
Abstract
Background
Development of the valves and septa of the heart depends on the formation and remodeling of the endocardial cushions in the atrioventricular canal and outflow tract. These cushions are populated by mesenchyme produced from the endocardium by epithelial-mesenchymal transition (EMT). The endocardial cushions are remodeled into the valves at post-EMT stages via differentiation of the mesenchyme and changes in the extracellular matrix (ECM). Transforming growth factor β (TGFβ) signaling has been implicated in both the induction of EMT in the endocardial cushions and the remodeling of the valves at post-EMT stages. We previously identified the RNA binding protein muscleblind-like 1 (MBNL1) as a negative regulator of TGFβ signaling and EMT in chicken endocardial cushions ex vivo. Here, we investigate the role of MBNL1 in endocardial cushion development and valvulogenesis in Mbnl1 ∆E3/∆E3 mice, which are null for MBNL1 protein.
Methods
Collagen gel invasion assays, histology, immunohistochemistry, real-time RT-PCR, optical coherence tomography, and echocardiography were used to evaluate EMT and TGFβ signaling in the endocardial cushions, and morphogenesis, ECM composition, and function of the heart valves.
Results
As in chicken, the loss of MBNL1 promotes precocious TGFβ signaling and EMT in the endocardial cushions. Surprisingly, this does not lead to the production of excess mesenchyme, but later valve morphogenesis is aberrant. Adult Mbnl1 ∆E3/∆E3 mice exhibit valve dysmorphia with elevated TGFβ signaling, changes in ECM composition, and increased pigmentation. This is accompanied by a high incidence of regurgitation across both inflow and outflow valves. Mbnl1 ∆E3/∆E3 mice also have a high incidence of ostium secundum septal defects accompanied by atrial communication, but do not develop overt cardiomyopathy.
Conclusions
Together, these data indicate that MBNL1 plays a conserved role in negatively regulating TGFβ signaling, and is required for normal valve morphogenesis and homeostasis in vivo.
Similar content being viewed by others


Background
Valve defects are among the most common congenital heart defects, and are associated with significant health problems and mortality throughout life [1]. Their exact incidence is unknown because many valve defects are not diagnosed until adulthood [2]. Aortic valve defects alone occur in 1-2 % of the population [3]. The prevalence of adults with valvular heart disease is increasing, due to improved surgical treatments and increases in lifespan that may be accompanied by valve degeneration [4]. Congenital valve defects and adult valve disease may not be distinct, since structural or biomechanical abnormalities in the valves may be present but not readily appreciated at birth. The heart valves form by remodeling embryonic structures called endocardial cushions. Outgrowth of the cushions occurs via localized expansion of extracellular matrix (ECM) between the endocardium and myocardium in the atrioventricular canal (AVC) and outflow tract (OFT), after which the cushions are cellularized by invasion of a subpopulation of endocardial cells undergoing epithelial-mesenchymal transition (EMT) [5]. At post-EMT stages, this cushion mesenchyme differentiates into valve interstitial cells, specialized fibroblasts that produce the highly stratified matrix of the mature valves [2, 3]. The mitral and tricuspid valves form from the AVC endocardial cushions, while the aortic and pulmonary valves arise from the OFT cushions [6].
Transforming growth factor β (TGFβ) signaling induces endocardial cushion EMT in both birds and mammals, although the roles of specific TGFβ paralogs differ [7–9]. In chick, TGFβ2 is required for activation of the endocardial cells, in which cell polarity and cell-cell contacts are lost, and TGFβ3 is required for invasion of the transformed mesenchymal cells into the cushion matrix [8, 9]. TGFβ1 is not expressed in the developing cushions. Both TGFβ2 and TGFβ3 are produced in the myocardium prior to the onset of EMT, and TGFβ3 is found in the endocardium during stages of active EMT [8, 10, 11]. Autocrine production of TGFβ3 in the cushion endocardium is essential for induction of invasive mesenchyme [11, 12]. In mouse, TGFβ3 is not expressed in the heart until E11.5, well after EMT is underway, and is restricted to the mesenchymal cells of the cushions [13]. No cardiac malformations have been found in Tgfb3-null mice [14, 15]. TGFβ2 has an expression pattern in the mouse heart that is similar to that of TGFβ3 in chick, and inhibition of TGFβ2 blocks invasion in mouse AVC explants [13, 16]. Tgfb2-null mice exhibit impaired EMT, hypocellular cushions, and a variety of valve and septal defects [7, 17, 18]. Thus, in mouse TGFβ2 plays a role orthologous to that of chick TGFβ3. TGFβ1 is also expressed in mouse AVC endocardium, and although it is not essential for endocardial cushion EMT in vivo, Tgfb1-null mice have disorganized valves [19].
We previously identified a role for muscleblind-like 1 (MBNL1) in the regulation of TGFβ-dependent EMT in the endocardial cushions in chick [20, 21]. MBNL1 is a member of the MBNL family of RNA binding proteins that regulate pre-mRNA alternative splicing, alternative polyadenylation, microRNA biogenesis, mRNA stability and localization [22–25]. We previously reported that in the embryonic chicken heart MBNL1 transcripts are found in the atrial and ventricular myocardium, and in the endocardium of the AVC and OFT endocardial cushions during EMT and immediately prior to its onset [20, 21]. We demonstrated that knockdown of MBNL1 in chick AVC or OFT explants promotes invasive mesenchyme formation ex vivo [20, 21]. This effect is dependent upon TGFβ3, and is accompanied by precocious and elevated secretion of active TGFβ3 by the endocardial cells [20, 21]. Early exposure to excess TGFβ3 is sufficient to induce precocious and elevated invasion in AVC explants [20]. These data suggested that MBNL1 negatively regulates EMT in the endocardial cushions by limiting the levels and timing of inductive, autocrine TGFβ signaling.
In this study, we investigate the role of MBNL1 in mouse endocardial cushion development and valvulogenesis. We demonstrate that high expression of MBNL1 in the endocardial cushions is conserved in mouse. Using Mbnl1 ∆E3/∆E3 mice, which are null for MBNL1 protein, we show that loss of MBNL1 promotes invasive mesenchyme production in AVC explants ex vivo and precocious TGFβ signaling and EMT in the AVC in vivo, but does not lead to an excess in total mesenchyme production. In the fetal Mbnl1 ∆E3/∆E3 heart, however, valve morphogenesis is aberrant. In the adult heart, Mbnl1 ∆E3/∆E3 mice continue to exhibit valve dysmorphia, accompanied by elevated TGFβ signaling and changes in ECM composition. Significant regurgitation was observed across both inflow and outflow valves. Mbnl1 ∆E3/∆E3 mice also have a high incidence of ostium secundum atrial septal defects and increased valve pigmentation. Taken together, these data indicate that MBNL1 is required for normal heart valve development and function in vivo.
Results
MBNL1 is expressed in the endocardial cushions and mature heart valves
To determine whether MBNL1 expression in the endocardial cushions is conserved in mammals, we performed immunohistochemistry on sections from mouse hearts at different stages of development. As in the chicken heart, MBNL1 was strongly detected in the myocardium and AVC endocardium at E9, prior to the onset of EMT (Fig. 1a, b). MBNL1 is broadly expressed throughout the embryo, but is not ubiquitous; for example, although MBNL1 protein is strongly detected in the ectodermal linings it is low or absent in the interior cephalic tissues of the mid-, fore-, and hindbrain (Additional file 1: Figure S1). At E11.5, when EMT is well underway, intense nuclear MBNL1 staining was observed in the endocardium and mesenchyme of the AVC and OFT cushions (Fig. 1c, d). Strong nuclear MBNL1 staining persists in the immature valves at late fetal stages (Fig. 1f) and in the mature valves of the adult heart (Fig. 1g). MBNL1 was also seen in the developing myocardium, where it is found in both the nucleus and cytoplasm (Fig. 1c, e, f). In the adult myocardium, MBNL1 staining is more diffuse and cytoplasmic (Fig. 1g), consistent with previous reports that MBNL1 is more strongly detected in cytoplasmic than nuclear extracts from whole hearts at postnatal stages [26, 27].

MBNL1 is highly expressed in the endocardial cushions and heart valves. Immunohistochemistry with an anti-MBNL1 antibody was performed on sagittal sections from wild type mouse embryos (a-f) and coronal sections of adult hearts (g). Representative sections from one of four embryos or one of two adult hearts are shown. a Close-up of the heart at E9 stained with the secondary antibody alone is shown as a negative control. b Close-up of the heart at E9 shows expression of MBNL1 in the early myocardium and endocardium of the nascent endocardial cushions just prior to the onset of EMT. c Close-up of the heart at E11.5 shows strong MBNL1 expression in the endocardial cushions of the AVC and OFT. Higher magnification views of the boxed regions in the endocardial cushion (d) and the ventricular wall (e) show intense nuclear staining in the cushion endocardial cells, but more diffuse staining in the myocardium. Intense nuclear MBNL1 staining persists in the fetal valves at E19.5 (f) and mature heart valves (g), while MBNL1 remains more diffusely distributed in the nucleus and cytoplasm in the myocardium. Asterisks highlight accumulations of melanin in the adult valve. h Western blot analysis confirms high MBNL1 expression in the adult heart, and suggests different cardiac tissues may express different MBNL1 isoforms. Protein integrity and loading were assessed by Ponceau S staining. A representative of three independent blots with similar results is shown. Atr = atrium, Vent = ventricle, EC = endocardial cushion, OFT = outflow tract, TC = tricuspid valve, Val = heart valves
By western blot, MBNL1 is expressed in adult heart valves at levels comparable or higher than levels in the chambers (Fig. 1h). Interestingly, slightly different MBNL1 isoforms were detected in the different compartments of the heart, with the slowest migrating isoform in the valves and the fastest in the ventricles. Alternative splicing of Mbnl1 transcripts can lead to the generation of several different MBNL1 protein isoforms, ranging in size from 35 to 43 kDa [28]. We and others previously reported that inclusion or skipping of exon 7 [28] affects the subcellular localization of MBNL1 protein such that the larger exon-included form is predominantly nuclear whereas the exon-skipped form is both nuclear and cytoplasmic [29–31]. Although preferential inclusion of exon 7 in the heart valves would explain both its strong nuclear localization and its slower migration, we found that exon 7 is in fact ~95 % skipped in adult mouse heart valves, atria, and ventricles (data not shown).
Loss of MBNL1 has a transient stimulatory effect on EMT in the endocardial cushions ex vivo
We previously demonstrated that knockdown of MBNL1 in chick H&H stage 14 AVC explants promotes invasive mesenchyme production ex vivo, indicating that MBNL1 is a negative regulator of endocardial cushion EMT [20, 21]. To determine if this role is conserved in mammals, invasive mesenchyme production was compared in AVC explants from wild type and Mbnl1 ∆E3/∆E3 hearts (Fig. 2a-c). Indeed, E9 explants from Mbnl1 ∆E3/∆E3 mice had more invaded cells than wild type explants. In chick, the effects of MBNL1 knockdown are temporally regulated, such that knockdown of MBNL1 induces increased invasion in stage 14 AVC explants, but has no significant effect in stage 16 AVC explants [20]. Likewise, the number of invaded cells in AVC explants from E9.5 Mbnl1 ∆E3/∆E3 mice was similar to that of wild type E9.5 explants (Fig. 2c). Thus, both the stimulatory effect of loss of MBNL1 on EMT and its temporal regulation are conserved between birds and mammals.

Loss of MBNL1 stimulates EMT ex vivo in a stage-specific manner. Images of wild type (a, a’) or Mbnl1 ∆E3/∆E3 (b, b’) E9 AVC explant cultures were taken in the plane of focus of the surface of the gel (a, b) and beneath the surface of the gel (a’, b’) to show invaded mesenchymal cells. Filled arrowheads indicate cells in the focal plane, whereas open arrowheads indicate the same cells out of focus in a different plane. c AVC regions from E9 or E9.5 wild type or Mbnl1 ∆E3/∆E3 hearts were explanted onto collagen gels. The number of explants per group is indicated. An asterisk indicates a significant difference from wild type (P ≤ 0.05). d Conditioned supernatants were collected from E9 wild type and Mbnl1 ∆E3/∆E3 AVC explants and the levels of active TGFβ proteins secreted into the media were assayed by ELISA
In chick stage 14 AVC explants, elevated levels of active TGFβ3, but not TGFβ2, are secreted into the medium following MBNL1 knockdown [20]. In chick, TGFβ3 is required for invasion of mesenchymal cells, whereas TGFβ2 is required for endocardial cell activation [32]. TGFβ signaling is also important for endocardial cushion EMT in mammals, but with some differences in ligand specificity [7–9]. To determine whether the levels of active TGFβ secreted by Mbnl1 ∆E3/∆E3 AVC cushions differed from those of wild type, we collected conditioned media from wild type and Mbnl1 ∆E3/∆E3 AVC explants and measured the levels of active TGFβ1, TGFβ2, and TGFβ3 by enzyme-linked immunosorbent assay (ELISA) (Fig. 2d). There was a trend towards higher levels of all three TGFβ proteins, but differences did not reach statistical significance. This may be attributable in part to the detection limits of the assay, as the measured levels of TGFβ proteins were very low, and we failed to detect any TGFβ in some supernatants from both wild type and Mbnl1 ∆E3/∆E3 explants.
Loss of MBNL1 leads to precocious EMT and TGFβ signaling in the endocardial cushions in vivo without a persistent increase in total mesenchyme production
In chick stage 14 AVC explants, knockdown of MBNL1 stimulates precocious and elevated levels of active TGFβ3 secreted into the medium, and early exposure to elevated TGFβ3 levels is sufficient to induce precocious invasion [20]. The level of invasion induced at the earlier stage by loss of MBNL1 is similar to that of control explants from the later stage in both chick and mouse [20], consistent with an accelerated induction of invasion. EMT normally begins in the mouse AVC cushions at E9.5 [33], and indeed no invaded cells were observed in heart sections from wild type embryos at E9.25 (Fig. 3a). In contrast, histological analysis of Mbnl1 ∆E3/∆E3 embryos revealed the presence of invaded cells within the AVC cushions already at E9 (Fig. 3b), suggesting that MBNL1 prevents precocious mesenchyme formation both ex vivo and in vivo. To determine whether TGFβ signaling is precociously activated in Mbnl1 ∆E3/∆E3 AVC cushions in vivo, we performed immunohistochemistry using an antibody against phosphorylated SMAD2 (pSMAD2), a mediator of canonical TGFβ signaling. In wild type hearts, high pSMAD2 levels were seen in a small number of AVC cushion endocardial cells at E9.25 (Fig. 3c). In Mbnl1 ∆E3/∆E3 hearts pSMAD2 was strongly detected in nearly all cells in the AVC endocardium (as well as the invaded mesenchyme) by E9 (Fig. 3d), resembling pSMAD2 staining in wild type hearts at E10 (data not shown). This indicates that the precocious production of invasive mesenchyme in the absence of MBNL1 is accompanied by precocious TGFβ signaling.

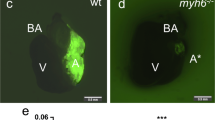
Mbnl1 E∆3/∆E3 mice have precocious EMT in the endocardial cushions without a persistent increase in total mesenchyme production. Close-up views of the hearts in sagittal sections of wild type E9.25 (a) and Mbnl1 ∆E3/∆E3 E9 (b) embryos stained with H&E reveal precocious invasion of mesenchymal cells (red arrowhead) into the AVC cushions of knockout mice. Sections from three individuals were evaluated for each group; representative images are shown. Close-up views of the AVC cushions in adjacent sections from wild type E9.25 (c) and Mbnl1 ∆E3/∆E3 E9 (d) embryos subjected to immunohistochemistry with an antibody against pSMAD2 reveal high levels of TGFβ signaling in only a few cells in wild type cushions (red arrowhead), but nearly all cells in the knockout cushions. H&E stained sagittal sections of wild type (e) and Mbnl1 ∆E3/∆E3 (f) hearts do not reveal noticeable hypercellularity in the AVC cushions at E10.5. Atr = atrium, vent = ventricle, EC = endocardial cushion, OFT = outflow tract. g The average relative number of mesenchymal cells was determined by counting the cells within the AVC cushions on serial, H&E-stained, sagittal sections of wild type and Mbnl1 ∆E3/∆E3 E10.5 embryos. Representative three-dimensional reconstructions of AVC cushions from optical sections of wild type (h) and Mbnl1 ∆E3/∆E3 (i) E10.5 hearts imaged using optical coherence tomography. Videos of the optical sections and reconstructions of these hearts are shown in Additional file 2: Movie S1 and Additional file 3: Movie S2, respectively; still images shown here were rotated to the same viewing angle
To determine whether the precocious induction of invasion leads to an over-production of cushion mesenchyme in vivo, the numbers of invaded cells in the AVC cushions were counted in serial sections from E10.5 wild type and Mbnl1 ∆E3/∆E3 embryos (Fig. 3e-g). Despite the early initiation of EMT in Mbnl1 ∆E3/∆E3 AVC cushions, there was no significant difference in the relative amount of AVC mesenchyme once EMT is well underway (P = 0.34). Imaging of E10.5 hearts using optical coherence tomography (OCT) confirmed that Mbnl1 ∆E3/∆E3 AVC cushions are indeed similar in size and shape to wild type (Fig. 3h, i and Additional file 2: Movie S1 and Additional file 3: Movie S2). Overt differences in cushion cellularity were also not observed in sections of wild type and Mbnl1 ∆E3/∆E3 hearts at E11.5, E12.5, or E14.5 (data not shown). Thus, although MBNL1 restricts the timing of EMT initiation in the AVC cushions, it is not essential for regulating the total amount of mesenchyme produced in vivo.
Loss of MBNL1 has little effect on EMT in the AVC cushions at E10.5
At E10.5, abundant pSMAD2 staining is seen throughout the endocardium and mesenchyme of both wild type and Mbnl1 ∆E3/∆E3 AVC cushions (data not shown). To evaluate EMT-associated gene expression in the Mbnl1 ∆E3/∆E3 AVC cushions at this stage, the levels of 84 genes that either change their expression during EMT or that regulate gene expression changes during EMT were assessed by real-time RT-PCR on total RNA from wild type or Mbnl1 ∆E3/∆E3 E10.5 AVC cushions using Epithelial to Mesenchymal Transition RT2 Profiler PCR Arrays (Additional file 4: Table S1). TGFβ transcript levels were not affected (Fig. 4a), although it should be noted that increases in the levels of active TGFβ3 secreted by chick AVC explants following MBNL1 knockdown occurred without an accompanying increase in TGFB3 transcript levels [20]. Consistent with equivalent amounts of mesenchyme production, both endothelial markers such as versican (Vcan) and E-cadherin (Cdh1), which are down-regulated during EMT, and mesenchymal markers such as fibronectin (Fn1) and matrix metallopeptidase 2 (Mmp2), which are up-regulated during EMT, did not differ between wild type and Mbnl1 ∆E3/∆E3 cushions. Important transcriptional regulators of EMT such as Snai1, Snai2 (Slug), and Twist1 also do not differ between wild type and Mbnl1 ∆E3/∆E3 cushions. Only four genes (Zeb1, Mitf, Bmp7, and Spp1) differed significantly between wild type and Mbnl1 ∆E3/∆E3 cushions by at least two-fold (Fig. 4b, c). Zinc-finger E-box binding homeobox 1 (ZEB1), a transcription factor that interacts with activated SMAD proteins to promote EMT in response to TGFβ signaling [34], is down-regulated more than three-fold. Microphthalmia-associated transcription factor (MITF), which negatively regulates SMAD-dependent TGFβ signaling in mast cells [35], is up-regulated approximately two-fold. Bone morphogenetic protein 7 (Bmp7) levels are also doubled in Mbnl1 ∆E3/∆E3 cushions. While neither Bmp7 nor Bmp6 alone are essential for endocardial cushion development, double-knockout mice lacking both Bmp7 and Bmp6 exhibit delayed formation of the OFT cushions and defects in valve morphogenesis [36]. Finally, the glycoprotein secreted phosphoprotein 1 (Spp1), also known as osteopontin, is elevated more than three-fold. Transcription of Spp1 is induced by both TGFβ and BMP signaling [37], so its up-regulation could be due to elevated TGFβ signaling or to the up-regulation of BMP7 or both. Repression of Spp1 is required for prevent matrix mineralization in the developing heart valves [38], and elevated SPP1 levels are associated with calcific valve disease in mice and humans [39–41]. Thus, while these results suggest that TGFβ signaling is precociously activated in the Mbnl1 ∆E3/∆E3 cushions, it is unclear whether elevated TGFβ signaling is maintained once EMT is underway. Unfortunately, the small size and lack of cellularity in the cushions at E9-9.5 makes collecting enough AVC cushion tissue for a similar analysis during the initiation of EMT untenable.

Epithelial to Mesenchymal Transition RT2 Profiler PCR Arrays reveal few changes in gene expression in Mbnl1 E∆3/∆E3 AVC cushions at E10.5. a-c) The expression of genes involved in EMT was compared in E10.5 wild type and Mbnl1 ∆E3/∆E3 AVC endocardial cushions using the Epithelial to Mesenchymal Transition RT2 Profiler PCR Array (Qiagen). a Tgfb1, Tgfb2, and Tgfb3 transcript levels in the knockout cushions did not differ from wild type. b Clustered heat map of transcripts that showed a ≥ 2 fold change in Mbnl1 ∆E3/∆E3 cushions with P ≤ 0.05. c Transcript levels of the four genes that showed a ≥ 2 fold change in Mbnl1 ∆E3/∆E3 cushions. An asterisk indicates significant difference from wild type (P ≤ 0.05). Error bars represent 95 % confidence intervals
Heart valve morphogenesis is aberrant in fetal Mbnl1 ∆E3/∆E3 hearts
The continued expression of MBNL1 in the endocardial cushions and heart valves at post-EMT stages (Fig. 1f-h) suggests that MBNL1 may play additional roles in heart valve development beyond regulating the timing of the onset of EMT. Transformation of the endocardial cushions into the heart valves is a complex process involving expansion and condensation of the mesenchyme, differentiation of mesenchymal cells into valve interstitial cells, and changes in the composition and organization of the extracellular matrix [2, 3, 5]. Heart valve remodeling in the mouse occurs between E10.5 and E18.5. To determine whether heart valve morphogenesis is altered in Mbnl1 ∆E3/∆E3 mice, we compared the three-dimensional structures of fetal valves in E19.5 wild type and Mbnl1 ∆E3/∆E3 hearts using OCT. We chose to focus on the pulmonary valve because this valve is the most accessible at E19.5 for imaging by this method. In the E19.5 wild type heart, the pulmonary valve is globular (Fig. 5a and Additional file 5: Movie S3). Although the boundaries of the individual leaflets cannot be resolved due to their tight apposition, three distinct lobes roughly aligned in the same plane are readily appreciated. In contrast, in the E19.5 Mbnl1 ∆E3/∆E3 heart, the pulmonary valve is highly dysmorphic and its leaflets are not aligned within a single horizontal plane (Fig. 5b and Additional file 6: Movie S4).

Loss of MBNL1 leads to valve dysmorphia in the fetal heart. Three-dimensional representations of the pulmonary valve at E19.5 reconstructed from optical sections of a representative wild type (a) and three different Mbnl1 ∆E3/∆E3 (b) hearts imaged using optical coherence tomography. Videos of the optical sections and reconstructions of the wild type heart in (a) and the leftmost of the Mbnl1 ∆E3/∆E3 hearts in (b) are shown in Additional file 5: Movie S3 and Additional file 6: Movie S4, respectively. Representative high magnification views of the pulmonary (c, d), aortic (f, g), mitral (i, j), and tricuspid (l, m) valves of E18.5 wild type (c, d, i, l) and Mbnl1 ∆E3/∆E3 (d, g, j, m) heart sections stained with hematoxylin and eosin. Yellow asterisks indicate eosinophilic tissue connecting the valve leaflets to the vessel or chamber wall. Nearby sections through the pulmonary (e), aortic (h), mitral (k), and tricuspid (n) valves of the Mbnl1 ∆E3/∆E3 hearts shown in (d, g, j, m) were stained with Movat’s pentachrome stain. Scale bars in panels C-N represent 100 μm
Next, the pulmonary, aortic, mitral, and tricuspid valves were compared in sections from wild type and Mbnl1 ∆E3/∆E3 E18.5 hearts by histology (Fig. 5c-n). In four of eight Mbnl1 ∆E3/∆E3 E18.5 hearts, but none of three wild type hearts, anucleate, eosinophilic tissue connecting the valve leaflets to the vessel or chamber wall was observed in one or more valves by hematoxylin and eosin staining (Fig. 5 and data not shown). In nearby sections, this eosinophilic connective tissue stained bright red with Movat’s pentachrome stain, indicative of fibrin. Consistent with the OCT results, apparent misalignment of valve leaflets was also seen to a variable degree in Mbnl1 ∆E3/∆E3, but not wild type, hearts. Thus, in the absence of MBNL1 the remodeling of the endocardial cushions into the heart valves is aberrant in the fetal heart.
Loss of MBNL1 results in changes in the matrix composition and TGFβ signaling in adult heart valves
Histological analysis also revealed valve dysmorphia in adult Mbnl1 ∆E3/∆E3 mice. Whereas wild type inflow valves stain predominantly blue with Masson’s trichrome stain due to their high collagen content, collagen-poor swellings sparsely populated with small, unstained, stellate cells were seen in some sections from Mbnl1 ∆E3/∆E3 hearts (Fig. 6a). Although these swellings occurred in both the mitral and tricuspid valves, they were larger and more common in the tricuspid valve. These swellings are reminiscent of those seen in periostin-null mice [42], although other defects characteristic of periostin-null valves such as loss of collagen, failure of delamination, and improper remodeling of the associated myocardium were not observed. In the mature valve, the ECM is stratified into three highly organized layers: the collagen-rich fibrosa, proteoglycan-rich spongiosa, and elastin-rich atrialis (or ventricularis in the outflow valves) [43]. Although this stratification appears relatively normal in the Mbnl1 ∆E3/∆E3 valves by Movat’s pentachrome staining, there is reduced mucin staining in the spongiosa layer in the regions of the swellings, indicative of a loss or dilution of the proteoglycans in these sites (Fig. 6b). Using an antibody against chrondroitin sulfate, lower levels of total chondroitin sulfate proteoglycan (CSPG) expression were observed in Mbnl1 ∆E3/∆E3 valves (Fig. 6c). When CSPG transcript levels were compared by real time RT-PCR, however, aggrecan, versican, decorin, and biglycan showed no difference, whereas brevican and neurocan were actually higher in Mbnl1 ∆E3/∆E3 valves (Fig. 6d). It is possible that the apparent loss of CSPGs in the Mbnl1 ∆E3/∆E3 valves is due to reduced production or attachment of the sulfated glycosaminoglycan side chains to the core proteins rather than a decline in proteoglycan expression per se. The ECM protein fibronectin is highly expressed in the endocardial cushions and valve precursors, but is down-regulated during maturation [44, 45]. Although fibronectin transcript levels did not differ between wild type and Mbnl1 ∆E3/∆E3 cushions at E10.5 (Additional file 4: Table S1), fibronectin transcript levels were elevated in adult Mbnl1 ∆E3/∆E3 valves by real-time RT-PCR (Fig. 6d). Elevated levels of fibronectin protein were also seen in adult Mbnl1 ∆E3/∆E3 valves by western blot (Fig. 6c), and accumulations of fibronectin were seen in the tricuspid valve by immunohistochemistry (Fig. 6e).

Loss of MBNL1 leads to changes in matrix composition and TGFβ signaling in the heart valves. a High magnification view of the septal leaflet of the tricuspid valve on a coronal section of an adult Mbnl1 ∆E3/∆E3 heart stained with Masson’s trichrome reveals swellings of collagen-poor tissue in the valve (black arrowhead). The inset shows the corresponding valve leaflet in a wild type heart. Histological analysis was performed on sections from both male and female wild type (n = 7) and knockout (n = 12) mice ranging in age from 4 to 15 months. Representative images shown are from 5 month-old mice. b High magnification view of the mural leaflet of the tricuspid valve on a coronal section of an adult Mbnl1 ∆E3/∆E3 heart stained with Movat’s pentachrome reveals normal stratification but reduced mucin staining indicative of glycosaminoglycans in the regions of swelling. The inset shows a comparable wild type leaflet. Movat’s pentachrome staining was performed on heart sections from three mice for each group ranging from 10 to 15 months old; representative images shown are from 15 month-old mice. c Western blot analysis confirms loss of MBNL1 protein, and shows a decrease in chondroitin sulfate proteoglycans (CSPG) and increases in fibronectin, phosphorylated SMAD2 (pSMAD2), and MBNL2 in Mbnl1 ∆E3/∆E3 heart valves compared to wild type. Protein integrity and loading were also confirmed by Ponceau S staining (data not shown). Representatives of three independent sets using samples from 5–6 month-old male mice are shown. d Transcript levels in Mbnl1 ∆E3/∆E3 heart valves were compared to those of wild type heart valves by real-time RT-PCR (n = 5 to 6 each, ~10 month-old males). e Accumulations of fibronectin (red arrowheads) were seen in the Mbnl1 ∆E3/∆E3 tricuspid valve by immunohistochemistry. The inset shows a tricuspid valve leaflet from a wild type heart. Immunohistochemistry was performed on heart sections from three mice for each group ranging from 4 to 10 months old; representative images shown are from 4 month-old mice
Western blot analyses also revealed higher levels of pSMAD2 in adult Mbnl1 ∆E3/∆E3 valves compared to wild type (Fig. 6c), indicating elevated levels of TGFβ signaling in the adult valves. Unlike precocious activation of TGFβ signaling in the endocardial cushions, this is accompanied by an up-regulation of Tgfb1, though not Tgfb2 or Tgfb3, mRNA levels in the knockout valves (Fig. 6d).
Loss of MBNL1 leads to valve regurgitation and atrial septal defects
To determine whether heart valve function is compromised by loss of MBNL1, transthoracic echocardiography was performed on approximately 22 week old wild type and Mbnl1 ∆E3/∆E3 mice. Color-flow and 2D-pulsed Doppler revealed a high incidence of regurgitation across the pulmonary, aortic, and mitral valves in Mbnl1 ∆E3/∆E3 mice (Table 1 and Additional file 7: Figure S2); blood flow across the tricuspid valve was not assessed. In contrast, no regurgitation was observed across the aortic or mitral valves in wild type mice. Although mild regurgitation was observed across the pulmonary valve in some wild type animals, it was less common and less severe than that observed in the knockout animals (Table 1 and data not shown).
In addition to regurgitation, atrial communication was observed in several of the Mbnl1 ∆E3/∆E3 mice, but only one of their wild type counterparts, by color and pulsed Doppler (Table 1 and Additional file 7: Figure S2). Anatomical dissection of male and female mice ranging in age from 10 weeks to one year-old confirmed the presence of a patent foramen ovale in 50 % of Mbnl1 ∆E3/∆E3 hearts, but less than 10 % of wild type hearts (Fig. 7a and Table 2). Even when successfully closed, the foramen ovale was noticeably enlarged in some Mbnl1 ∆E3/∆E3 hearts, but not in wild type (Fig. 7b and Table 2). Since atrial communication would not occur in these cases, however, there is unlikely to be any functional defect. Closure of the foramen ovale occurs in mice within the first week of postnatal life when the septum primum fuses with the septum secundum [46, 47]. When examined at late fetal stages, the foramen ovale was markedly enlarged in sections of some Mbnl1 ∆E3/∆E3 hearts (Fig. 7c-d). The infolding of the atrial roof at the superior margin (septum secundum) and the flap valve (derived from the septum primum) appear normal in these hearts, but the muscular base of the primary atrial septum where the flap valve is anchored appears reduced. Ostium secundum defects can result from either a failure to fully form the septum secundum, or from a failure of the septum primum to cover the entire oval fossa [47]. This histological analysis suggests the latter, not the former, is the reason for the high incidence of foramen ovale patency and atrial communication in the knockout mice. In other fetal Mbnl1 ∆E3/∆E3 hearts, the foramen ovale and its associated structures look similar to wild type (data not shown). No other septal defects were observed in wild type or knockout hearts.

Mbnl1 E∆3/∆E3 mice exhibit ostium secundum atrial septal defects. a The heart of a one year-old adult Mbnl1 ∆E3/∆E3 mouse is shown with the atrial appendages removed. Patency of the foramen ovale is demonstrated by the unobstructed passage of a coarse bristle. b Excision of the atrial septum from adult hearts shows failure of the foramen ovale to close in some Mbnl1 ∆E3/∆E3 mice (left), while the foramen ovale is closed but enlarged in others (right). Representative images of the wild type and patent Mbnl1 ∆E3/∆E3 foramen ovales shown are from 10 month-old mice; the image of the closed, but enlarged, foramen ovale is from a 2.5-month old Mbnl1 ∆E3/∆E3 mouse. Compared to a wild type counterpart (c), the foramen ovale (bracketed by the green lines) is enlarged in an E19.5 Mbnl1 ∆E3/∆E3 heart (d) as shown in close-ups of coronal sections stained with Masson’s trichrome. SS = septum secundum, FV = flap valve, FO = foramen ovale
Although MBNL1 is also expressed in the myocardium (Fig. 1), myocardial dysfunction has not been observed in Mbnl1 ∆E3/∆E3 mice. Lee and colleagues reported that Mbnl1 ∆E3/∆E3 hearts perform similarly to wild type in echocardiographic and electrocardiographic tests of muscle function [48]. We also evaluated myocardial function in Mbnl1 ∆E3/∆E3 mice by M-mode echocardiography of 8 week old mice (Table 3). Although slight increases in anterior and posterior wall thickness were observed during diastole, there were no differences during systole. There was a trend towards small reductions in ejection fraction and fractional shortening (to ~90 % of wild type values), but these did not reach statistical significance, consistent with previous reports [48]. Total heart size, measured either as a percentage of total body weight or as heart weight normalized to tibia length, also did not differ between wild type and Mbnl1 ∆E3/∆E3 mice at four months of age (Table 4).
It has previously been reported that the closely related paralog, MBNL2, can compensate for loss of MBNL1 in the myocardium. MBNL2 protein levels are elevated in the hearts of Mbnl1 ∆E3/∆E3 mice [48]. MBNL1 and MBNL2 recognize similar RNA elements [49, 50], and MBNL2 binding to MBNL targets increases when MBNL1 is absent [48]. Restoring MBNL2 to near wild type levels in Mbnl1-null mice by removal of one Mbnl2 allele was sufficient to induce atrial dilation, ventricular hypertrophy, and conduction defects [48]. In these compound knockout mice MBNL2 levels were assessed in whole heart, however, leaving it unclear whether MBNL2 was elevated specifically within the myocardium, which comprises the majority of the heart by volume, or in all cells that normally express MBNL1. We tested whether loss of MBNL1 results in increased MBNL2 levels within the heart valves by western blot and real-time RT-PCR (Fig. 6c, d). We found that MBNL2 protein levels were higher in Mbnl1 ∆E3/∆E3 valves, although Mbnl2 transcript levels remained unchanged, suggesting post-transcriptional regulation at the level of translation or protein stability. Thus, the up-regulation of MBNL2 is not restricted to muscle tissue. The presence of valve dysmorphia and regurgitation in Mbnl1 ∆E3/∆E3 mice, however, indicates that unlike in the myocardium, higher levels of MBNL2 are insufficient to fully compensate for loss of MBNL1 in the heart valves.
Pigmentation is increased in Mbnl1 ∆E3/∆E3 heart valves
During dissection and collection of heart valves from adult wild type and Mbnl1 ∆E3/∆E3 hearts, we observed a greater amount of pigment in valves from the knockout mice (Fig. 8). The presence of cardiac melanocytes in murine heart valves has been well documented [51–56]. Lineage tracing studies have demonstrated that these melanocytes are derived from invading neural crest cells, and normally populate the tricuspid, mitral, and aortic valves, but not the pulmonary valve [53, 55]. We saw an increase in the presence of pigmentation in both inflow and outflow valves in Mbnl1 ∆E3/∆E3 hearts, including one instance of pigment in the pulmonary valve (Fig. 8a-c). In order to compare the amount of pigment in wild type and Mbnl1 ∆E3/∆E3 valves, we developed a pigmentation scoring system (Additional file 8: Figure S3) in which each valve was given a score of 0 (no melanin) to 3 (abundant melanin). In addition to an increased incidence of pigmentation, we also saw an increase in the amount of pigment within pigmented valves (Fig. 8d). The level of pigmentation has been shown to correlate with coat color in mice [55]. All of our wild type and Mbnl1 ∆E3/∆E3 mice are on the B6129 background, a hybrid of C57BL/6 black mice and 129 albino mice, and have either brown or black coats. The incidence and relative amount of pigmentation in wild type or Mbnl1 ∆E3/∆E3 valves were the same regardless of coat color (data not shown), so data from brown and black mice were pooled in the graphs in Fig. 8. Despite these differences in pigmentation, wild type and Mbnl1 ∆E3/∆E3 valves express similar levels of dopachrome tautomerase (DCT), a marker of the melanoblast lineage found in melanocytes and their precursors, and tyrosinase related protein 1 (TYRP1), a marker of differentiated melanocytes (Fig. 8e).

Mbnl1 ∆E3/∆E3 heart valves exhibit increased valve pigmentation. a A top view of a 10 month-old adult Mbnl1 ∆E3/∆E3 heart with the major vessels and atrial appendages removed is shown. Pigment can be seen in all four valves of this heart. TC = tricuspid valve, mit = mitral valve, ao = aortic valve, pulm = pulmonary valve. b The accumulation of pigment is readily apparent in a representative high magnification view of the mitral valve on an unstained coronal section of a 10 month-old adult Mbnl1 ∆E3/∆E3 heart. c The percentage of each valve with visible pigment was calculated for wild type (n = 26) and Mbnl1 ∆E3/∆E3 (n = 16) hearts from male and female mice ranging from 2.5 to 10 months old. No differences were observed between the sexes. d The extent of pigmentation was compared for each valve using a scoring system of 0 (no melanin) to 3 (abundant melanin) in the hearts from (c); see Additional file 8: Figure S3 for examples. An asterisk indicates a significant difference from wild type (P ≤ 0.05). e Western blots analysis shows no difference in the expression of melanocyte markers in wild type versus Mbnl1 ∆E3/∆E3 heart valves. Each lane represents a pool of all four valves (tricuspid, mitral, aortic, and pulmonary) from one individual. A representative of two blots (n = 2 or 3 independent samples on each blot from 5–6 month-old males) is shown. Protein integrity and equivalent loading were confirmed by Ponceau S staining (data not shown)
Discussion
The present study demonstrates that MBNL1 is required for normal heart valve development and function. Mbnl1 ∆E3/∆E3 mice, which lack MBNL1 protein, have valve dysmorphia, alterations in ECM composition, and regurgitation across both inflow and outflow valves. Loss of MBNL function has been implicated in the pathogenesis of myotonic dystrophy (dystrophia myotonica, DM), a multisystemic genetic disorder characterized by a number of cardiac, muscle, endocrine, and neurological defects [57]. DM is caused by the expression of mutant RNAs containing expanded CUG (DM1) or CCUG (DM2) repeats, which leads to the dysregulation of other transcripts in part through the sequestration of MBNL proteins (for a detailed review of DM pathogenesis see [57]). Notably, DM patients exhibit a variety of cardiac abnormalities, including a high incidence of mitral valve prolapse [58]. Mbnl1 ∆E3/∆E3 mice recapitulate many of the features of DM, including skeletal muscle myotonia and cataracts [59], but a valve phenotype had not previously been described. Valve dysfunction in DM patients has generally been assumed to be a consequence of myopathy, caused by weakness of the papillary muscles to which the inflow valves connect [58]. Although Mbnl1 ∆E3/∆E3 mice do not exhibit overt cardiomyopathy, loss of MBNL1 function in the papillary muscles could predispose the mice to tricuspid and mitral valve failure. This would not explain differences in function of the aortic and pulmonary valves in Mbnl1 ∆E3/∆E3 mice, however, which are free standing. The expression of both MBNL1 and MBNL2 in wild type adult heart valves argues instead for a direct function of MBNL proteins in heart valve homeostasis. Suggestively, MBNL2 is one of sixteen genes within a locus on chromosome 13 that has been genetically linked with mitral valve prolapse [60], although to date no studies have corroborated a role for MBNL2 in valve disease. Both nuclear and cytoplasmic roles of MBNL proteins have been implicated in DM pathogenesis, including regulation of pre-mRNA alternative splicing, alternative polyadenylation, microRNA biogenesis, mRNA localization and stability [22, 24, 25, 29]. It remains to be determined which of these roles are essential in the developing valves.
We previously showed that knockdown of MBNL1 in chicken endocardial cushion explants enhanced autocrine TGFβ3 and invasive mesenchyme production ex vivo in a stage-dependent manner [20, 21]. Here, we demonstrate that loss of MBNL1 has a similar effect in mouse endocardial cushion explants ex vivo. In chick explants, activation (i.e., separation of cells from the endothelial monolayer) and invasion (migration into the collagen gel) are easily distinguishable steps of EMT; unfortunately, in mouse endocardial cushion explants activation is not readily apparent, as cells appear to invade directly from the explant into the collagen gel [13]. Thus, although we saw a stage-dependent increase in the number of invaded cells in Mbnl1 ∆E3/∆E3 explants, we cannot say whether invasion per se is specifically stimulated as we observed in the chick [20, 21]. Loss of MBNL1 stimulates precocious TGFβ signaling and EMT in mouse endocardial cushions in vivo, which is also consistent with our previous finding that knockdown of MBNL1 in chick endocardial cushion explants stimulates precocious secretion of active TGFβ3 in vitro [20]. This effect is temporary in the mice, however, as the amount of total mesenchyme produced is normalized by E10.5, and few changes in EMT gene expression are observed at this stage. Taken together, these data suggest that MBNL1 regulates the initiation of EMT, but not its progression. The dysmorphia and presence of abnormal connective tissue in the fetal valves indicates a developmental role for MBNL1 in valvulogenesis; if not regulating the amount of cushion mesenchyme, then perhaps influencing its fate during valve maturation.
The stratification of the adult valve suggests that matrix remodeling is relatively unimpaired in Mbnl1 ∆E3/∆E3 mice. The presence of collagen/proteoglycan-poor swellings sparsely populated with stellate cells, however, indicates localized increases in matrix with an atypical composition. Indeed, while CSPGs levels are reduced, the ECM protein fibronectin is strongly up-regulated. The maintenance of stratification and the specific changes in ECM composition observed in the adult Mbnl1 ∆E3/∆E3 valves are not characteristic of either myxomatous valve degeneration or calcific valve disease. The persistence of fibronectin in adult Mbnl1 ∆E3/∆E3 valves could be indicative of incomplete or defective differentiation of the cushion mesenchyme in the knockout mice. MBNL1 has recently been shown to regulate alternative splicing changes that are important for stem cell differentiation [61, 62], and MBNL1 has been implicated in the differentiation of skeletal muscle, blood, lens, and adipose cells [63–67]. Alternatively, elevated fibronectin expression in the adult valves could be indicative of injury to the valves, perhaps due to changes in shear stress caused by altered valve biomechanics. Valve interstitial cells have been shown to up-regulate fibronectin in response to injury in primary cultures [68]. The accumulation of fibronectin in the adult valve may in turn then have a detrimental effect on valve function by altering the valve’s elasticity or stiffness.
The higher levels of fibronectin in adult Mbnl1 ∆E3/∆E3 valves could also potentially have a protective effect. We found that both Tgfb1 transcript levels and TGFβ signaling (as indicated by pSMAD2) are elevated in the adult knockout valves. Elevated TGFβ1 levels are associated with calcification in stenotic valves, and addition of TGFβ1 is sufficient to induce calcification in valve interstitial cells in culture [69]. We have observed no signs of overt calcification in Mbnl1 ∆E3/∆E3 valves, however, even in animals as old as 90 weeks (data not shown). Notably, fibronectin has been shown to protect cultured valve interstitial cells from TGFβ1-induced calcification [70]. The mechanisms by which TGFβ1 and fibronectin are up-regulated in adult Mbnl1 ∆E3/∆E3 valves, and the extent to which MBNL1 exerts its effects in the mature valve through modulation of the ECM and/or TGFβ activity, remain to be determined.
The relevance of increased pigmentation in the Mbnl1 ∆E3/∆E3 heart valves is unclear. Pigmented cells were first observed in the mouse heart over fifty years ago [54]. Although gene expression profiling suggests these cardiac melanocytes are more similar to cardiomyocytes than canonical melanocytes found in the skin, these neural crest-derived cells express melanocyte markers and produce melanin in non-albino mice [51–53, 55, 56]. DCT-expressing cells have also been reported in human heart valves, although these cells lack TYRP1 and are not pigmented [56]. The purpose of these cells is not well understood. One suggestion has been that the enzymes within the melanin synthesis pathway buffer calcium and reactive species in the heart [56]. Melanin is synthesized from the amino acid tyrosine in a multistep process in which DCT converts the intermediate dopachrome into 5,6-dihydroxyindole carboxylic acid (DHICA), which is then converted into eumelanin by TYRP1 [71]. The normal levels of DCT and TYRP1 in the Mbnl1 ∆E3/∆E3 valves suggests that the increase in pigmentation is not due to an increase in the number of melanocytes in the valves, or an up-regulation in these steps of melanin production. Little is known about melanin turnover. The accumulation of pigment in the heart valves is associated with stiffening of the valves, regurgitation, and stenosis in patients with alkaptonuria, a rare autosomal recessive disorder of tyrosine metabolism characterized by ochronosis, the accumulation of pigment within the connective tissues [72–75]. An ex vivo nanomechanical study of mouse tricuspid valves reported that pigmented regions of the tricuspid leaflet are stiffer than regions lacking pigment [76]. Thus, increased pigment in the valves of Mbnl1 ∆E3/∆E3 mice could contribute to their functional impairment by altering their biomechanical properties.
The impact of loss of MBNL1 in the heart is not confined to the developing valves. Many Mbnl1 ∆E3/∆E3 mice had ostium secundum defects. A strong association between ostium secundum defects and mitral valve prolapse has been documented [77, 78]. Examination of the fetal heart prior to closure revealed that the septum secundum, which forms from infolding of the atrial roof at the superior margin, and the septum primum, which forms the flap valve, appear normal in the knockout mice. The muscular base to which the flap valve is anchored, however, is smaller in some Mbnl1 ∆E3/∆E3 hearts. The muscular base of the atrial septum arises from myocardialization of mesenchyme within the dorsal mesenchymal protrusion (DMP), a derivative of the secondary heart field, not the endocardial cushions [47]. Underdevelopment of the DMP can be caused by reduced proliferation, increased apoptosis, or premature myocardialization of the DMP mesenchyme [47]. Mutations in several genes, including cardiac transcription factors such as NKX2.5, GATA4, and TBX20, have been linked to atrial septal defects and foramen ovale patency in humans [79–81], but their precise roles in the development and maturation of the DMP are not known. Often, defects in the DMP are associated with more severe atrioventricular septal defects, but these, or other types of septal defects that arise from defective remodeling of the endocardial cushions, are not observed in Mbnl1 ∆E3/∆E3 mice. Although atrial communication can lead to right atrial overload and other cardiac complications, stand-alone ostium secundum defects are often clinically benign [47].
The lack of overt cardiomyopathy in Mbnl1 ∆E3/∆E3 mice despite both atrial communication and poor valve function might initially be surprising, but it should be noted that these mice also experience progressive skeletal muscle myotonia [59]. As Mbnl1 ∆E3/∆E3 mice age and their myotonia worsens, their level of activity decreases, which may help reduce their cardiac burden. The up-regulation of MBNL2 may also help the strained heart muscle compensate [48]. It has also been reported, however, that Mbnl1-knockout mice on a pure 129 sv genetic background (as opposed to Mbnl1 ∆E3/∆E3 mice, which are on a hybrid background of C57/BL6 and129 strains) exhibit progressive cardiac dysfunction, cardiac hypertrophy, and myocardial cell death and fibrosis despite a similar up-regulation of MBNL2 [82]. The difference in heart muscle pathogenesis between the mouse strains suggests the existence of modifier genes that modulate the function of MBNL1 in the heart. MBNL1-dependent heart valve phenotypes have not been investigated on a pure 129 background.
Conclusions
MBNL1 is highly expressed in the endocardial cushions and heart valves. It plays a conserved role in regulating the onset of TGFβ signaling and EMT in the endocardial cushions, but is not essential for limiting the total amount of cushion mesenchyme production in vivo. Mice lacking MBNL1 exhibit defects in valve morphogenesis, ECM composition, function, and pigmentation, indicating that MBNL1 is required for normal post-EMT valve development.
Methods
Mbnl1 ∆E3/∆E3 Mice
Mouse studies were conducted in strict accordance with the recommendations of the American Veterinary Medical Association and under the approval of the Cleveland Clinic Institutional Animal Care and Use Committee (Protocols 2011–0547 and 2014–1201). All efforts were made to minimize pain and distress during mouse animal husbandry and euthanasia. Mbnl1 ∆E3/∆E3 mice have a targeted deletion of Mbnl1 exon 3, which contains the canonical translation start codon, resulting in loss of MBNL1 expression [59]. Mice were maintained in a B6129 hybrid background. Embryos at specific stages were obtained by timed mating. In general, midday following observation of a copulation plug was considered embryonic day 0.5 (E0.5). Stages of embryos collected from E9 through E10.5 were confirmed by counting the number of somites of each individual embryo (E9 = 19–20 somites, E9.5 = 22–23 somites, E10.5 = 34–36 somites).
Immunohistochemistry and western blotting
For immunohistochemistry, whole embryos or postnatal hearts were fixed overnight by immersion in 4 % paraformaldehyde (embryonic stages) or 10 % neutrally buffered formalin (adult), and then embedded in paraffin. Immunohistochemistry was performed on 10 μm sections. The sections were immersed in xylene, rehydrated through a 100 %, 95 %, and 80 % ethanol series, and then washed in water. Antigen retrieval was performed by boiling slides in citrate buffer (10 mM citric acid, 0.05 % Tween 20, at pH 6.0) for 30 min. The slides were washed three times in 1x Tris-buffered saline (TBS) and blocked in 3 % goat serum in 1x TBS for 1 h at room temperature. The primary antibody was added at a 1:50 to 1:400 dilution and incubated overnight at 4 °C. The slides were washed in TBS as before, incubated in 0.3 % hydrogen peroxide for 30 min at room temperature, and washed in 1x TBS. The secondary antibody was added at a 1:400 dilution and incubated for one hour at room temperature. Slides were washed 3 times in 1X TBS and developed using DAB Plus Substrate Kit (Invitrogen) according to the manufacturer’s instructions. The slides were dehydrated through a 25 %, 50 %, 75 %, and 100 % ethanol series, cleared with xylene, and mounted with Permount (Fisher Scientific). Negative controls lacking primary antibody were performed in parallel for every experiment.
Heart tissues were homogenized in protein loading buffer and subjected to western blotting as previously described [30]. Protein integrity and equivalent loading were verified by Ponceau S staining. For western blots on adult heart valves, all four valves (tricuspid, mitral, aortic, and pulmonary) from one to three individuals were pooled together for each sample. All westerns were performed on three or more independent pooled samples from each group.
Primary antibodies used for immunohistochemistry and western blotting were: MBNL1 = rabbit polyclonal anti-MBNL1 (Aviva Systems Biology, catalog number ARP41227_P050) for immunohistochemistry, and mouse monoclonal anti-MBNL1, 3A4 [83] or rabbit polyclonal anti-MBNL1, A2764 (generously provided by Charles A. Thornton, University of Rochester, NY) for western blotting; pSMAD2 = rabbit polyclonal anti-phospho-Smad2 (Ser465/467) (Cell Signaling Technology, catalog number 3101); CSPG = mouse monoclonal anti-chondroitin sulfate, CS-56 (Sigma, catalog number C8035); Fibronectin = rabbit polyclonal anti-fibronectin (Sigma, catalog number F3648); GAPDH = mouse monoclonal anti-GAPDH, 6G5 (Biogenesis, catalog number 4699–9555); DCT = goat polyclonal anti-TRP2, D-18 (Santa Cruz, catalog number sc-10451); TYRP1 = rabbit polyclonal anti-TRP1, H-90 (Santa Cruz, catalog number sc-25543); MBNL2 = mouse monoclonal anti-MBNL2, 3B4 (Santa Cruz, catalog number sc-136167). Secondary antibodies used were: goat anti-rabbit-HRP (Calbiochem, catalog number 401393); goat anti-mouse-HRP (Calbiochem, catalog number DC02L); donkey anti-goat-HRP (Santa Cruz, sc-2056).
Explants
Collagen gels were prepared as previously described [21]. Collagen gels were incubated in Hydration Medium [Opti-MEM supplemented with 1 % each ITS (Invitrogen), fetal bovine serum, and pen/strep antibiotic solution] for 30 min at 37 °C prior to use, which was then removed before explants were placed on the gel. The AVC regions were excised from E9 or E9.5 embryos, cut open, placed endothelial-side down on the collagen gels, and incubated at 37 °C in 5 % CO2. After 12 h, Mouse M199+ [M199 supplemented with 1 % each ITS (Invitrogen), fetal bovine serum, and pen/strep antibiotic solution] was added to each well. Explants were fixed after 48 h of culture in 4 % paraformaldehyde for 45 min at room temperature, and stored in PBS at 4 °C. Invaded cells were counted on a Leica DMIRB microscope fitted with Hoffman Modulation Contrast optics. Explants were imaged using QCapture Pro 6.0 software.
For enzyme-linked immunosorbent assay (ELISA) experiments, 200 μl of Mouse M199+ medium was added to each explant at 12 h, and the conditioned medium was collected at 48 h. Secreted active TGFβ levels in the conditioned media were measured by sandwich ELISA as previously described [20]. A pan-TGFβ antibody (R&D Systems, catalog number MAB1835) was used for capture, and a specific biotinylated anti-TGFβ1 (R&D Systems, catalog number BAF240), anti-TGFβ2 (R&D Systems, catalog number BAF302), or anti-TGFβ3 (R&D Systems, catalog number BAF243) was used for detection. Serial dilutions of recombinant TGFβ1, TGFβ2, and TGFβ3 (R&D Systems, catalog numbers 240-B-002, 302-B2, and 243-B3, respectively) were used to generate standard curves.
Gene expression and alternative splicing
Total RNA was extracted using Trizol Reagent (Thermo Scientific) and quantified using a NanoDrop 1000. Endocardial cushions were dissected out of wild type and Mbnl1 ∆E3/∆E3 E10.5 embryos by cutting open the heart tube and separating the cushions from the underlying myocardium using an electrolytically-sharpened tungsten needle [84]. Both the superior and inferior AVC cushions were collected from each embryo, and the cushions from 10–14 embryos (one to two litters) were combined and treated as a single biological replicate. Three such pooled samples were collected for each group. The expression levels of 84 genes that either change their expression during EMT or regulate gene expression changes during EMT were assessed by real-time RT-PCR using the Epithelial to Mesenchymal Transition RT2 Profiler PCR Array (Qiagen) according to the manufacturer’s instructions. Transcript levels were normalized to Gusb; similar results were obtained when data were normalized against Gapdh (data not shown). Error bars represent 95 % confidence intervals.
For gene expression changes in adult heart valves, each sample represents a pool of all four heart valves (tricuspid, mitral, aortic, and pulmonary) from a single individual. Transcript levels were determined by real-time RT-PCR using SYBR Green Master Mix (Applied Biosystems) and normalized to Gapdh as previously described [85]. Primer sequences for aggrecan, versican, brevican, neurocan, decorin, bigylcan, and scleraxis were taken from Barnette et al. [86]; sequences of the remaining primers are shown in Table 5. Samples from five to six individuals per group were each run in triplicate. Error bars represent standard error of the mean values for the biological replicates.
Histology
Whole embryos or postnatal hearts were fixed overnight by immersion in 4 % paraformaldehyde (embryonic stages) or 10 % neutrally buffered formalin (adult). Fixed embryos or hearts were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E), Masson’s trichrome, or Movat’s pentachrome stain. To determine the relative number of mesenchymal cells at E10.5, 10 μm sections were taken through the entire heart, those sections containing AVC cushions were identified, and six sections taken 10 μm apart (every other section) from the center of the AVC region were stained with H&E, imaged on a Leica DM 2500 microscope using QCapture Pro 6.0 software, and counted. Invaded cells were counted in the cushions of four embryos per group.
Optical coherence tomography (OCT)
Wild type and Mbnl1 ∆E3/∆E3 embryos were collected at E10.5 (n = 10 and 6, respectively) or E19.5 (n = 3 each), fixed overnight in 4 % paraformaldehyde, dehydrated through a 25, 50, 75, 100 % methanol series in PBSw (PBS with 0.1 % Tween-20), and stored in 100 % methanol at −20 °C. Embryos were later rehydrated through a 100, 75, 50, 25 % methanol series in PBSw followed by transfer into PBS. The hearts were removed under a dissecting microscope. E19.5 hearts were cleared prior to imaging using the Clear T method described for whole dissected brains (E16-P11) [87], which has also been used for embryonic day 8 quail embryo hearts [88], to improve penetration depth. Embryos were imaged using a custom-built Fourier domain OCT system with a quasi-telecentric scanner, linear-in-wavenumber spectrometer and a line-scan camera with a line rate of 47 kHz [89, 90]. The axial resolution as well as the lateral resolution is approximately 10 μm in tissue. Thus OCT imaging offers high spatial resolution and good penetration depth (1–3 mm in cardiac tissues) for phenotyping embryonic hearts [91]. Custom MATLAB programs (MathWorks; Natick, MA) were utilized to create OCT images from the raw data. Amira software (FEI Visualization Sciences Group; Burlington, MA) was used to visualize OCT data.
Heart function and size
Cardiac structural and functional changes were assessed in vivo by echocardiography performed on age- and sex-matched wild type and Mbnl1 ∆E3/∆E3 mice anesthetized with isofluorane gas (1-2 % with 2 lpm oxygen) using a GE Vivid7 Ultrasound system equipped with a 14 MHz iL13 probe. Two-dimensional and M-mode echocardiographic measurements of the anterior wall, posterior wall, and left ventricular diameter were recorded in the parasternal short axis view of the left ventricle at the level of the papillary muscles. Modified short-axis views were recorded to assess the possibility of atrial or ventricular septal defects, and to obtain pulmonary, aortic, and mitral valve Doppler flow measurements. Functional parameters were calculated using standard formulas [92]. To control for differences in body size, heart size was evaluated both as the percentage of total body weight that is heart weight and as a ratio of heart weight to tibia length.
Statistics
Data are reported as the mean + standard error of the mean unless otherwise noted. Comparisons between means were performed via t-test assuming unequal variances. Comparisons between proportions were performed via z-test. Differences were considered statistically significant when P ≤ 0.05.
Abbreviations
- AVC:
-
Atrioventricular canal
- CSPG:
-
Chondroitin sulfate proteoglycan
- DCT:
-
Dopachrome tautomerase
- DMP:
-
Dorsal mesenchymal protrusion
- E:
-
Embryonic day
- ECM:
-
Extracellular matrix
- ELISA:
-
Enzyme-linked immunosorbent assay
- EMT:
-
Epithelial-mesenchymal transition
- GAP:
-
GTPase activating protein
- MBNL:
-
Muscleblind-like
- OCT:
-
Optical coherence tomography
- OFT:
-
Outflow tract
- SPP1:
-
Secreted phosphoprotein 1
- TGFβ:
-
Transforming growth factor β
- TYRP1:
-
Tyrosinase related protein 1
References
Pierpont ME, Basson CT, Benson Jr DW, Gelb BD, Giglia TM, Goldmuntz E, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):3015–38.
Lincoln J, Yutzey KE. Molecular and developmental mechanisms of congenital heart valve disease. Birth Defects Res A Clin Mol Teratol. 2011;91(6):526–34.
Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Ann Rev Physiol. 2011;73:29–46.
Lahaye S, Lincoln J, Garg V. Genetics of valvular heart disease. Curr Cardiol Rep. 2014;16(6):487.
Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. Int Rev Cytol. 2005;243:287–335.
Kirby ML. Cardiac Development. New York: Oxford University Press; 2007.
Azhar M, Runyan RB, Gard C, Sanford LP, Miller ML, Andringa A, et al. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev Dyn. 2009;238(2):431–42.
Boyer AS, Ayerinskas II, Vincent EB, McKinney LA, Weeks DL, Runyan RB. TGFbeta2 and TGFbeta3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev Biol. 1999;208(2):530–45.
Potts JD, Dagle JM, Walder JA, Weeks DL, Runyan RB. Epithelial-mesenchymal transformation of embryonic cardiac endothelial cells is inhibited by a modified antisense oligodeoxynucleotide to transforming growth factor beta 3. Proc Natl Acad Sci U S A. 1991;88(4):1516–20.
Ghosh S, Brauer PR. Latent transforming growth factor-beta is present in the extracellular matrix of embryonic hearts in situ. Dev Dyn. 1996;205(2):126–34.
Nakajima Y, Yamagishi T, Nakamura H, Markwald RR, Krug EL. An autocrine function for transforming growth factor (TGF)-beta3 in the transformation of atrioventricular canal endocardium into mesenchyme during chick heart development. Dev Biol. 1998;194(1):99–113.
Ramsdell AF, Markwald RR. Induction of endocardial cushion tissue in the avian heart is regulated, in part, by TGFbeta-3-mediated autocrine signaling. Dev Biol. 1997;188(1):64–74.
Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA, et al. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol. 2002;248(1):170–81.
Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11(4):415–21.
Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11(4):409–14.
Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111(1):131–43.
Azhar M, Brown K, Gard C, Chen H, Rajan S, Elliott DA, et al. Transforming growth factor Beta2 is required for valve remodeling during heart development. Dev Dyn. 2011;240(9):2127–41.
Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124(13):2659–70.
Letterio JJ, Geiser AG, Kulkarni AB, Roche NS, Sporn MB, Roberts AB. Maternal rescue of transforming growth factor-beta 1 null mice. Science. 1994;264(5167):1936–8.
LeMasters KE, Blech-Hermoni Y, Stillwagon SJ, Vajda NA, Ladd AN. Loss of muscleblind-like 1 promotes invasive mesenchyme formation in endocardial cushions by stimulating autocrine TGFbeta3. BMC Dev Biol. 2012;12(1):22.
Vajda NA, Brimacombe KR, LeMasters KE, Ladd AN. Muscleblind-like 1 is a negative regulator of TGF-beta-dependent epithelial-mesenchymal transition of atrioventricular canal endocardial cells. Dev Dyn. 2009;238(12):3266–72.
Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell. 2014;56(2):311–22.
Ho T, Charlet-B N, Poulos M, Singh G, Swanson M, Cooper T. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–12.
Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, et al. Transcriptome-wide Regulation of Pre-mRNA Splicing and mRNA Localization by Muscleblind Proteins. Cell. 2012;150(4):710–24.
Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol. 2011;18(7):840–5.
Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A. 2008;105(51):20333–8.
Ladd A, Stenberg M, Swanson M, Cooper T. A dynamic balance between activation and repression regulates pre-mRNA alternative splicing during heart development. Dev Dyn. 2005;233(3):783–93.
Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74(2–3):65–80.
Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15(13):2087–97.
Terenzi F, Ladd AN. Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biol. 2010;7(1):43–55.
Kino Y, Washizu C, Kurosawa M, Oma Y, Hattori N, Ishiura S, et al. Nuclear localization of MBNL1: splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum Mol Genet. 2015;24(3):740–56.
Mercado-Pimentel ME, Runyan RB. Multiple transforming growth factor-beta isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs. 2007;185(1–3):146–56.
DeLaughter DM, Saint-Jean L, Baldwin HS, Barnett JV. What chick and mouse models have taught us about the role of the endocardium in congenital heart disease. Birth Defects Res A Clin Mol Teratol. 2011;91(6):511–25.
Gheldof A, Hulpiau P, van Roy F, De Craene B, Berx G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell Mol Life Sci. 2012;69(15):2527–41.
Funaba M, Ikeda T, Murakami M, Ogawa K, Tsuchida K, Sugino H, et al. Transcriptional activation of mouse mast cell Protease-7 by activin and transforming growth factor-beta is inhibited by microphthalmia-associated transcription factor. J Biol Chem. 2003;278(52):52032–41.
Kim RY, Robertson EJ, Solloway MJ. Bmp6 and Bmp7 are required for cushion formation and septation in the developing mouse heart. Dev Biol. 2001;235(2):449–66.
Hullinger TG, Pan Q, Viswanathan HL, Somerman MJ. TGFbeta and BMP-2 activation of the OPN promoter: roles of smad- and hox-binding elements. Exp Cell Res. 2001;262(1):69–74.
Peacock JD, Huk DJ, Ediriweera HN, Lincoln J. Sox9 transcriptionally represses Spp1 to prevent matrix mineralization in maturing heart valves and chondrocytes. PLoS One. 2011;6(10):e26769.
Sainger R, Grau JB, Poggio P, Branchetti E, Bavaria JE, Gorman 3rd JH, et al. Dephosphorylation of circulating human osteopontin correlates with severe valvular calcification in patients with calcific aortic valve disease. Biomarkers. 2012;17(2):111–8.
Grau JB, Poggio P, Sainger R, Vernick WJ, Seefried WF, Branchetti E, et al. Analysis of osteopontin levels for the identification of asymptomatic patients with calcific aortic valve disease. Ann Thorac Surg. 2012;93(1):79–86.
Cheek JD, Wirrig EE, Alfieri CM, James JF, Yutzey KE. Differential activation of valvulogenic, chondrogenic, and osteogenic pathways in mouse models of myxomatous and calcific aortic valve disease. J Mol Cell Cardiol. 2012;52(3):689–700.
Norris RA, Moreno-Rodriguez RA, Sugi Y, Hoffman S, Amos J, Hart MM, et al. Periostin regulates atrioventricular valve maturation. Dev Biol. 2008;316(2):200–13.
Hinton Jr RB, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98(11):1431–8.
Bouchey D, Argraves WS, Little CD. Fibulin-1, vitronectin, and fibronectin expression during avian cardiac valve and septa development. Anat Rec. 1996;244(4):540–51.
Kruithof BP, Krawitz SA, Gaussin V. Atrioventricular valve development during late embryonic and postnatal stages involves condensation and extracellular matrix remodeling. Dev Biol. 2007;302(1):208–17.
Cole-Jeffrey CT, Terada R, Neth MR, Wessels A, Kasahara H. Progressive anatomical closure of foramen ovale in normal neonatal mouse hearts. Anat Rec. 2012;295(5):764–8.
Briggs LE, Kakarla J, Wessels A. The pathogenesis of atrial and atrioventricular septal defects with special emphasis on the role of the dorsal mesenchymal protrusion. Differentiation. 2012;84(1):117–30.
Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med. 2013;5(12):1887–900.
Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, et al. Muscleblind-like 2-Mediated Alternative Splicing in the Developing Brain and Dysregulation in Myotonic Dystrophy. Neuron. 2012;75(3):437–50.
Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, et al. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci Rep. 2012;2:209.
Brito FC, Kos L. Timeline and distribution of melanocyte precursors in the mouse heart. Pigment Cell Melanoma Res. 2008;21(4):464–70.
Mjaatvedt CH, Kern CB, Norris RA, Fairey S, Cave CL. Normal distribution of melanocytes in the mouse heart. Anat Rec A Discov Mol Cell Evol Biol. 2005;285(2):748–57.
Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98(12):1547–54.
Nichols Jr SE, Reams Jr WM. The occurrence and morphogenesis of melanocytes in the connective tissues of the PET/MCV mouse strain. J Embryol Exp Morphol. 1960;8:24–32.
Yajima I, Larue L. The location of heart melanocytes is specified and the level of pigmentation in the heart may correlate with coat color. Pigment Cell Melanoma Res. 2008;21(4):471–6.
Levin MD, Lu MM, Petrenko NB, Hawkins BJ, Gupta TH, Lang D, et al. Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J Clin Invest. 2009;119(11):3420–36.
Schoser B, Timchenko L. Myotonic dystrophies 1 and 2: complex diseases with complex mechanisms. Curr Genomics. 2010;11(2):77–90.
Chaudhry SP, Frishman WH. Myotonic dystrophies and the heart. Cardiol Rev. 2012;20(1):1–3.
Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302(5652):1978–80.
Nesta F, Leyne M, Yosefy C, Simpson C, Dai D, Marshall JE, et al. New locus for autosomal dominant mitral valve prolapse on chromosome 13: clinical insights from genetic studies. Circulation. 2005;112(13):2022–30.
Han H, Irimia M, Ross PJ, Sung HK, Alipanahi B, David L, et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013;498(7453):241–5.
Venables JP, Lapasset L, Gadea G, Fort P, Klinck R, Irimia M, et al. MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat Commun. 2013;4:2480.
Miller J, Urbinati C, Teng-umnuay P, Stenberg M, Byrne B, Thornton C, et al. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000;19(17):4439–48.
Redmond LC, Dumur CI, Archer KJ, Haar JL, Lloyd JA. Identification of erythroid-enriched gene expression in the mouse embryonic yolk sac using microdissected cells. Dev Dyn. 2008;237(2):436–46.
Shang CA, Thompson BJ, Teasdale R, Brown RJ, Waters MJ. Genes induced by growth hormone in a model of adipogenic differentiation. Mol Cell Endocrinol. 2002;189(1–2):213–9.
Coleman SM, Prescott AR, Sleeman JE. Transcriptionally correlated subcellular dynamics of MBNL1 during lens development and their implication for the molecular pathology of myotonic dystrophy type 1. Biochem J. 2014;458(2):267–80.
Cheng AW, Shi J, Wong P, Luo KL, Trepman P, Wang ET, et al. Muscleblind-like 1 (Mbnl1) regulates pre-mRNA alternative splicing during terminal erythropoiesis. Blood. 2014;124(4):598–610.
Fayet C, Bendeck MP, Gotlieb AI. Cardiac valve interstitial cells secrete fibronectin and form fibrillar adhesions in response to injury. Cardiovasc Pathol. 2007;16(4):203–11.
Jian B, Narula N, Li QY, Mohler 3rd ER, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75(2):457–65. discussion 465–456.
Gwanmesia P, Ziegler H, Eurich R, Barth M, Kamiya H, Karck M, et al. Opposite effects of transforming growth factor-beta1 and vascular endothelial growth factor on the degeneration of aortic valvular interstitial cell are modified by the extracellular matrix protein fibronectin: implications for heart valve engineering. Tissue Eng Part A. 2010;16(12):3737–46.
Sulaimon SS, Kitchell BE. The biology of melanocytes. Vet Dermatol. 2003;14(2):57–65.
Aquaron R. Alkaptonuria: a very rare metabolic disorder. Indian J Biochem Biophys. 2013;50(5):339–44.
Kenny D, Ptacin MJ, Bamrah VS, Almagro U. Cardiovascular ochronosis: a case report and review of the medical literature. Cardiology. 1990;77(6):477–83.
Butany JW, Naseemuddin A, Moshkowitz Y, Nair V. Ochronosis and aortic valve stenosis. J Card Surg. 2006;21(2):182–4.
Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, et al. Natural history of alkaptonuria. New Engl J Med. 2002;347(26):2111–21.
Balani K, Brito FC, Kos L, Agarwal A. Melanocyte pigmentation stiffens murine cardiac tricuspid valve leaflet. J R Soc Interface. 2009;6(40):1097–102.
Nagata S, Nimura Y, Sakakibara H, Beppu S, Park YD, Kawazoe K, et al. Mitral valve lesion associated with secundum atrial septal defect. Analysis by real time two dimensional echocardiography. Br Heart J. 1983;49(1):51–8.
Leachman RD, Cokkinos DV, Cooley DA. Association of ostium secundum atrial septal defects with mitral valve prolapse. Am J Cardiol. 1976;38(2):167–9.
Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424(6947):443–7.
Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, et al. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A. 2005;135(1):47–52.
Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81(2):280–91.
Dixon DM, Choi J, El-Ghazali A, Park SY, Roos KP, Jordan MC, et al. Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci Rep. 2015;5:9042.
Mankodi A, Teng-Umnuay P, Krym M, Henderson D, Swanson M, Thornton CA. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol. 2003;54(6):760–8.
Brady J. A simple technique for making very fine, durable dissecting needles by sharpening tungsten wire electrolytically. Bull World Health Organ. 1965;32(1):143–4.
Dasgupta T, Stillwagon SJ, Ladd AN. Gene expression analyses implicate an alternative splicing program in regulating contractile gene expression and serum response factor activity in mice. PLoS One. 2013;8(2):e56590.
Barnette DN, Hulin A, Ahmed AS, Colige AC, Azhar M, Lincoln J. Tgfbeta-Smad and MAPK signaling mediate scleraxis and proteoglycan expression in heart valves. J Mol Cell Cardiol. 2013;65:137–46.
Kuwajima T, Sitko AA, Bhansali P, Jurgens C, Guido W, Mason C. ClearT: a detergent- and solvent-free clearing method for neuronal and non-neuronal tissue. Development. 2013;140(6):1364–8.
Karunamuni G, Gu S, Doughman YQ, Noonan AI, Rollins AM, Jenkins MW, et al. Using optical coherence tomography to rapidly phenotype and quantify congenital heart defects associated with prenatal alcohol exposure. Dev Dyn. 2015;244(4):607–18.
Hu Z, Rollins AM. Fourier domain optical coherence tomography with a linear-in-wavenumber spectrometer. Opt Lett. 2007;32(24):3525–7.
Karunamuni G, Gu S, Doughman YQ, Peterson LM, Mai K, McHale Q, et al. Ethanol exposure alters early cardiac function in the looping heart: a mechanism for congenital heart defects? Amer J Physiol Heart Circ Physiol. 2014;306(3):H414–21.
Jenkins MW, Watanabe M, Rollins AM. Longitudinal Imaging of Heart Development with Optical Coherence Tomography. IEEE J Sel Topics Quantum Electron. 2012;18(3):1166–75.
Hartley C, Taffet G, Reddy A, Entman M, Michael L. Noninvasive cardiovascular phenotyping in mice. ILAR J. 2002;43(3):147–58.
Acknowledgements
We thank Michiko Watanabe for helpful discussions of the project, Oliver Wessely and members of his laboratory for their assistance with immunohistochemistry protocols, Shi Gu for helping write the code for processing OCT images, and Andrew Rollins for allowing us to use his OCT system. This work was supported by grants from the National Institutes of Health (R01HL089376 to ANL; R21HL115373 to MWJ; R01AR046799 and P01NS058901 to MSS). AG was supported by a postdoctoral fellowship from the American Heart Association. The funding bodies played no role in the study design; collection, analysis, or interpretation of data; writing of the manuscript; or in the decision to publish.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ANL is responsible for the overall study design, assisted with experiments, analyzed and interpreted data, and drafted the bulk of the manuscript. RJC and SJS performed the majority of the experiments, analyzed and interpreted data, drafted the Methods section, and edited the manuscript. AG performed the echocardiography and Doppler ultrasound experiments, analyzed and interpreted functional data, and helped revise the manuscript. MWJ helped design and performed the OCT experiments, analyzed and interpreted OCT data, and helped revise the manuscript. MSS provided the Mbnl1 ∆E3/∆E3 mice and technical support for the project, and helped revise the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1: Figure S1.
MBNL1 expression in the embryonic brain. Immunohistochemistry with an anti-MBNL1 antibody was performed on sagittal sections from E9 wild type mouse embryos. Close-up of the head shows strong MBNL1 expression in the ectoderm lining the entrance to Rathke’s pouch (Rp), forebrain (FB), midbrain (MB), and hindbrain (HB) with little to no detectable staining within the interior encephalic tissues (highlighted by asterisks). A representative section from one of four embryos is shown. (PDF 1247 kb)
Additional file 2: Movie S1.
Imaging of the AVC cushions in a representative wild type heart at E10.5 by OCT. Optical sections through a representative E10.5 wild type heart are shown. The AVC cushions are highlighted in purple, and a three-dimensional reconstruction of the AVC cushions is shown. A still image of this 3D reconstruction is shown in Fig. 3h, but has been rotated and scaled to match the orientation and magnification of the Mbnl1 ∆E3/∆E3 image in Fig. 3i. (MOV 2333 kb)
Additional file 3: Movie S2.
Imaging of the AVC cushions in a representative Mbnl1 ∆E3/∆E3 heart at E10.5 by OCT. Optical sections through a representative E10.5 Mbnl1 ∆E3/∆E3 heart are shown. The AVC cushions are highlighted in purple, and a three-dimensional reconstruction of the AVC cushions is shown. A still image of this 3D reconstruction is shown in Fig. 3i. (MOV 1197 kb)
Additional file 4: Table S1.
Epithelial to Mesenchymal Transition RT2 Profiler PCR Array data for Mbnl1 ∆E3/∆E3 versus wild type E10.5 AVC cushions. (XLS 42 kb)
Additional file 5: Movie S3.
Imaging of the pulmonary valve in a representative wild type heart at E19.5 by OCT. A three-dimensional reconstruction of a representative wild type E19.5 heart is shown with the pulmonary valve highlighted in purple, followed by optical sections through the heart. The pulmonary valve is indicated in purple, and a three-dimensional reconstruction of the valve is shown. A still image of this 3D reconstruction is shown in Fig. 5a. (MOV 3438 kb)
Additional file 6: Movie S4.
Imaging of the pulmonary valve in a representative Mbnl1 ∆E3/∆E3 heart at E19.5 by OCT. A three-dimensional reconstruction of a representative Mbnl1 ∆E3/∆E3 E19.5 heart is shown with the pulmonary valve highlighted in purple, followed by optical sections through the heart. The pulmonary valve is indicated in purple, and a three-dimensional reconstruction of the valve is shown. A still image of this 3D reconstruction is shown in Fig. 5b. (MOV 3542 kb)
Additional file 7: Figure S2.
Color and pulsed Doppler ultrasound revealed valve regurgitation and atrial communication in Mbnl1 E∆3/∆E3 mice. (A) Sequential color Doppler ultrasound images show regurgitation across the aortic valve of an Mbnl1 ∆E3/∆E3 mouse. Arrows indicate the direction of blood flow. LV = left ventricle, Ao = aorta, AV = aortic valve. Pulsed Doppler ultrasound revealed regurgitation across Mbnl1 ∆E3/∆E3 (B) pulmonary, (C) aortic, and (D) mitral valves. Arrows indicate regurgitation peaks. Atrial communication was observed in some Mbnl1 ∆E3/∆E3 mice by (E) color and (F) pulsed wave Doppler ultrasound. RA = right atrium, LA = left atrium, RV = right ventricle, LV = left ventricle. (PDF 8654 kb)
Additional file 8: Figure S3.
Scoring system for heart valve pigmentation. To evaluate the extent of pigmentation of the valves, a scoring system was developed in which each valve was given a score of 0 (no melanin), 1 (sparse melanin), 2 (moderate melanin), or 3 (abundant melanin). Representative inflow and outflow valves for each category are shown. (PDF 3893 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Coram, R.J., Stillwagon, S.J., Guggilam, A. et al. Muscleblind-like 1 is required for normal heart valve development in vivo . BMC Dev Biol 15, 36 (2015). https://doi.org/10.1186/s12861-015-0087-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12861-015-0087-4