phylogeny of iguanian lizards inferred from 29 nuclear loci, and a comparison of concatenated and species-tree approaches for an ancient, rapid radiation. Mol Phylogenet Evol. 2011;61:363–80.
Glor RE, Kolbe JJ, Powell R, Larson A, Losos JB. Phylogenetic analysis of ecological and morphological diversification in Hispaniolan trunk-ground anoles (Anolis cybotes group). Evolution. 2003;57:2383–97.
Glor RE, Losos JB, Larson A. Out of Cuba: overwater dispersal and speciation among lizards in the Anolis carolinensis subgroup. Mol Ecol. 2005;14:2419–32.
Ree RH, Moore BR, Webb CO, Donoghue MJ. A likelihood framework for inferring the evolution of geographic range on phylogenetic trees. Evolution. 2005;59:2299–311.
Ree RH, Smith SA. Maximum-likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst Biol. 2008;57:4–14.
Denyer P, Alvarado GE, Aguilar T. Historia geológica. In: Denyer P, Kussmaul S, editors. Geología de Costa Rica. Cartago, Costa Rica: Editorial Tecnológica de Costa Rica; 2000. p. 155–67.
Bagley JC, Johnson JB. Phylogeography and biogeography of the lower Central American Neotropics: diversification between two continents and between two seas. Biol Rev. 2014;89:767–90.
Pinto-Sánchez NR, Ibáñez R, Madriñán S, Sanjur OI, Bermingham E, Crawford AJ. The Great American Biotic Interchange in frogs: Multiple and early colonization of Central America by the South American genus Pristimantis (Anura: Craugastoridae). Mol Phylogenet Evol. 2012;62:954–72.
Robertson JM, Duryea MC, Zamudio KR. Discordant patterns of evolutionary differentiation in two Neotropical treefrogs. Mol Ecol. 2009;18:1375–95.
Taylor EH. A review of the lizards of Costa Rica. Univ Kansas Sci Bull. 1956;38:3–322.
Losos JB. The evolution of convergent community structure in Caribbean Anolis communities. Syst Biol. 1992;41:403–20.
Losos JB. Lizards in an evolutionary tree: ecology and adaptive radiation of anoles. Berkeley, CA: University of California Press; 2009.
Harmon LJ, Kolbe JJ, Cheverud JM, Losos JB. Convergence and the multidimensional niche. Evolution. 2005;59:409–21.
Losos JB, Glor RE, Kolbe JJ, Nicholson KE. Adaptation, speciation, and convergence: a hierarchical analysis of adaptive radiation in Caribbean Anolis lizards. Ann Mo Bot Gard. 2006;93:24–33.
Johnson MA, Revell LJ, Glor RE, Losos JB. Behavioral convergence and adaptive radiation: effects of habitat use on territorial behavior in Anolis lizards. Evolution. 2010;64:1151–9.
Malhotra A, Thorpe RS. The dynamics of natural selection and vicariance in the Dominican anole: patterns of within-island molecular and morphological divergence. Evolution. 2000;54:245–58.
Thorpe RS, Stenson AG. Phylogeny, paraphyly and ecological adaptation of the colour and pattern in the Anolis roquet complex on Martinique. Mol Ecol. 2003;12:117–32.
Thorpe RS, Surget-Groba Y, Johansson H. Genetic tests for ecological and allopatric speciation in anoles on an island archipelago. PLoS Genet. 2010;6:e1000929.
Ng J, Glor RE. Genetic differentiation among populations of a Hispaniolan trunk anole that exhibit geographical variation in dewlap colour. Mol Ecol. 2011;20:4302–17.
Brown WM, George M, Wilson AC. Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A. 1979;76:1967–71.
Castellana S, Vicario S, Saccone C. Evolutionary patterns of the mitochondrial genome in Metazoa: exploring the role of mutation and selection in mitochondrial protein coding genes. Genome Biol Evol. 2011;3:1067–79.
Levin L, Blumberg A, Barshad G, Mishmar D. Mito-nuclear co-evolution: the positive and negative sides of functional ancient mutations. Front Genet. 2014;5:448.
Melnick DJ, Hoelzer GA. Differences in male and female macaque dispersal lead to contrasting distributions of nuclear and mitochondrial DNA variation. Int J Primatol. 1992;13:379–93.
Jockusch EL, Wake DB. Falling apart and merging: diversification of slender salamanders (Plethodontidae: Batrachoseps) in the American West. Biol J Linn Soc. 2002;76:361–91.
Stenson AG, Malhotra A, Thorpe RS. Population differentiation and nuclear gene flow in the Dominican anole (Anolis oculatus). Mol Ecol. 2002;11:1679–88.
Johansson H, Surget-Groba Y, Thorpe RS. Microsatellite data show evidence for male-biased dispersal in the Caribbean lizard Anolis roquet. Mol Ecol. 2008;17:4425–32.
Harmon LJ, Schulte II JA, Larson A, Losos JB. Tempo and mode of evolutionary radiation in iguanian lizards. Science. 2003;301:961–4.
Jackman TR, Irschick DJ, de Queiroz K, Losos JB, Larson A. Molecular phylogenetic perspective on evolution of lizards of the Anolis grahami series. J Exp Zool B Mol Dev Evol. 2002;294:1–16.
Nicholson KE, Glor RE, Kolbe JJ, Larson A, Hedges SB, Losos JB. Mainland colonization by island lizards. J Biogeogr. 2005;32:929–38.
Acknowledgements
We would like to thank S. Burton, J. Gubler, D. Laurencio, L. Obando, J. Steffen, D. Steen, J. Sunyer, Y. Svec and J. Townsend for assistance in the collection of specimens and tissues from the field. K. de Queiroz (USNM) and G. Köhler (Senckenberg Museum) provided additional tissues. B. Kubicki, and many other landowners allowed us access to collect specimens on their property. We would like to thank C. Moritz, and two anonymous reviewers at Axios Review for comments on the manuscript. R. Bonett reviewed an earlier draft of this manuscript and provided invaluable feedback. R. Bonett and S. Martin gave technical advice on conducting analyses, and G. Louthan at the Tandy Super Computer in Tulsa, OK provided assistance in supercomputing. Funding was provided by NSF grant DEB 0949359 (KEN), Central Michigan University internal funds from the Office of Research and Sponsored Programs (KEN, JGP, and SH), and the Central Michigan University Honors Program (JGP). Specimens were collected in Costa Rica under Resolucíones 239-2008-SINAC, 040–2009-SINAC, 017-2010-SINAC and 005–2011-SINAC and MINAET Permit no. 029–2011-SINAC, in Nicaragua under DGPN/DB-10–2010 and in Panama under ANAM Scientific Permit No. SEX/A-50-12. We would like to thank all permitting agencies for assisting us in this study. We complied with all applicable Animal Care guidelines (CMU-IACUC # 10–02) and all federal and international permits are on file and available from KEN.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KEN conceived the ideas. JGP, JD, CG and KEN collected specimens. JGP and SH conducted labwork. JGP and KEN analyzed the data, JGP led the writing, JD, CG and KEN contributed significantly to the writing. All authors read and approved the final manuscript.
Additional files
Additional file 1: Appendix S1.
Locality and sequence data for Norops humilis species group sensu Savage and Guyer [31]. ITS accession numbers are not applicable to all specimens, as many taxa were only sequenced for the mtDNA region.
Additional file 3: Appendix S3.
Populations used in the SAMOVA analysis. With a graphical representation of the FCT and FSC values for a range of genetic clusters as defined in the spatial analysis of molecular variance.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Phillips, J.G., Deitloff, J., Guyer, C. et al. Biogeography and evolution of a widespread Central American lizard species complex: Norops humilis, (Squamata: Dactyloidae). BMC Evol Biol 15, 143 (2015). https://doi.org/10.1186/s12862-015-0391-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-015-0391-4