
Abstract
Background
Recent advances in genome sequencing technologies and the cost drop in high-throughput sequencing continue to give rise to a deluge of data available for downstream analyses. Among others, evolutionary biologists often make use of genomic data to uncover phenotypic diversity and adaptive evolution in protein-coding genes. Therefore, multiple sequence alignments (MSA) and phylogenetic trees (PT) need to be estimated with optimal results. However, the preparation of an initial dataset of multiple sequence file(s) (MSF) and the steps involved can be challenging when considering extensive amount of data. Thus, it becomes necessary the development of a tool that removes the potential source of error and automates the time-consuming steps of a typical workflow with high-throughput and optimal MSA and PT estimations.
Results
We introduce LMAP_S (Lightweight Multigene Alignment and Phylogeny eStimation), a user-friendly command-line and interactive package, designed to handle an improved alignment and phylogeny estimation workflow: MSF preparation, MSA estimation, outlier detection, refinement, consensus, phylogeny estimation, comparison and editing, among which file and directory organization, execution, manipulation of information are automated, with minimal manual user intervention. LMAP_S was developed for the workstation multi-core environment and provides a unique advantage for processing multiple datasets. Our software, proved to be efficient throughout the workflow, including, the (unlimited) handling of more than 20 datasets.
Conclusions
We have developed a simple and versatile LMAP_S package enabling researchers to effectively estimate multiple datasets MSAs and PTs in a high-throughput fashion. LMAP_S integrates more than 25 software providing overall more than 65 algorithm choices distributed in five stages. At minimum, one FASTA file is required within a single input directory. To our knowledge, no other software combines MSA and phylogeny estimation with as many alternatives and provides means to find optimal MSAs and phylogenies. Moreover, we used a case study comparing methodologies that highlighted the usefulness of our software. LMAP_S has been developed as an open-source package, allowing its integration into more complex open-source bioinformatics pipelines. LMAP_S package is released under GPLv3 license and is freely available at https://lmap-s.sourceforge.io/.
Similar content being viewed by others


Background
Recent advances in genome sequencing technologies and the cost drop in high-throughput sequencing, allowed a new era of genome science, widening the amount of data available for downstream analyses [1, 2]. As the genomes become completely sequenced and assembled, they are subsequently released to public databases, such as Ensembl [3] and/or NCBI Genbank [4]. This allows other researchers to easily build datasets to their own object of study [5]. Evolutionary biologists often make use of such (nucleotide) data to uncover phenotypic diversity and adaptive evolution in protein-coding genes [6,7,8,9,10]. However, to perform such studies, multiple sequence alignments (MSA) and phylogenetic trees (PT) need to be estimated. In fact, the MSA is of central importance in molecular biology, many bioinformatics analyses and other areas of study, such as comparative sequence analyses, functional motif or domain characterization, prediction of secondary or tertiary structures, sequence-based structural alignment (e.g., [11, 12]), detection of key functional residues and homology searches [13,14,15,16]. With such importance, MSAs raise relevant questions concerning their accuracy [14, 15, 17, 18] or uncertainty [19,20,21], which can negatively influence downstream analyses, starting with phylogeny estimations [16, 18, 22, 23].
To date several software has been developed (with different objectives and approaches [15, 16, 24]) to improve MSA estimation (e.g., MULTAN [25], MUSCLE [26], PRANK [27], MO-SAStrE [28]) and refinement (e.g., Gblocks [29], GUIDANCE [14], TrimAl [30], MaxAlign [31]). Despite the efforts for reaching optimal solutions, uncertainty and confidence in the result persists, with co-optimal solutions not being the “true” alignment and/or the “true” alignment possibly being suboptimal [14].
Beyond adaptive evolution analyses, PTs are also of great importance for various biological research, for instance, the inference of trait evolution, protein structure and function [32] or in other phylogenomics areas, e.g. gene family evolution [9, 10]. Likewise, they suffer from identical uncertainty [13, 19, 33] issues (additionally aggravated by the MSA issues [13,14,15,16,17, 19, 20, 23, 34]). This has taken to the development of several improvements in algorithms and heuristics leading to alternatives, such as PAUP [35], PHYLIP [36], PhyML [37], RaxML [38], FastTree [39], or MrBayes [40].
In fact, the investment in improving and developing novel MSA and/or phylogeny estimators, has reached a level where it becomes difficult to the researcher to select the appropriate MSA [24] and/or phylogeny software. For the interested reader, during our literature survey, we have encountered more than 30 MSA software published solely respecting DNA multiple alignment (a few of them also enabling other data types); beyond other cases like amino acid and RNA, exceeding in total 80 software [24]. Many of them are presently unavailable or discontinued.
Considering the large amount of data (genomes) currently and in the future available, with the evolutionary biologist requiring the analyses of datasets with multiple genes (including cases of large gene families). Additionally, considering the preparation of an initial multiple sequence file(s) (MSF) dataset and the several steps involved to achieve the result of optimal MSAs and PTs, it becomes necessary the development of a tool that automates the time-consuming steps and accelerates estimations. For a single gene, the steps involved typically include, (i) the preparation of the initial protein-coding gene sequences MSF (e.g., EASER [5]), (ii) MSA estimation, (iii) MSA refinement, (iv) MSA substitution saturation detection (e.g., DAMBE [41, 42]), (v) detection of data fitting evolutionary model (e.g., JModelTest [43, 44], MrAIC [45]), (vi) phylogeny estimation (using the detected best model) and any (vii) phylogenetic tree posterior editing.
Several bioinformatics tools have been developed that consider MSA estimation and/or phylogeny estimation. They can be organized in two categories: (i) Command-line Interfaces (CLI), such as M-coffee [24], SATé [46], POTION [47], ETE [48] and, (ii) Graphical user interfaces (GUI), such as DAMBE [42], StatAlign [34], Bosque [49], PALM [50], Seaview [51], Armadillo [52]. Still, to our knowledge and from the available literature, none of them covers the aforementioned steps in an automated fashion, with the purpose of (i) enabling MSA and phylogeny high-throughput estimations, (ii) providing optimal estimation strategies, and (iii) including the additional characteristic of generating reproducible experiments [53, 54].
Here we present LMAP_S (Lightweight Multigene Alignment and Phylogeny eStimation), a high-throughput, versatile and user-friendly software package developed in Perl [55], built on top of our recent LMAP [7] package platform. LMAP_S was designed to handle in seven stages: (i) the input nucleotide (MSF) data pre-processing (NDP); (ii) the MSA estimation (AE); (iii) the MSA outlier detection (AOD); (iv) the MSA refinement and consensus (ARC); (v) the phylogeny estimation (PE); (vi) the phylogeny comparison and consensus (PCC) and (vii) phylogeny data post-processing (PDP). LMAP_S package consists of a single application, lmap-s.pl, which executes the aforementioned stages in a systematic fashion and depending on the user requirements. LMAP_S conveniently requires input nucleotide datasets, thus enabling (among others) further downstream evolutionary analyses [13]. With these objectives in mind, LMAP_S integrates various software covering all stages, except in (i) and (vii).
To enable trial and testing, we adapt the example dataset from LMAP [7] to the current case, consisting of the mitochondrial DNA of 20 freshwater and terrestrial turtles and provide it in LMAP_S archive. Additionally, this is complemented with a case study on mitochondrial genes from a previously studied Cephalopoda dataset [56].
In the following sections, we present LMAP_S development and scheduling of tasks executions; integrated software in relation with stage organization and file identification; phylogeny estimation and the criteria for evolutionary model detection and alternative approach to typical substitution saturation detection; and lastly, the PCC method. Next, we present the functioning of LMAP_S, discuss integrated software options, the PCC method and potential future developments. Finally, we introduce (i) the example dataset used to perform the benchmarking tests; and (ii) the case study, explored to demonstrate the usefulness of LMAP_S.
Implementation
LMAP_S development
LMAP_S package was implemented in Perl [55] and has been tested in Linux/UNIX. It consists of one command-line/interactive application, lmap-s.pl. Additionally, seven specific LMAP_S library modules (MyUtil.pm, MyISWU.pm, MyNotify.pm, MyPhyloInfo.pm, MyPPMSF.pm, MyMMAP.pm and MyPhylo.pm) were developed to support its functionality.
LMAP_S requires the Comprehensive Perl Archive Network (CPAN) [57] modules in four cases: (i) in MyPhylo.pm, for parsing and editing of Newick tree files (BioPerl [58] module); (ii) in MyMMAP.pm, for interactive monitoring of parallel executions (for which is required the UNIX screen [59] utility program); (iii) in MyNotify.pm, for email notifications (for which is required the UNIX sendmail [60] utility program); and (iv) in all, for handling files and directories.
The MyMMAP.pm module was adapted and improved from the mmap.pl application of LMAP [7] to allow the parallel execution of the diversity of software here integrated and to cope with the several stages of LMAP_S execution. Its functioning was largely maintained and is re-described in Additional file 1: Section 1. Other modules adapted from LMAP package are the MyUtil.pm, MyPhylo.pm and the MyNotify.pm.
LMAP_S includes an additional application, RYcode.pl, to enable the RY-coding of MSAs (see following section). Beyond being part of LMAP_S, it was also designed to enable independent operation from our package.
LMAP_S integrated software, stage organization and file identification
In this section, we list LMAP_S integrated software, to show how they are organized into stages and subsequently how in relation file identification was designed.
With exception of the first (NDP) and last (PDP) stages, Table 1, lists the integrated software for remaining stages.
This software is the result of criteria, whose main goal was to ensure they could be properly integrated and function correctly in the necessary conditions. Three examples of such “pipeline-friendly” criteria are: (i) command-line options enabling automation, (ii) facilitated accessibility to additional input dependencies, and (iii) program termination.
All the stages provide options that enable algorithm selection, except AOD and PCC (Additional file 2 and 3). The “Default” options besides being frequently preferred [15] were also designed to allow customization by the interested researcher.
File identification has been implemented to help the researcher to recall the algorithms that have manipulated the dataset genes [19] and to further allow any comparisons. This is done in a stage-by-stage fashion by using the algorithms identification (Additional file 3). Hence, the general format for MSA file identification is [GeneName]_[STAGE2AL]_[STAGE4AL].fas and for PT file is [GeneName]_[STAGE2AL]_[STAGE4AL]_[STAGE5AL]_[STAGE7ED].nwk (without brackets). For more information on these topics, please see the LMAP_S Manual.
LMAP_S phylogeny estimation and evolutionary model detection
Here we present how we integrate the data-fitting evolutionary model detection step, required prior to Maximum Likelihood (ML) PT estimation.
Evolutionary model selection involves testing all substitution models available (e.g., Table 1 in [43]) and select the best according to criteria, such as Akaike Information Criterion (AIC) [89] or Bayesian Information Criterion (BIC) [90]. This typically requires the researcher to run a priori software, such as JModelTest [43, 44] or MrAIC [45], which would add further complexity to LMAP_S workflow. However, with recent advances it becomes straightforward to have both evolutionary model detection and consecutive phylogeny estimation in the same software. For this reason, we have included IQ-TREE [80] and SMS [84].
LMAP_S phylogeny estimation and alternative to substitution saturation detection
Following the previous section, here we present the alternative solution for the substitution saturation detection, with its foundation, and the reasoning behind the presented solution.
Substitution saturation is a mutational process that (when present) negatively affects the information contained in molecular sequences of the MSA. This process affects the codon positions and takes to a decrease in phylogenetic information/signal. This phylogenetic signal is thus important for a reliable well-defined phylogeny estimation [41, 91].
Substitution saturation test [41] is a step typically performed to detect saturation in the MSA. This is done to ensure it contains sufficient phylogenetic information/signal before the phylogenetic tree estimation [23, 41, 91]. To perform the mutational saturation test, the DAMBE [42] software is available. However, its integration in LMAP_S bears a few difficulties, for instance, its incompatibility with Linux/UNIX systems. On the other hand, metrics for estimating phylogenetic signal are available and provide valid results [92]. Still, we have not found any suitable software.
To overcome this adversity, we have devised a methodology that gathers (i) the most relevant character coding (CC) methods [93] and (ii) phylogeny comparison methods. Together they help decide under different conditions which phylogeny provides a better resolution and is thus optimal.
Firstly, among the possible character coding methods, those frequently employed consist of plain nucleotide (DNA), 1st and 2nd codon positions only (DNA12), 3rd positions only (DNA3), puRine and pYrimidine coding (RY), degeneracy coding (DEG), codon (CDN) and amino acid (AA) [93]. According to Simmons [93] conclusions and for our methodology, the final selected CC methods are DNA, RY, DEG, AA and CDN, which we have implemented with IQ-TREE algorithms (Table 1). The specific CC methods DEG and RY are accomplished by combining IQ-TREE DNA data type with Degen [85, 86] and RYcode (Table 1 and Additional file 2: Figure S2). For these two cases, MSA coding is performed prior to IQ-TREE execution.
Secondly, we employ phylogeny comparison algorithms (Table 1). They are included to attempt to capture the phylogeny strategy that is consensually inferred to be the optimal among all selected CC cases (see following section). Henceforth, we refer to strategy, as the chain of algorithms applied to a specific gene, since AE Stage (see also section LMAP_S Integrated software, stage organization and file identification). This methodology was inspired and adapted to IQ-TREE [80] following the analyses performed by Simmons [93].
LMAP_S phylogeny comparison and consensus (PCC) method
Here we describe the implemented procedures devised to allow the comparison of several PTs (per gene) both topologically and statistically. Next, we describe how their combination is achieved to identify optimal consensus strategies (c.f. previous section; Additional file 4). This includes a total of six LMAP_S reports.
Phylogeny comparison is accomplished with two distinct approaches. One approach consists in statistical analyses, employing the site-wise log-likelihoods (SWLH) produced by several phylogeny estimators (e.g., PAUP [35], PHYML [37], TREE-PUZZLE [94]). Another approach, consists in the topological analyses, employing methods that target comparison of tree structure, nodes, branches and leafs (e.g., Robinson-Foulds (R-F) [95] and MatchingPair (MP) [88]). For the former case, we have integrated the CONSEL [87] package (which consists of three programs executed in the following order: (1) makermt, (2) consel and (3) catpv). For the latter we have integrated the TreeCmp [88] package. From both approaches three reports are generated (Additional file 5: Tables S3-S5), one from the statistical analyses and two from the topological analyses (including TreeCmp MP and R-F_C methods). To achieve these reports, LMAP_S processes data (SWLHs and PTs) generated by the different IQ-TREE algorithms (PE Stage; Table 1 and Additional file 4). The comparison of multiple SWLHs, requires their agglutination into a single file. Here, due to the discrepancies with resulting SWLHs, a heurist was implemented (Additional file 1: Section 2). This file is then served as input to CONSEL (i.e., to makermt). Likewise, TreeCmp input requires a single file containing all Newick formatted topologies. At this point, TreeCmp is ready for comparison. Hereafter, we describe how the results from both approaches (Additional file 1: Section 3) are combined to achieve the consensus.
An initial consensus report (Additional file 5: Table S6) is formed by using both CONSEL and TreeCmp MP results (Additional file 4). This is accomplished by locating in the MP report both (i) the top ranking statistical strategy and (ii) the corresponding optimal topological support. In detail, when such strategy match is found, the TreeCmp MP matrix row/column is searched for the best topological result (where the MP score is zero). When both criteria are met, the corresponding strategy is doubly marked, otherwise only once. LMAP_S further summarizes these results by successively deriving two additional reports. The fifth report is a condensed matrix discarding the unmarked strategies (Additional file 5: Table S7). Whereas, the last report, shows the total topological score (TTS) associated with each consensus strategy (Additional file 5: Table S8). To calculate the TTS of the consensus strategy, LMAP_S proceeds by counting all related zero values from the MP scores. Hence, the optimal consensus strategy (i.e., optimal underlying chain of algorithms), is one with the maximum TTS. For more details, please see the LMAP_S Manual.
Results
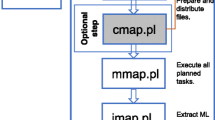
The LMAP_S package consists of a single main application lmap-s.pl that executes the workflow comprising seven stages (Fig. 1). Only AE Stage is mandatory and thus LMAP_S gives the possibility to apply any of the remaining stages, provided the data dependencies are satisfied. Hereafter, LMAP_S functionalities are described in a stage-by-stage fashion.

LMAP_S workflow. Flowchart exhibiting the lmap-s.pl workflow where stages are organized in a sequential fashion. The omission or inclusion of certain Stages helps devise specific workflows based on researcher requirements. Gray boxes reflect optional stages. Stages 3 (AOD) and 6 (PCC) produce reports only, seven in total. Stage 4 (ARC) additionally produces one report. NDP – Nucleotide Data Pre-processing, AE – MSA Estimation, AOD – MSA Outlier Detection, ARC – MSA Refinement and Consensus, PE – PT Estimation, PCC – PT Comparison and Consensus, PDP – PT Data Post-processing
Stage 1 – NDP
This stage provides two modes of functioning, (i) a default mode and (ii) data treatment mode. In the default mode, it is responsible for the creation of the directory structure and placement of MSFs (expected in FASTA format). In the data treatment mode, beyond the default mode operations, the extra functionalities are available with additional arguments (Additional file 2: Figures S1 and S6). Possible data treatments are included for not-ready MSFs and ready MSFs. We consider the not-ready datasets, as the MSF(s) not expected to be grouped in files by gene homology.
Stage 2 – AE
With all the MSFs ready and organized in the directory structure, this stage enables the alignment of every gene by all the algorithms here selected, thus estimating a different MSA version for each gene MSF. This requires the selection among 32 MSA algorithms (Table 1, Additional file 2: Figure S3 and Additional file 3). After completion, an additional procedure ensures that all MSAs have the same taxa order for the following stages.
Stage 3 – AOD
Here, LMAP_S enables the identification and possible removal of divergent sequences in MSAs from the AE Stage (Table 1 and Additional file 3). It enables the generation of the outlier report (Additional file 5: Table S1), containing the results from two software, OD-Seq [73] and EvalMSA [74], gathered for further result complementarity [74]. This stage report or results will not interfere with further stages (Fig. 1).
Stage 4 – ARC
This stage targets the employment of several algorithms for MSA refinement and consensus (Table 1, Additional file 2: Figure S4 and Additional file 3). The refinement algorithms (13 in total) have the purpose to improve each MSA phylogenetic signal by either removing, masking or duplicating MSA eventual ambiguous regions [32]. Whereas, the consensus algorithms (3 in total) enable the combination of several MSAs from different sources. In this case, it is possible to find the best result (TrimAl “compareset” [30]) or a result that gathers the best characteristics among all MSAs [20, 76]. To enable the quick identification of which original MSA was selected for the TrimAl “compareset” option, an additional CSV report is produced (Additional file 5: Table S2).
Stage 5 – PE
Here, a variety of algorithms can be selected among 22 possibilities (Table 1, Additional file 2: Figure S5 and Additional file 3) for the estimation of phylogenies.
This stage currently provides two ML, one Maximum Parsimony (MP), and one Neighbor-Joining (NJ) approaches. The NJ is available with Ninja [83] software, the MP with MPBoot [82], and ML with SMS [84] and IQ-TREE [80]. Any of the phylogeny estimation algorithms can be used together without interfering with each other or with other stages.
Stage 6 – PCC
This stage enables the comparison of phylogenies estimated by (two or more) IQ-TREE algorithms and of optimal consensus strategies (Additional file 4 and Additional file 5: Tables S3-S8). For more details, please see Implementation section LMAP_S Phylogeny comparison and consensus (PCC) method.
Stage 7 – PDP
The last stage applies to the phylogenies resulting from the PE Stage. Similarly, to the NDP Stage data treatment mode, this allows the application of three phylogeny-editing options (Additional file 2: Figure S1). These enable preparations for further downstream analyses.
LMAP_S described stages are subject to monitoring (Fig. 2) of integrated software executions until lmap-s.pl terminates. This is similar to mmap.pl application from LMAP package (see also Fig. 3 in [7]).

LMAP_S interactive functioning. a default or main “Run Status” screen presenting the currently running tasks; by pressing “2”, shows the “Task Status” screen, showing (b) the tasks that will be running next (first ten) and (c) the tasks currently finished (last ten) (press “1” to go back to (a)); (d) when interrupting the execution of lmap-s.pl (by typing “Ctrl-c” or “Ctrl-\”), beyond the choice of quitting, the user has also the choice to proceed to the built-in process manager here presented, allowing the termination of specific tasks. In this case, it is possible to terminate a group of tasks by typing “G:MMAPID” or a single task “P:PROCID”. The identifiers for MMAPID and PROCID are shown in the table, in the respective columns
Discussion
LMAP_S implementation options and general remarks
The integrated software (Table 1) is the result of aforementioned criteria. Even though, we have not integrated a few relevant software in ARC and PE Stages. In the ARC Stage, the most striking is GUIDANCE [14] a widely-used software in which we have found a significant limitation. This software only works with MSAs from PRANK [27], CLUSTALW [62] and MAFFT [68]. Hence, its integration could limit LMAP_S functionality. In terms of phylogeny estimation, we have not integrated software such as RAxML [38], FastTree [39] or MrBayes [40]. We recognize their relevance and wide spread utilization, however, including them would go against the design established (see also section LMAP_S Phylogeny estimation and evolutionary model detection). Here, we require to minimize the number of stages in the workflow, hereby reducing workflow complexity. This can only be accomplished by SMS and IQ-TREE software, which incorporate the automated selection of the evolutionary best-fit models. Otherwise, if we were to integrate this software we would have to additionally integrate, for instance, JModelTest [44] and/or MrAIC [45]. Thus, another stage would be necessary previous to PE Stage. Additionally, both RAxML and FastTree provide limited selection of nucleotide models, which does not enable a well-supported justification for model selection [93]. Although RAxML supplies several alternatives, all are based on the GTR model. We understand that GTR is the most complex and successful model to date being selected for the most cases [96]. However, the analysis may become limited, if a different model is found as best fit. FastTree, additionally provides Jukes-Cantor nucleotide model [39]. Even though, both solutions are quite limited. On the other hand, MrBayes provides several models that can be employed. However, the complexity of automatically managing the commands that need to be specified/provided depends on each researcher and dataset, making it very hard to integrate.
Regarding the PCC Stage method, this software would have to provide SWLH data. RAxML is compatible (but does not support codon analysis [93]); the authors of FastTree provide an additional Perl script to convert into PAUP format (but still does not support DEG and RY-coding [39]), and MrBayes does not provide such information. However, as described by the authors of CONSEL, it only works with the matrices produced with ML methods [87]. Nevertheless, they could still be useful and attractive alternatives to complement the existing ones when only applied for the phylogeny estimations (PE Stage). Thus, considering the CC options and straightforward compatibility with CONSEL [87], IQ-TREE is the only ML integrated software that makes the PCC method possible (see also section LMAP_S Phylogeny estimation and evolutionary model detection).
This method has been implemented to enable inference of reliable and well-defined phylogeny estimations. In the statistical approach, this is ensured by compiling SWLHs for the same (i) gene, (ii) CC method and (iii) possible refinement algorithms. Otherwise, collecting different CC methods or refinement algorithms SWLHs, would deteriorate the conditions for same site-wise lengths. In the topological approach, it is ensured by gathering the several topologies for the same gene. Finally, for each gene, the top statistical results are mapped into the wider range of topological results. Beyond being an alternative to substitution saturation detection (see section LMAP_S Phylogeny estimation and alternative to substitution saturation detection), this procedure additionally uncovers optimal consensus strategies supported by both the statistical and topological agreement (Additional file 5: Tables S6-S8). Specifically, a PT with highest TTS score has an optimal phylogenetic signal, resolution and underlying strategy. Thus, this method hereby addresses a very important topic, not always undertaken by researchers. Its importance stems from the fact that researchers frequently opt for a widely used MSA software or that is found as being better [18]. In fact, the same software is usually applied to all genes (or gene family), thus neglecting the possibility that other software might produce better results with specific genes [18, 24, 97]. Through the PCC method, example dataset and case study, LMAP_S shows that there may be different or better MSA choices for different or specific genes (Fig. 3, Additional file 5: Table S8 and Additional file 6). In fact, the works [18, 24, 97] support the notion that neither the dataset nor the algorithms define the optimal MSA strategy. Furthermore, some authors regard the impact of the phylogeny estimation as an alternative way to evaluating MSA methods [13, 98]. This perspective provided by our software is also partly supported by Beiko et al. [97] in the case of the “shotgun” MSA approach.

Pie charts exhibiting the optimal consensus strategies (with highest TTS). Illustration of the optimal results for the provided dataset derived from LMAP_S consensus histogram report (Additional file 5: Table S8). Each pie chart presents the results for each gene showing at the center the highest TTS value in parenthesis. The arc lines surrounding each pie chart highlights the amount of optimal consensus strategies. The fraction at the bottom right corner of each chart shows the number of consensus strategies with equal highest TTS over the total number of strategies. The three squared cases (genes ATP8, COX2 and ND4L) are the only ones showing a unique consensus strategy with highest TTS. These optimal strategies are “ATP8_MAFFTF2_TRIMALS_DNA_UB”, “COX2_PRANKCDF_MAXALIGN_DNA_UB” and “ND4L_MAFFTEI_MAXALIGN_DNA_UB”, from where it is clearly visible the different optimal algorithms (from AE and ARC Stages). Notably the optimal CC option (DNA) was the same in all three. For the remaining cases, it is possible to take any of the consensus strategies as optimal as long as they have the highest TTS
Our software, makes possible to execute many different analyses and with different extensions. For instance, at minimum it is possible to estimate MSAs (with possible NDP Stage data treatments), and to a maximum extent, it is possible to have phylogenies ready for any downstream analyses (e.g., adaptive evolution). With the several integrated software and options, we foresee LMAP_S can have potential application in several scenarios. Among which we mention, (i) preliminary data study, (ii) finalized data for downstream analyses, (iii) benchmarking purposes and algorithms comparison, (iv) study of optimal strategies for each gene (MSA and PT estimations), (v) large-scale gene and (phylo) genomic analyses, and also to (vi) serve the input for LMAP [7] and/or IMPACT_S [6] packages.
Comparatively, LMAP_S extends the mentioned software in CLI and GUI categories in several ways e.g., high-throughput, MSA refinement, phylogeny comparisons. We found that Bosque [49] and PALM [50], present workflows most similar to our program. Contrarily to LMAP_S, they include less algorithm choices, provide client-server functioning and GUI interfaces, which although being possibly more user-friendly, always depend on external resources availability and may disrupt pipeline integration. Compared to other methods, LMAP_S does not intend to provide consensus PTs or MSAs for each gene. Instead, it is intended to provide high-throughput estimations and by the PCC method infer optimal phylogeny estimation strategies (Fig. 3 and Additional file 6), whereby the underlying chain of algorithms and methods reflected in the phylogenies are also optimal.
Presently, LMAP_S has been developed to gather the most software alternatives around nucleotide data type necessary for evolutionary analyses [13]. LMAP_S does not provide options for the concatenation of genes enabling multi-gene inferences. Still, it can help to estimate the necessary MSAs. We understand the relevance of such process, which we plan to implement soon. Furthermore, we recognize that LMAP_S may lack a stage (close to ARC Stage) where software can be employed to determine highest scoring MSAs for the next stages [17, 99], but it is discussed that the highest scoring MSAs are not necessarily the “true” MSA [14, 16,17,18]. Anyhow, we found that the software available (e.g., FASTSP [33]) often depended on a reference MSA (exception made for MUMSA [17]). To our understanding, the reference MSA is considered as the “true” MSA [13, 99], which for reasons mentioned before, becomes a contradictory possibility. This required feature poses several problems, for instance, when the data at hand (e.g., from newly assembled genomes) does not “readily” enable a priori reference MSA (usually available from the benchmark databases [14, 99, 100]). Hence, how can one determine it (or from a set of alternate nucleotide alignments of the same sequences)?
Additionally, LMAP_S could benefit from the integration of additional algorithms, for instance, in MSA masking (e.g., ZORRO [21], SR [23]) and other phylogeny estimation tools (e.g., MrBayes [40]). LMAP_S is not applicable in Windows OS due to its main dependency on the screen [59] utility program.
Example dataset and benchmarking
An example dataset is provided in LMAP_S archive to help users explore and experience the workflow of the package. Except for the TMConc2 concatenated MSA, here we reuse the dataset explored in LMAP [7]. The folder (“ExampleDataset”) contains two directories, one for ready MSFs and the other for not-ready MSFs. The “Ready” folder contains the sequences organized by gene and the “NotReady” folder contains the sequences organized by genomes as downloaded from NCBI. In both cases, the sequences contain stop codons. To demonstrate the performance of LMAP_S, full command-lines are provided in LMAP_S archive in the “lmap-s.command” file, from where we have executed the “NotReady” one. Its output originated 3264 MSA and 30,392 PT files that took 4 days, 2 h, 45 min and 38 s to complete (5925 min and 38 s). This was measured in the UNIX time [101] utility program, by using a single workstation configured with 64GB of RAM and two Intel Xeon E5-2683v4 processors, which together yield a total of 64 hyper-threading cores. In contrast, using a single core, the same instances would take 14,628,152 s (more than five months). To summarize, our package does not interfere in the execution time required by each software, but instead mitigates how much the researcher spends overseeing each step of the workflow, from the moment the input files are ready to be analyzed, which may be none or minimal.
Case study with Cephalopoda mitochondrial genes
To provide further insight on the usefulness of our software, we have employed a previously published dataset of 13 Cephalopoda mitochondrial genes [56]. As described, all the alignments were performed with MUSCLE [26]. Improvement over phylogenetic signal and resolution, considered the concatenation and RY-coding (3rd codon position). By employing LMAP_S we test two possible outcomes: (i) if the same strategy (MUSCLE and RY-coding) is inferred for all genes and (ii) if the optimal consensus strategies convey topology improvements when compared to the concatenated ML topology from the study.
The applied methods, results and discussion are presented in Additional file 6.
In conclusion, the application of a software with similar characteristics as LMAP_S in this study, would have been highly beneficial. With the differences found among strategies, these results support the application of a more precise chain of algorithms for each gene. Additionally, the fact that the LMAP_S topologies show better average scores, confirms that employing our software can provide more reliable phylogeny estimations and avoid performing gene concatenation and related analyses.
With this and previous example dataset, we show compelling results demonstrating that different strategies should be applied for different genes and that multi-gene concatenation methods are not the most powerful solution [102].
Conclusions
We have developed a simple, versatile and highly customizable package named, Lightweight Multigene/Multi-core Alignment and Phylogeny eStimation (LMAP_S), that readily enables the application of several MSA (33) and PT (22) estimation algorithms. Beyond the central stages, it also enables MSF editing (NDP), AOD (2), and ARC (16) algorithms. With two algorithms, it enables phylogeny statistical and topological comparison and the combination of both to reach consensus in optimal phylogeny and consequently in the underlying MSA algorithms applied from the beginning. Finally, resulting phylogenies can be automatically edited for further downstream analyses. To our knowledge, no other software combines MSA and phylogeny estimation with as many alternatives and provides means to find optimal MSAs and phylogenies. Additionally, we have supplied evidence that LMAP_S is well-supported and useful in methodologies of alignments and phylogenies estimations.
At minimum, one MSF is required with the gene sequences to be analyzed within a single input directory. From this moment, LMAP_S automatically creates, organizes, executes, manipulates and extracts the necessary information from the integrated algorithms results to provide additional information and high-throughput estimations. Furthermore, LMAP_S enables at all times, monitoring and control of software and tasks, and email notification when the job is done. LMAP_S has been developed as an open-source command-line and interactive package, allowing its integration into more complex open-source bioinformatics pipelines.
Availability and requirements
Project Name: LMAP_S.
Project Home Page: https://lmap-s.sourceforge.io/
Operating System: Linux/UNIX.
Programming Language: Perl.
Other Requirements: integrated software from Table 1, CPAN modules (IO::All, Email::MIME, Email::Sender, Sys::Info, Term::Readkey, Thread::Semaphore, Bio::TreeIO, File::Copy, File::Copy::Recursive), screen and sendmail UNIX command-line utilities.
License: GNU General Public License, version 3.0 (GPLv3).
Any restrictions to use by non-academics: no restrictions except the ones stated in GPLv3.
Installation
The LMAP_S package provides two additional applications to facilitate LMAP_S functionality and installation: (i) the install.pl (requires sudo command) to enable the installation of LMAP_S dependencies from Linux repositories, such as CPAN modules, integrated software (Table 1) and UNIX utilities and (ii) the configure.pl to enable the configuration of LMAP_S package (lmap-s.pl and LMAP_S library). A manual with detailed instructions is included in the archive to allow LMAP_S user-friendly installation and application.
Availability of data and materials
All additional files that support the findings of the current study are available as a collection in the figshare repository, https://doi.org/10.6084/m9.figshare.c.4743515.v2 [103]. Others are included in the LMAP_S software archive, which is available to download from the project home page. The archive version here revised (LMAP_S version 1.0.0) is also available from the project home page or by request.
Abbreviations
- AE:
-
MSA Estimation (stage 2)
- AIC:
-
Akaike Information Criterion
- AOD:
-
MSA Outlier Detection (stage 3)
- ARC:
-
MSA Refinement and Consensus (stage 4)
- BIC:
-
Bayesian Information Criterion
- CC:
-
Character Coding
- CLI:
-
Command-Line Interface
- CPAN:
-
Comprehensive Perl Archive Network
- CSV:
-
Comma-Separated Values
- DAMBE:
-
Data Analysis in Molecular Biology and Evolution
- EASER:
-
Ensembl Easy Sequence Retriever
- ETE:
-
Environment for Tree Exploration
- FSA:
-
Fast Statistical Alignment
- GUI:
-
Graphical User Interface
- GUIDANCE:
-
GUIDe tree–based AligNment ConfidencE
- IMPACT_S:
-
Integrated Multiprogram Platform to Analyze and Combine Tests of Selection
- LMAP:
-
Lightweight Multigene Analyses in PAML
- LMAP_S:
-
Lightweight Multigene Alignment and Phylogeny eStimation
- MACSE:
-
Multiple Alignment of Coding SEquences
- MAFFT:
-
Multiple Alignment using Fast Fourier Transform
- ML:
-
Maximum Likelihood
- MO-SAStrE:
-
Multiobjective Optimizer for Sequence Alignments based on Structural Evaluations
- MP (TreeCmp):
-
MatchingPair
- MP:
-
Maximum Parsimony
- MSA:
-
Multiple Sequence Alignment
- MSF:
-
Multiple Sequence File
- MUSCLE:
-
MUltiple Sequence Comparison by Log-Expectation
- NCBI:
-
National Center for Biotechnology Information
- NDP:
-
Nucleotide Data Pre-processing (stage 1)
- NJ:
-
Neighbor-Joining
- NNI:
-
Nearest-Neighbor Interchange
- PALM:
-
Phylogenetic reconstruction by Automatic Likelihood Model selector
- PAML:
-
Phylogenetic Analysis by Maximum Likelihood
- PAUP:
-
Phylogenetic Analysis Using Parsimony
- PCC:
-
PT Comparison and Consensus (stage 6)
- PDP:
-
PT Data Post-processing (stage 7)
- PE:
-
PT Estimation (stage 5)
- PHYLIP:
-
PHYLogenetic Inference Package
- POTION:
-
POsitive selecTION
- PSAR:
-
Probabilistic Sampling-based Alignment Reliability
- PT:
-
Phylogenetic Tree
- R-F_C:
-
Robinson-Foulds_Cluster
- SATé:
-
Simultaneous Alignment and Tree estimation
- SMS:
-
Smart Model Selection
- SPR:
-
Subtree Prunning and Regrafting
- SR:
-
Signal Refinement
- SWLH:
-
Site-Wise Log-Likelihood
- T-COFFEE:
-
Tree-based Consistency Objective Function For alignmEnt Evaluation
- TCS:
-
Transitive Consistency Score
- TTS:
-
Total Topological Score
References
KCoS G. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Heredity. 2009;100(6):659–74. https://doi.org/10.1093/jhered/esp086.
Koepfli KP, Paten B, Kcos G, O'Brien SJ. The genome 10K project: a way forward. Annu Rev Anim Biosci. 2015;3:57–111. https://doi.org/10.1146/annurev-animal-090414-014900.
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–D61. https://doi.org/10.1093/nar/gkx1098.
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Ostell J, Pruitt KD, et al. GenBank. Nucleic Acids Res. 2018;46(D1):D41–D7. https://doi.org/10.1093/nar/gkx1094.
Maldonado E, Khan I, Philip S, Vasconcelos V, Antunes A. EASER: Ensembl easy sequence retriever. Evol Bioinformatics Online. 2013;9:487–90. https://doi.org/10.4137/EBO.S11335.
Maldonado E, Sunagar K, Almeida D, Vasconcelos V, Antunes A. IMPACT_S: integrated multiprogram platform to analyze and combine tests of selection. PLoS One. 2014;9(10):e96243. https://doi.org/10.1371/journal.pone.0096243.
Maldonado E, Almeida D, Escalona T, Khan I, Vasconcelos V, Antunes A. LMAP: lightweight multigene analyses in PAML. BMC bioinformatics. 2016;17(1):354. https://doi.org/10.1186/s12859-016-1204-5.
Luo SJ, Johnson WE, Martenson J, Antunes A, Martelli P, Uphyrkina O, et al. Subspecies genetic assignments of worldwide captive tigers increase conservation value of captive populations. Curr Biol. 2008;18(8):592–6. https://doi.org/10.1016/j.cub.2008.03.053.
Khan I, Maldonado E, Vasconcelos V, O'Brien SJ, Johnson WE, Antunes A. Mammalian keratin associated proteins (KRTAPs) subgenomes: disentangling hair diversity and adaptation to terrestrial and aquatic environments. BMC Genomics. 2014;15:779. https://doi.org/10.1186/1471-2164-15-779.
Khan I, Yang Z, Maldonado E, Li C, Zhang G, Gilbert MT, et al. Olfactory receptor subgenomes linked with broad ecological adaptations in Sauropsida. Mol Biol Evol. 2015;32(11):2832–43. https://doi.org/10.1093/molbev/msv155.
Pereira SR, Vasconcelos VM, Antunes A. The phosphoprotein phosphatase family of Ser/Thr phosphatases as principal targets of naturally occurring toxins. Crit Rev Toxicol. 2011;41(2):83–110. https://doi.org/10.3109/10408444.2010.515564.
Pereira SR, Vasconcelos VM, Antunes A. Computational study of the covalent bonding of microcystins to cysteine residues--a reaction involved in the inhibition of the PPP family of protein phosphatases. FEBS J. 2013;280(2):674–80. https://doi.org/10.1111/j.1742-4658.2011.08454.x.
Morrison DA. Multiple sequence alignment for phylogenetic purposes. Aust Syst Bot. 2006;19(6):479–539. https://doi.org/10.1071/SB06020.
Penn O, Privman E, Landan G, Graur D, Pupko T. An alignment confidence score capturing robustness to guide tree uncertainty. Mol Biol Evol. 2010;27(8):1759–67. https://doi.org/10.1093/molbev/msq066.
Pais FS, Ruy PC, Oliveira G, Coimbra RS. Assessing the efficiency of multiple sequence alignment programs. Algorithms Mol Biol. 2014;9(1):4. https://doi.org/10.1186/1748-7188-9-4.
Ezawa K. Characterization of multiple sequence alignment errors using complete-likelihood score and position-shift map. BMC bioinformatics. 2016;17:133. https://doi.org/10.1186/s12859-016-0945-5.
Lassmann T, Sonnhammer EL. Automatic assessment of alignment quality. Nucleic Acids Res. 2005;33(22):7120–8. https://doi.org/10.1093/nar/gki1020.
Kemena C, Taly JF, Kleinjung J, Notredame C. STRIKE: evaluation of protein MSAs using a single 3D structure. Bioinformatics. 2011;27(24):3385–91. https://doi.org/10.1093/bioinformatics/btr587.
Wong KM, Suchard MA, Huelsenbeck JP. Alignment uncertainty and genomic analysis. Science. 2008;319(5862):473–6. https://doi.org/10.1126/science.1151532.
Herman JL, Novak A, Lyngso R, Szabo A, Miklos I, Hein J. Efficient representation of uncertainty in multiple sequence alignments using directed acyclic graphs. BMC bioinformatics. 2015;16:108. https://doi.org/10.1186/s12859-015-0516-1.
Wu M, Chatterji S, Eisen JA. Accounting for alignment uncertainty in phylogenomics. PLoS One. 2012;7(1):e30288. https://doi.org/10.1371/journal.pone.0030288.
Hohl M, Ragan MA. Is multiple-sequence alignment required for accurate inference of phylogeny? Syst Biol. 2007;56(2):206–21. https://doi.org/10.1080/10635150701294741.
Rajan V. A method of alignment masking for refining the phylogenetic signal of multiple sequence alignments. Mol Biol Evol. 2013;30(3):689–712. https://doi.org/10.1093/molbev/mss264.
Wallace IM, O'Sullivan O, Higgins DG, Notredame C. M-coffee: combining multiple sequence alignment methods with T-coffee. Nucleic Acids Res. 2006;34(6):1692–9. https://doi.org/10.1093/nar/gkl091.
Bains W. MULTAN: a program to align multiple DNA sequences. Nucleic Acids Res. 1986;14(1):159–77. https://doi.org/10.1093/nar/14.1.159.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. https://doi.org/10.1093/nar/gkh340.
Loytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 2005;102(30):10557–62. https://doi.org/10.1073/pnas.0409137102.
Ortuno FM, Valenzuela O, Rojas F, Pomares H, Florido JP, Urquiza JM, et al. Optimizing multiple sequence alignments using a genetic algorithm based on three objectives: structural information, non-gaps percentage and totally conserved columns. Bioinformatics. 2013;29(17):2112–21. https://doi.org/10.1093/bioinformatics/btt360.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–52. https://doi.org/10.1093/oxfordjournals.molbev.a026334.
Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25(15):1972–3. https://doi.org/10.1093/bioinformatics/btp348.
Gouveia-Oliveira R, Sackett PW, Pedersen AG. MaxAlign: maximizing usable data in an alignment. BMC bioinformatics. 2007;8:312. https://doi.org/10.1186/1471-2105-8-312.
Eisen JA. Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998;8(3):163–7. https://doi.org/10.1101/gr.8.3.163.
Mirarab S, Warnow T. FastSP: linear time calculation of alignment accuracy. Bioinformatics. 2011;27(23):3250–8. https://doi.org/10.1093/bioinformatics/btr553.
Novak A, Miklos I, Lyngso R, Hein J. StatAlign: an extendable software package for joint Bayesian estimation of alignments and evolutionary trees. Bioinformatics. 2008;24(20):2403–4. https://doi.org/10.1093/bioinformatics/btn457.
Swofford DL. PAUP*: Phylogenetic analysis using parsimony (*and other methods). Version 4.0. ed: Sinauer Associates, Sunderland.; 2002.
Felsenstein J. PHYLIP - phylogeny inference package (version 3.2). Cladistics. 1989;5:164–6. https://doi.org/10.1111/j.1096-0031.1989.tb00562.x.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21. https://doi.org/10.1093/sysbio/syq010.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–3. https://doi.org/10.1093/bioinformatics/btu033.
Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. https://doi.org/10.1371/journal.pone.0009490.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–42. https://doi.org/10.1093/sysbio/sys029.
Xia X, Xie Z, Salemi M, Chen L, Wang Y. An index of substitution saturation and its application. Mol Phylogenet Evol. 2003;26(1):1–7. https://doi.org/10.1016/S1055-7903(02)00326-3.
Xia X. DAMBE7: new and improved tools for data analysis in molecular biology and evolution. Mol Biol Evol. 2018. https://doi.org/10.1093/molbev/msy073.
Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25(7):1253–6. https://doi.org/10.1093/molbev/msn083.
Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. https://doi.org/10.1038/nmeth.2109.
Nylander JAA. MrAIC.pl. Program distributed by the author. 2004. Evolutionary Biology Centre, Uppsala University. https://github.com/nylander/MrAIC.
Liu K, Raghavan S, Nelesen S, Linder CR, Warnow T. Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees. Science. 2009;324(5934):1561–4. https://doi.org/10.1126/science.1171243.
Hongo JA, de Castro GM, Cintra LC, Zerlotini A, Lobo FP. POTION: an end-to-end pipeline for positive Darwinian selection detection in genome-scale data through phylogenetic comparison of protein-coding genes. BMC Genomics. 2015;16:567. https://doi.org/10.1186/s12864-015-1765-0.
Huerta-Cepas J, Serra F, Bork P. ETE 3: reconstruction, analysis, and visualization of Phylogenomic data. Mol Biol Evol. 2016;33(6):1635–8. https://doi.org/10.1093/molbev/msw046.
Ramirez-Flandes S, Ulloa O. Bosque: integrated phylogenetic analysis software. Bioinformatics. 2008;24(21):2539–41. https://doi.org/10.1093/bioinformatics/btn466.
Chen SH, Su SY, Lo CZ, Chen KH, Huang TJ, Kuo BH, et al. PALM: a paralleled and integrated framework for phylogenetic inference with automatic likelihood model selectors. PLoS One. 2009;4(12):e8116. https://doi.org/10.1371/journal.pone.0008116.
Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27(2):221–4. https://doi.org/10.1093/molbev/msp259.
Lord E, Leclercq M, Boc A, Diallo AB, Armadillo MV. 1.1: an original workflow platform for designing and conducting phylogenetic analysis and simulations. PLoS One. 2012;7(1):e29903. https://doi.org/10.1371/journal.pone.0029903.
Kjer KM, Gillespie JJ, Ober KA. Opinions on multiple sequence alignment, and an empirical comparison of repeatability and accuracy between POY and structural alignment. Syst Biol. 2007;56(1):133–46. https://doi.org/10.1080/10635150601156305.
Blair C, Murphy RW. Recent trends in molecular phylogenetic analysis: where to next? J Heredity. 2011;102(1):130–8. https://doi.org/10.1093/jhered/esq092.
The Perl Programming Language. www.perl.org. Accessed 8 Oct 2015.
Almeida D, Maldonado E, Vasconcelos V, Antunes A. Adaptation of the mitochondrial genome in cephalopods: enhancing proton translocation channels and the subunit interactions. PLoS One. 2015;10(8):e0135405. https://doi.org/10.1371/journal.pone.0135405.
The Comprehensive Perl Archive Network. http://www.cpan.org/. Accessed 8 Oct 2015.
Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12(10):1611–8. https://doi.org/10.1101/gr.361602.
Screen User's Manual. https://www.gnu.org/software/screen/manual/screen.html. Accessed 8 Oct 2015.
Open Source - Sendmail.com. http://www.sendmail.com/sm/open_source/. Accessed 8 Oct 2015.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol Syst Biol. 2011;7:539. https://doi.org/10.1038/msb.2011.75.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80. https://doi.org/10.1093/nar/22.22.4673.
Subramanian AR, Kaufmann M, Morgenstern B. DIALIGN-TX: greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol Biol. 2008;3:6. https://doi.org/10.1186/1748-7188-3-6.
Bradley RK, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, et al. Fast statistical alignment. PLoS Comput Biol. 2009;5(5):e1000392. https://doi.org/10.1371/journal.pcbi.1000392.
Russell DJ, Otu HH, Sayood K. Grammar-based distance in progressive multiple sequence alignment. BMC bioinformatics. 2008;9:306. https://doi.org/10.1186/1471-2105-9-306.
Lassmann T, Frings O, Sonnhammer EL. Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 2009;37(3):858–65. https://doi.org/10.1093/nar/gkn1006.
Ranwez V, Harispe S, Delsuc F, Douzery EJ. MACSE: multiple alignment of coding SEquences accounting for frameshifts and stop codons. PLoS One. 2011;6(9):e22594. https://doi.org/10.1371/journal.pone.0022594.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–80. https://doi.org/10.1093/molbev/mst010.
Wheeler TJ, Kececioglu JD. Multiple alignment by aligning alignments. Bioinformatics. 2007;23(13):i559–68. https://doi.org/10.1093/bioinformatics/btm226.
Roshan U, Livesay DR. Probalign: multiple sequence alignment using partition function posterior probabilities. Bioinformatics. 2006;22(22):2715–21. https://doi.org/10.1093/bioinformatics/btl472.
Do CB, Mahabhashyam MS, Brudno M, Batzoglou S. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15(2):330–40. https://doi.org/10.1101/gr.2821705.
Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–17. https://doi.org/10.1006/jmbi.2000.4042.
Jehl P, Sievers F, Higgins DG. OD-seq: outlier detection in multiple sequence alignments. BMC bioinformatics. 2015;16:269. https://doi.org/10.1186/s12859-015-0702-1.
Chiner-Oms A, Gonzalez-Candelas F. EvalMSA: a program to evaluate multiple sequence alignments and detect outliers. Evol Bioinformatics Online. 2016;12:277–84. https://doi.org/10.4137/EBO.S40583.
Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56(4):564–77. https://doi.org/10.1080/10635150701472164.
Collingridge PW, Kelly S. MergeAlign: improving multiple sequence alignment performance by dynamic reconstruction of consensus multiple sequence alignments. BMC bioinformatics. 2012;13:117. https://doi.org/10.1186/1471-2105-13-117.
Dress AW, Flamm C, Fritzsch G, Grunewald S, Kruspe M, Prohaska SJ, et al. Noisy: identification of problematic columns in multiple sequence alignments. Algorithms Mol Biol. 2008;3:7. https://doi.org/10.1186/1748-7188-3-7.
Kim J, Ma J. PSAR-align: improving multiple sequence alignment using probabilistic sampling. Bioinformatics. 2014;30(7):1010–2. https://doi.org/10.1093/bioinformatics/btt636.
Chang JM, Di Tommaso P, Notredame C. TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction. Mol Biol Evol. 2014;31(6):1625–37. https://doi.org/10.1093/molbev/msu117.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–74. https://doi.org/10.1093/molbev/msu300.
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35(2):518–22. https://doi.org/10.1093/molbev/msx281.
Hoang DT, Vinh LS, Flouri T, Stamatakis A, von Haeseler A, Minh BQ. MPBoot: fast phylogenetic maximum parsimony tree inference and bootstrap approximation. BMC Evol Biol. 2018;18(1):11. https://doi.org/10.1186/s12862-018-1131-3.
Wheeler TJ. Large-scale neighbor-joining with NINJA. Berlin: springer Berlin Heidelberg; 2009.
Lefort V, Longueville JE, Gascuel O. SMS: smart model selection in PhyML. Mol Biol Evol. 2017;34(9):2422–4. https://doi.org/10.1093/molbev/msx149.
Regier JC, Shultz JW, Zwick A, Hussey A, Ball B, Wetzer R, et al. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010;463(7284):1079–83. https://doi.org/10.1038/nature08742.
Zwick A, Regier JC, Zwickl DJ. Resolving discrepancy between nucleotides and amino acids in deep-level arthropod phylogenomics: differentiating serine codons in 21-amino-acid models. PLoS One. 2012;7(11):e47450. https://doi.org/10.1371/journal.pone.0047450.
Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17(12):1246–7. https://doi.org/10.1093/bioinformatics/17.12.1246.
Bogdanowicz D, Giaro K. Comparing phylogenetic trees by matching nodes using the transfer distance between partitions. J Comput Biol. 2017;24(5):422–35. https://doi.org/10.1089/cmb.2016.0204.
Akaike H. Information theory and an extension of the maximum likelihood principle. In: Petrov BN, Csáki F, editors. 2nd international symposium on information theory; September 2–8, 1971; Tsahkadsor, Armenia, USSR. Budapest: Akadémiai Kiadó; 1973. p. 267–81.
Schwarz G. Estimating the dimension of a model. Ann Stat. 1978;6(2):461–4. https://doi.org/10.1214/aos/1176344136.
Nabholz B, Uwimana N, Lartillot N. Reconstructing the phylogenetic history of long-term effective population size and life-history traits using patterns of amino acid replacement in mitochondrial genomes of mammals and birds. Genome Biol Evol. 2013;5(7):1273–90. https://doi.org/10.1093/gbe/evt083.
Diniz-Filho JA, Santos T, Rangel TF, Bini LM. A comparison of metrics for estimating phylogenetic signal under alternative evolutionary models. Genet Mol Biol. 2012;35(3):673–9. https://doi.org/10.1590/S1415-47572012005000053.
Simmons MP. Relative benefits of amino-acid, codon, degeneracy, DNA, and purine-pyrimidine character coding for phylogenetic analyses of exons. J Syst Evol. 2017;55(2):85–109. https://doi.org/10.1111/jse.12233.
Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18(3):502–4. https://doi.org/10.1093/bioinformatics/18.3.502.
Robinson DF, Foulds LR. Comparison of phylogenetic trees. Math Biosci. 1981;53(1):131–47. https://doi.org/10.1016/0025-5564(81)90043-2.
Zhou X, Shen XX, Hittinger CT, Rokas A. Evaluating fast maximum likelihood-based phylogenetic programs using empirical Phylogenomic data sets. Mol Biol Evol. 2018;35(2):486–503. https://doi.org/10.1093/molbev/msx302.
Beiko RG, Chan CX, Ragan MA. A word-oriented approach to alignment validation. Bioinformatics. 2005;21(10):2230–9. https://doi.org/10.1093/bioinformatics/bti335.
Warnow T. Large-scale multiple sequence alignment and phylogeny estimation. In: Chauve C, El-Mabrouk N, Tannier E, editors. Models and algorithms for genome evolution. London: Springer London; 2013. p. 85–146.
Kececioglu J, DeBlasio D. Accuracy estimation and parameter advising for protein multiple sequence alignment. J Comput Biol. 2013;20(4):259–79. https://doi.org/10.1089/cmb.2013.0007.
Thompson JD, Koehl P, Ripp R, Poch O. BAliBASE 3.0: latest developments of the multiple sequence alignment benchmark. Proteins. 2005;61(1):127–36. https://doi.org/10.1002/prot.20527.
time - GNU Project - Free Software Foundation (FSF). http://www.gnu.org/software/time/. Accessed 8 Oct 2015.
Salichos L, Rokas A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature. 2013;497(7449):327–31. https://doi.org/10.1038/nature12130.
Maldonado E, Antunes A. LMAP_S Additional files 1 to 6 [Internet]. figshare; 2019. [cited 2019 Dec20]. Available from: https://figshare.com/collections/LMAP_S_Additional_files_1_to_6/4743515/2. https://doi.org/10.6084/m9.figshare.c.4743515.v2.
Acknowledgements
We are thankful to Dr. Imran Khan from the University of Helsinki, Institute of Biotechnology for helpful discussions. To Dr. Bui Quang Minh, from the Australian National University; to Dr. Salvador Capella-Gutiérrez, from the Barcelona Supercomputing Centre (BSC); to Dr. Jaebum Kim, from the Konkuk University; to Dr. Cedric Notredame, from the Centre of Genomic Regulation (CRG) of Barcelona; and to Dr. Xuhua Xia from University of Ottawa, for all their helpful discussions and assistance concerning their bioinformatics software. We are thankful for the comments provided by the editors Alison Cuff, Jijun Tang and Danielle Talbot and two anonymous reviewers, which helped to improve a previous version of the manuscript.
Funding
AA was partially supported by the Strategic Funding UID/Multi/04423/2019 through national funds provided by FCT and European Regional Development Fund (ERDF) in the framework of the programme PT2020, and the FCT project PTDC/AAG-GLO/6887/2014 (POCI-01-0124-FEDER-016845) and PTDC/CTA-AMB/31774/2017 (POCI-01-0145-FEDER/031774/2017). The funding body did not play any roles in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
EM conceived and participated in the design, carried out implementation of the software, contributed with additional functionalities, debugging and software testing phases and drafted the manuscript and the manual. AA participated in the initial design and coordination, contributed with materials and computational resources and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Additional implementation and algorithms. (Section 1): LMAP_S Scheduling of tasks executions. (Section 2): Description and reasoning of SWLHs heuristic involved in the PCC method. (Section 3): Description of LMAP_S statistical and topological reports involved in the PCC method.
Additional file 2.
Figures exhibiting LMAP_S applications options and stage arguments. (Figure S1): command-line options for lmap-s.pl application. (Figure S2): command-line options for RYcode.pl application. (Figure S3): lmap-s.pl arguments for algorithm selection in Stage 2 (AE). (Figure S4): lmap-s.pl arguments for algorithm selection in Stage 4 (ARC). (Figure S5): lmap-s.pl arguments for algorithm selection in Stage 5 (PE). (Figure S6): lmap-s.pl arguments for translation table selection. (Figure S7): lmap-s.pl display of available integrated software.
Additional file 3.
Table extending Table 1 information. Shows absolute identification assigned to algorithms and respective code abbreviation, which enables their selection into LMAP_S stages.
Additional file 4.
Flowchart illustrating the PCC method. Shows the several steps of the method starting with the PE Stage data until final consensus reports.
Additional file 5:
Resulting CSV reports compiled from LMAP_S execution of the included example dataset. (Table S1): Stage 3 (AOD) outlier detection report. (Table S2): Stage 4 (ARC) TrimAl “compareset” report. (Table S3): Stage 6 (PCC) CONSEL report. (Table S4): Stage 6 (PCC) TreeCmp MP reports. (Table S5): Stage 6 (PCC) TreeCmp R-F_C reports. (Table S6): Stage 6 (PCC) consensus reports. (Table S7): Stage 6 (PCC) consensus brief reports. (Table S8): Stage 6 (PCC) consensus histogram reports.
Additional file 6.
LMAP_S case study analyses of the Cephalopoda mitochondrial genes. (File 1): Description of experiments, results and discussion. (File 2): LMAP_S and TreeCmp command-lines with additional benchmarking. (File 3): Tables with LMAP_S consensus histogram reports from CephaResults. (File 4): Tables with LMAP_S consensus histogram reports from CephaResultsARC. (File 5): Figures showing side-by-side consensus strategies charts comparisons. (File 6): Tables with results of the topological comparisons. (File 7): Final and original LMAP_S results (PTs and Reports). (File 8): Bash scripts used to generate the TreeCmp input files.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Maldonado, E., Antunes, A. LMAP_S: Lightweight Multigene Alignment and Phylogeny eStimation. BMC Bioinformatics 20, 739 (2019). https://doi.org/10.1186/s12859-019-3292-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-019-3292-5