
Abstract
Background
Drug repositioning refers to the identification of new indications for existing drugs. Drug-based inference methods for drug repositioning apply some unique features of drugs for new indication prediction. Complementary information is provided by these different features. It is therefore necessary to integrate these features for more accurate in silico drug repositioning.
Results
In this study, we collect 3 different types of drug features (i.e., chemical, genomic and pharmacological spaces) from public databases. Similarities between drugs are separately calculated based on each of the features. We further develop a fusion method to combine the 3 similarity measurements. We test the inference abilities of the 4 similarity datasets in drug repositioning under the guilt-by-association principle. Leave-one-out cross-validations show the integrated similarity measurement IntegratedSim receives the best prediction performance, with the highest AUC value of 0.8451 and the highest AUPR value of 0.2201. Case studies demonstrate IntegratedSim produces the largest numbers of confirmed predictions in most cases. Moreover, we compare our integration method with 3 other similarity-fusion methods using the datasets in our study. Cross-validation results suggest our method improves the prediction accuracy in terms of AUC and AUPR values.
Conclusions
Our study suggests that the 3 drug features used in our manuscript are valuable information for drug repositioning. The comparative results indicate that integration of the 3 drug features would improve drug-disease association prediction. Our study provides a strategy for the fusion of different drug features for in silico drug repositioning.
Similar content being viewed by others
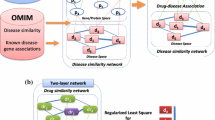

Background
Despite continuous advances in modern technologies, the process of traditional drug discovery is still extremely time-consuming and costly. According to a recent study [1], it takes over 10 years and more than $2 billion to bring a new drug to market. Moreover, the risk of failure during drug discovery is significantly high. Most drug leads could not pass beyond the early stage of development because of toxicity, and lack of efficacy or adverse side-effects could further prevent testing drugs from entering clinical trials. Therefore, improving research and development (R&D) productivity becomes the most important priority for the global pharmaceutical industry [2].
Drug repositioning [3], which aims to find new indications for approved or investigational drugs, has emerged as an important alternative to the traditional drug discovery. As it uses de-risked drug compounds, drug repositioning has the potential to reduce development time and increase success ratio compared to developing an entirely new drug for disease treatment [4]. Some successful examples of drug repositioning have been reported. A well-known instance is sildenafil, which has been repurposed from an antihypertensive drug to the treatment of erectile dysfunction. Existing antivirals, such as baloxavir, azvudine and darunavir, are repurposed to fight the current COVID-19 pandemic [5].
With the accumulation of biomedical data, computational approaches exploiting multi-source information for drug repositioning have been continuously proposed [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27]. These methods can be roughly categorized as drug-based and disease-based (see Review [28] for more details). Drug-based approaches are preferred when rich chemical or pharmacological data for drugs are available. For example, under the principle that drugs with chemical similarities could suggest shared biological activity, Keiser et al. [7] applied a similarity ensemble approach (SEA) to evaluate the 2D structural similarity of drugs to identify new drug–target interactions for drug repositioning. Based on the hypothesis that the mechanism of actions (MoA) of two drugs would be same if they induced the same side effects, Yang and Agarwal [8] used clinical side-effects of drugs as features to build Naive Bayes models to predict indications for diseases. Because protease is a common target for SARS-CoV-2, HIV-1 and hepatitis C viral (HCV) strains. FDA approved HIV-1 protease inhibitors and HCV protease inhibitors have been screened to be potential effective drugs against the COVID-19 [27]. Considering the fact that a drug usually acts on multiple targets, Rutherford et al. [14] extracted drug-disease associations for drug repositioning using the interactions between disease-related genes and drug targets. For these methods, different drug features are applied to address the drug repositioning problem from different angles.
Generally, these drug-based approaches compare some unique signature of a drug against that of another one. The signature of a drug could be mainly derived from three categories of data: chemical structures, genomic data and adverse event profiles. As we know, collection bias and noise may exist in these data and some are even not complete. Meanwhile, complementary information exists in these different types of data. Therefore, it is necessary to combine these data for a comprehensive understanding of drug’s MoA. However, integrating these different kinds of data to improve in silico drug repositioning is an open question till now.
In this paper, we first collect 3 types of drug data (i.e., drug substructures, drug targets and drug side-effects) from public databases. Drug–drug similarities are then calculated based on each of the three types of features. A method using propagation to integrate the three similarity measurements is proposed. Under the guilt-by-association principle, we finally test their ability to infer drug-disease associations for drug repositioning. Experimental results based on cross-validations and case studies show that the integrated similarity measurement outperforms each of the 3 similarity measurements. We also compare our fusion method with 3 state-of-the-art similarity-integration methods and our method shows superior prediction performance in drug repositioning.
Results
Evaluation metrics
In order to evaluate the prediction performance of the 4 similarity measurements, we implement leave-one-out cross-validations (LOOCVs) on the 548 drugs. For each drug, we consider it as a new one and leave it out once as the testing data. We remove all the associated diseases of the testing drug from our dataset. The remaining 547 drugs with indication information and similarity measurement are taken as the training data.
For each drug, we prioritize the whole candidate diseases according to the scores derived from Eq. (8) (see “Methods”). When the score of a predicted association exceeds a given threshold, we consider it as a positive prediction; otherwise, a negative prediction. True positive rate (TPR), false positive rate (FPR), Precision (P) and Recall (R) are calculated by varying the thresholds to plot ROC and PR curves. Area under ROC curve (AUC) values and area under precision-recall curve (AUPR) values are computed for performance comparison.
Furthermore, comprehensive drug-disease association predictions using all known information as training set are conducted. We analyse the top-ranked results for the 548 drugs by searching evidence from public databases.
Prediction performance comparison
We report in Table 1 the average AUC values and AUPR values received by LOOCVs on the 548 drugs from the 4 similarity measurements. As shown in Table 1, IntegratedSim receives the highest average values of AUC and AUPR and performs best in the 4 similarity measurements. The average AUC value for IntegratedSim increases by 0.0659, 0.0310 and 0.0536 than these for the other 3 measurements, respectively. Meanwhile, the average AUPR value for IntegratedSim is 0.1474, 0.0586 and 0.1289 higher compared with these for the other 3 measurements, respectively. The overall results of LOOCVs for all 4 similarity measurements are illustrated by ROC curves and PR curves in Figs. 1 and 2, respectively.

ROC curves of the 4 similarity measurements to predict drug-disease associations by leave-one-out cross-validation tests

PR curves of the 4 similarity measurements to predict drug-disease associations by leave-one-out cross-validation tests
We conduct paired t-tests to measure whether the AUC values and AUPR values obtained by IntegratedSim across the 548 drugs are significantly higher than these in the other 3 datasets. The calculated p-values are available at Table 2. We can discover from the statistical results that IntegratedSim achieves significantly better performance than all the other 3 measurements at the significance level 0.05.
We show the precision and recall values across the 548 drugs in the 4 similarity datasets within the top k (k = 5, 10, 15 and 20) candidates in Figs. 3 and 4, respectively. Because higher values of precision and recall within the top k predictions indicate that more real drug indications are successfully inferred. We can conclude from the two figures that IntegratedSim consistently outperforms the other 3 measurements at different k cutoffs.

Comparison of average precision values in the top-k predictions for the 548 drugs in the 4 similarity datasets by leave-one-out cross-validations

Comparison of average recall values in the top-k predictions for the 548 drugs in the 4 similarity datasets by leave-one-out cross-validations
Effects of parameters k and t in similarity fusion on drug repositioning
There are two parameters k and t in our method for similarity fusion. The parameter k is the number of neighbours and t is the number of iterations. We comprehensively set their values in the range of [1, 30] and list the average AUC values and AUPR values in Tables 3 and 4, respectively. We find from the 2 tables that the best inference performance can be achieved when the values of both parameters are set to be 5.
Comprehensive prediction of novel drug–disease associations
After extensive comparison, we choose the best-performed similarity measurement IntegratedSim to conduct comprehensive drug-disease association predictions. In this inference process, all known information including associations and similarity measurement are used as the training set. We rank the unknown pairs according to their scores derived from Eq. (8). The list of the top 20 predicted results can be seen in Additional file 1.
We check the top 20 predicted results according to the public database CTD [29], a knowledgebase that contains information for chemicals, genes, phenotypes, diseases, and exposures to advance our understanding about human health. Literature-based drug-disease associations are downloaded from this database to validate our predictions. For the predicted results in IntegratedSim, we discover that 158, 612, 1006 and 1575 predictions from the top 1, top 5, top 10 and top 20 results for the 548 drugs are verified in CTD, respectively. We also predict new drug-disease associations using the other 3 similarity measurements. Comparison of numbers of confirmed associations in the top k (k = 1, 5, 10 and 20) predictions is showed in Fig. 5. We receive the largest numbers of confirmed predictions from IntegratedSim in most cases. It should be noted that the top predictions that are not supported in CTD yet may also exist in reality.

The numbers of confirmed results in the top-k predictions in the 4 similarity datasets
Comparison with other similarity fusion methods
We compare our integration method with 3 latest similarity fusion methods. We refer to the 3 methods as Napolitano’s method [30], Oerton’s method [31] and Li’s method [32]. To make fair comparison, we apply the 3 fusion methods to our datasets for drug-disease association prediction. We also use leave-one-out cross validations to test their prediction abilities. The average AUC and AUPR values of these methods are listed in Table 5. We discover that our method performs best in the 4 fusion methods.
Discussion
Drug-based inference methods for drug repositioning make use of some unique drug features for matching. However, such information may be incomplete or contain noise. The incomplete or noisy data would produce biased results for drug repositioning.
We develop a method to combine 3 different drug features. We ensure in our integration method that a drug is more similar to itself than to other drugs throughout iterations, which results in more reliable drug-disease association predictions.
Note that the information of target proteins used in our manuscript is not complete. Meanwhile, according to a review [33], non-coding RNAs (ncRNAs) would be another new class of drug targets as they play significant roles in gene expression regulation and in disease progression. Integrating these ncRNAs with target proteins would make us know better about drug’s MoA. We therefore expect that the performance of our method would be improved when more experimentally supported drug targets are integrated.
In addition, our method could be easily extended when more drug features are available. This is useful because diverse categories of biomedical data are becoming available with recent advances in technologies. These biomedical data offer new potential for drug repositioning [34,35,36,37].
It should be noted that the performance of our similarity integration method depends on suitable parameter setting. Choosing proper parameters under different conditions for our method is a problem that needs to be properly addressed. Meanwhile, we only study the effects of drug features on drug repositioning. Recent repurposing approaches [38,39,40,41] are making using of both drug and disease data. Our previous study [42] demonstrated that the topology of drug-disease bipartite network is also a vital factor in predicting new indications for drugs. In the future, we plan to integrate more information to improve the prediction ability.
Conclusions
In this paper, we comprehensively study the effects of 3 drug features from chemical, genomic and pharmacological spaces on drug repositioning. Cross-validations and case studies suggest the 3 drug features are all predictive factors for drug repositioning. We further develop a fusion method to integrate these features for better in silico drug repositioning. Compared with 3 latest state-of-the-art methods, our fusion method shows improvements in prediction accuracy. We expect that our study will provide guidance in data integration for in silico drug repositioning.
Methods
Data preparation
In our manuscript, we collect and integrate 3 types of drug signatures for drug repositioning. The datasets used for performance evaluation and new drug indication prediction are downloaded from two references [43, 44].
In reference [43], Zhang et al. collected chemical structures of 1103 drugs from PubChem [45]. They used 881-dimensional binary fingerprint profiles to encode the presence or absence of substructures. Target proteins of 1007 drugs were obtained from DrugBank [46]. Each drug was represented by a 775-dimensional binary target profile. Side-effects of 888 drugs were received from SIDER [47]. They used 1385-dimensional binary profiles to encode the presence or absence of each side-effect keyword.
In reference [44], Li and Lu extracted therapeutic uses for 799 drugs from NDF-RT (http://www.nlm.nih.gov/research/umls/sourcereleasedocs/current/NDFRT/) and provided 3250 drug-disease relationships between the 799 drugs and 719 diseases. Finally, we receive 548 drugs which contain all information of chemical structures, target proteins, side-effects and indications.
Similarity calculation and fusion
As there are three types of drug features (chemical structures, target proteins and side-effects) in our study and these features are represented by binary profiles, we separately calculate the similarity between drugs in each feature set according to the Jaccard score. This strategy of similarity calculation is also applied in reference [48], in which the similarity score between two drugs based on the feature of chemical structures is computed as the size of the intersection over the union when viewing each chemical structure as specifying a set of elements. We refer to the 3 similarity datasets as chemSim, genoSim and pharSim.
Inspired by the successful work of reference [49] in shape/image retrieval and reference [50] in cancer subtype identification, we apply a diffusion method as follows to combine the 3 calculated similarity measurements. We refer to this integrated similarity as IntegratedSim.
For generality, we use an \(n \times n\) similarity matrix \(W\) with \(W(i,j)\) indicating the similarity between drug \(x_{i}\) and drug \(x_{j}\). We define a full and sparse kernel on the similarity matrix \(W\) and the full kernel is normalized as:
Let \(N_{i}\) represent a set of drug \(x_{i}\)’s neighbours. We use K nearest neighbours (KNN) to measure local affinity as:
Suppose there are 2 similarity datasets for fusion. We compute \(P^{(1)}\) and \(P^{(2)}\) according to Eq. (1) for the two similarity matrices; then the matrices \(S^{(1)}\) and \(S^{(2)}\) are calculated as in Eq. (2). Let \(P_{t = 0}^{(1)} = P^{(1)}\) and \(P_{t = 0}^{(2)} = P^{(2)}\) denote the initial two status matrices when t = 0. We propagate the similarity information through the common neighbourhood and update the two similarity matrices iteratively as follows:
After t steps, the final integrated similarity matrix is computed as
For the 3 similarity measurements in our study, we adjust Eq. (3) to
The final fused similarity matrix is calculated as
Drug-disease association prediction
Based on the guilt-by-association principle, we assume if a drug is prescribed to treat a disease, similar drugs might also be able to cure the disease (see Fig. 6). The same idea for association analysis has been used in some other bioinformatics fields [51,52,53].

The guilt-by-association principle behind our in silico drug repositioning. If a drug with unknown indication profile shares a similar property with another drug whose indication profile is known, the former may share the same indication profile with the latter
For an unknown drug-disease association (ri, dj), we calculate its inference score as,
where ri and dj denote drug i and disease j, \(Sim(r_{i} ,r_{l} )\) is the similarity value between drugs i and l, and \(a_{lj}\) = 1if there exists an association between drug l and disease j, otherwise \(a_{lj}\) = 0. The higher a score is received from Eq. (8), the higher with confidence a prediction is. The top predicted diseases are considered as new indications for drugs of interest.
Availability of data and materials
The datasets used and/or analysed during the current study are available from public databases. The links for these databases are as follows.
PubChem: https://pubchem.ncbi.nlm.nih.gov/
DrugBank: https://go.drugbank.com/
SIDER: http://sideeffects.embl.de/
CTD: http://ctdbase.org/
Abbreviations
- R&D:
-
Research and development
- SEA:
-
Similarity ensemble approach
- MoA:
-
Mechanism of actions
- KNN:
-
K nearest neighbours
- ncRNAs:
-
non-coding RNAs
References
Wouters OJ, McKee M, Luyten J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA. 2020;323(9):844–53.
Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug DISCOV. 2010;9(3):203–14.
Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3(8):673–83.
Pushpakom S, Iorio F, Eyers PA, Escott KJ, Hopper S, Wells A, Doig A, Guilliams T, Latimer J, McNamee C, et al. Drug repurposing: progress, challenges and recommendations. Nat REV Drug Discov. 2019;18(1):41–58.
Harrison C. Coronavirus puts drug repurposing on the fast track. Nat Biotechnol. 2020;38:379–81.
Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J, Subramanian A, Ross KN, et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–35.
Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, et al. Predicting new molecular targets for known drugs. Nature. 2009;462(7270):175–81.
Yang L, Agarwal P. Systematic drug repositioning based on clinical side-effects. PLoS ONE. 2011;6(12):e28025.
Rajendran V. Structural analysis of oncogenic mutation of isocitrate dehydrogenase 1. Mol Biosyst. 2016;12(7):2276–87.
Huang C, Ciou J, Chen S, Kok VC, Chung Y, Tsai JJP, Kurubanjerdjit N, Huang CF, Ng K. Identify potential drugs for cardiovascular diseases caused by stress-induced genes in vascular smooth muscle cells. PeerJ (San Francisco, CA). 2016;4:e2478.
Rajendran V, Gopalakrishnan C, Sethumadhavan R. Pathological role of a point mutation (T315I) in BCR-ABL1 protein-A computational insight. J Cell Biochem. 2017;119(1):918–25.
Chtita S, Belhassan A, Aouidate A, Belaidi S, Bouachrine M, Lakhlifi T. Discovery of potent SARS-CoV-2 inhibitors from approved antiviral drugs via docking and virtual screening. Comb Chem High Throughput Screen. 2020;23:1.
Bhardwaj VK, Singh R, Das P, Purohit R. Evaluation of acridinedione analogs as potential SARS-CoV-2 main protease inhibitors and their comparison with repurposed anti-viral drugs. Comput Biol Med. 2021;128:104117.
Rutherford KD, Mazandu GK, Mulder NJ. A systems-level analysis of drug–target–disease associations for drug repositioning. Brief Funct Genomics. 2018;17(1):34–41.
Singh R, Bhardwaj VK, Sharma J, Das P, Purohit R. Discovery and in silico evaluation of aminoarylbenzosuberene molecules as novel checkpoint kinase 1 inhibitor determinants. Genomics. 2020;113:707–15.
Wicks P, Vaughan TE, Massagli MP, Heywood J. Accelerated clinical discovery using self-reported patient data collected online and a patient-matching algorithm. Nat Biotechnol. 2011;29(5):411–4.
Singh R, Bhardwaj V, Das P, Purohit R. Natural analogues inhibiting selective cyclin-dependent kinase protein isoforms: a computational perspective. J Biomol Struct Dyn. 2020;38(17):5126–35.
Paik H, Chung A, Park H, Park RW, Suk K, Kim J, Kim H, Lee K, Butte AJ. Repurpose terbutaline sulfate for amyotrophic lateral sclerosis using electronic medical records. Sci Rep UK. 2015;5(1):8580.
Bhardwaj VK, Purohit R. A new insight into protein–protein interactions and the effect of conformational alterations in PCNA. Int J Biol Macromol. 2020;148:999–1009.
Singh R, Bhardwaj VK, Sharma J, Purohit R. Identification of novel and selective agonists for ABA receptor PYL3. Plant Physiol Biochem. 2020;154:387–95.
Bhardwaj VK, Purohit R. Targeting the protein-protein interface pocket of Aurora-A-TPX2 complex: rational drug design and validation. J Biomol Struct Dyn. 2020. https://doi.org/10.1080/07391102.2020.1772109.
Wang Z, Zhang H. Rational drug repositioning by medical genetics. Nat Biotechnol. 2013;31:1080–2.
Chen H, Zhang Z. A semi-supervised method for drug–target interaction prediction with consistency in networks. PLoS ONE. 2013;8(5):e62975.
KumarBhardwaj V, Singh R, Sharma J, Das P, Purohit R. Structural based study to identify new potential inhibitors for dual specificity tyrosine-phosphorylation-regulated kinase. Comput Methods Programs Biomed. 2020;194:105494.
Dakshanamurthy S, Issa NT, Assefnia S, Seshasayee A, Peters OJ, Madhavan S, Uren A, Brown ML, Byers SW. Predicting new indications for approved drugs using a proteochemometric method. J Med Chem. 2012;55(15):6832–48.
Bhardwaj VK, Singh R, Sharma J, Rajendran V, Purohit R, Kumar S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J Biomol Struct Dyn. 2020. https://doi.org/10.1080/07391102.2020.1766572
Nandi S, Kumar M, Saxena M, Saxena AK. The antiviral and antimalarial drug repurposing in quest of chemotherapeutics to combat COVID-19 utilizing structure-based molecular docking. Comb Chem High Throughput Screen. 2020. https://doi.org/10.2174/1386207323999200824115536.
Dudley JT, Deshpande T, Butte AJ. Exploiting drug-disease relationships for computational drug repositioning. Brief Bioinform. 2011;12(4):303–11.
Davis AP, Grondin CJ, Johnson RJ, Sciaky D, Wiegers J, Wiegers TC, Mattingly CJ. Comparative toxicogenomics database (CTD): update 2021. Nucleic Acids Res. 2021;49:D1138–D1143.
Napolitano F, Zhao Y, Moreira VM, Tagliaferri R, Kere J, D’Amato M, Greco D. Drug repositioning: a machine-learning approach through data integration. J Cheminform. 2013;5(1):30.
Oerton E, Roberts I, Lewis PSH, Guilliams T, Bender A. Understanding and predicting disease relationships through similarity fusion. Bioinformatics. 2019;35(7):1213–20.
Li J, Zhao Y, Zhou S, Zhou Y, Lang L. Inferring lncRNA functional similarity based on integrating heterogeneous network data. Front Bioeng Biotechnol. 2020;8:27.
Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12(11):847–65.
Yu L, Huang J, Ma Z, Zhang J, Zou Y, Gao L. Inferring drug-disease associations based on known protein complexes. BMC Med Genomics. 2015;8(S2):S2.
Chen H, Zhang Z. A miRNA-driven inference model to construct potential drug-disease associations for drug repositioning. Biomed Res Int. 2015;2015:1–9.
Wang Z, Monteiro CD, Jagodnik KM, Fernandez NF, Gundersen GW, Rouillard AD, Jenkins SL, Feldmann AS, Hu KS, McDermott MG, et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat Commun. 2016;7(1):12846.
Zeng X, Zhu S, Liu X, Zhou Y, Nussinov R, Cheng F. deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics. 2019;35(24):5191–8.
Luo H, Li M, Wang S, Liu Q, Li Y, Wang J. Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics. 2018;34(11):1904–12.
Zhang W, Yue X, Lin W, Wu W, Liu R, Huang F, Liu F. Predicting drug-disease associations by using similarity constrained matrix factorization. BMC Bioinform. 2018;19(1):233.
Yang M, Luo H, Li Y, Wang J. Drug repositioning based on bounded nuclear norm regularization. Bioinformatics. 2019;35(14):i455–63.
Chen H, Cheng F, Li J. iDrug: Integration of drug repositioning and drug-target prediction via cross-network embedding. PLoS Comput Biol. 2020;16(7):e1008040.
Chen H, Zhang H, Zhang Z, Cao Y, Tang W. Network-based inference methods for drug repositioning. Comput Math Methods Med. 2015;2015:1–7.
Zhang P, Wang F, Hu J, Sorrentino R. Exploring the relationship between drug side-effects and therapeutic indications. In: American medical informatics association annual symposium proceedings; 2013. p. 1568–1577.
Li J, Lu Z. A new method for computational drug repositioning using drug pairwise similarity. In: IEEE international conference on bioinformatics and biomedicine: 2012-01-01 2012. United States: IEEE; 2012. p. 1–4.
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47(D1):D1102–9.
Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–82.
Kuhn M, Letunic I, Jensen LJ, Bork P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016;44(D1):D1075–9.
Gottlieb A, Stein GY, Ruppin E, Sharan R. PREDICT: a method for inferring novel drug indications with application to personalized medicine. Mol Syst Biol. 2011;7(1):496.
Wang B, Jiang J, Wang W, Zhou Z, Tu Z. Unsupervised metric fusion by cross diffusion. In: 2012 IEEE conference on computer vision and pattern recognition (CVPR) 2012-01-01 2012. IEEE; 2012. p. 2997–3004.
Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M, Haibe-Kains B, Goldenberg A. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11(3):333–7.
Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4(2):293–305.
Chen H, Guo R, Li G, Zhang W, Zhang Z. Comparative analysis of similarity measurements in miRNAs with applications to miRNA-disease association predictions. BMC Bioinform. 2020;21(1):176.
Gloeckner CJ, Porras P. Guilt-by-association—functional insights gained from studying the LRRK2 interactome. Front Neurosci Switz. 2020;14:485.
Acknowledgements
We are grateful to Dr. Mengyun Yang at Central South University for useful discussions.
Funding
This work was supported by the National Natural Science Foundation of China under Grant number 61862026.
Author information
Authors and Affiliations
Contributions
HC and ZZ performed data preparation. HC, ZZ and JZ conceived and designed the experiments. HC performed all computational experiments. HC and JZ analyzed the results. HC and ZZ wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The top 20 predicted indications for the 548 drugs based on the similarity measurement IntegratedSim.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, H., Zhang, Z. & Zhang, J. In silico drug repositioning based on the integration of chemical, genomic and pharmacological spaces. BMC Bioinformatics 22, 52 (2021). https://doi.org/10.1186/s12859-021-03988-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-021-03988-x