
Abstract
Background
DNA-binding proteins perform important functions in a great number of biological activities. DNA-binding proteins can interact with ssDNA (single-stranded DNA) or dsDNA (double-stranded DNA), and DNA-binding proteins can be categorized as single-stranded DNA-binding proteins (SSBs) and double-stranded DNA-binding proteins (DSBs). The identification of DNA-binding proteins from amino acid sequences can help to annotate protein functions and understand the binding specificity.
In this study, we systematically consider a variety of schemes to represent protein sequences: OAAC (overall amino acid composition) features, dipeptide compositions, PSSM (position-specific scoring matrix profiles) and split amino acid composition (SAA), and then we adopt SVM (support vector machine) and RF (random forest) classification model to distinguish SSBs from DSBs.
Results
Our results suggest that some sequence features can significantly differentiate DSBs and SSBs. Evaluated by 10 fold cross-validation on the benchmark datasets, our prediction method can achieve the accuracy of 88.7% and AUC (area under the curve) of 0.919. Moreover, our method has good performance in independent testing.
Conclusions
Using various sequence-derived features, a novel method is proposed to distinguish DSBs and SSBs accurately. The method also explores novel features, which could be helpful to discover the binding specificity of DNA-binding proteins.
Similar content being viewed by others

Background
Proteins-DNA interaction is important for a great number of biological processes such as DNA replication, transcription, DNA repair and gene expression [1,2,3,4], etc. DNA-binding proteins contain essential protein-DNA binding domains, and they have specific or general affinities for either ssDNA or dsDNA [5,6,7]. Currently, X-ray crystallography, NMR and filter binding assays have been used to dissect structural features [8,9,10], multiple domain structures of SSBs [11], uncover the biological functions [12,13,14,15], etc. However, wet methods of identifying DSBs and SSBs are relatively expensive and time-consuming. Therefore, a reliable and effective computational method is an urgent task, and computational method plays a crucial role in protein function annotation and the identification of proteins. However, a great number of computational methods have been focused on analyzing the specific binding sites of DSBs [16,17,18,19,20,21,22], classification of DNA binding proteins [23,24,25,26,27,28] and protein-DNA binding specificities [29] etc. But few methods pay attention to the large-scale identification of DSBs and SSBs. In our previous work [30], we constructed a SVM prediction model to classify DSBs and SSBs based on the structure information. Although structure-based methods can produce high-accuracy performances, they can’t be applied in high-throughput function annotation because limited structures are known. In contrast, the prediction based on sequence information has more potential use in practice. In this work, we predict whether a protein binds ssDNA or dsDNA without relying on the geometry of the protein. The protein sequence can provide lots of information for predicting protein function [31]. At present, the most familiar methods for predicting protein function involve sequence features [32]. Many methods are employed to predict protein function classes, such as homology detection, sequence patterns, structural similarity, and so on. However, few computational works have studied the sequence features and identify SSBs and DSBs sequences. The recent study [8] shows that SSBs bind with specifically and non-specifically to ssDNA and SSBs have lower sequence conservation. Some DSBs with similar functions have common subsequences, and diverse DSBs involved in different functions seem to have lower conserved subsequences [33]. Recognizing DNA-binding protein sequences helps to realize the implications of properties of proteins and reveal the undiscovered protein features, which help to understand the mechanism of protein-DNA interactions. [34,35,36].
Here, we propose a novel method to predict DSBs or SSBs by using the SVM algorithm and random forest (RF) algorithm with various sequence-derived features. Specifically, consider a variety of sequence-derived features, including OAAC, PSSM, dipeptide composition, and physicochemical properties, which can provide diverse information to differentiate ssDNAs from dsDNAs. Fig. 1 shows the workflow of our method. In the computational experiments, our model achieves MCC of 0.647 (Matthew’s correlation coefficient), accuracy of 0.887, sensitivity of 0.908 and specificity of 0.788 based on 10-fold cross-validation, respectively. The results show that our method can perform well in predicting SSBs or DSBs for novel proteins.

The whole workflow of our method
Methods
Training datasets
In this study, DNA-binding proteins were obtained from UniProtKB/Swiss-Prot (www.uniprot.org). The dataset consists of 2136 DSBs and 339 SSBs which are extracted from literature and manually reviewed entries (Additional file 1). Then we used the CD_HIT toolkit [37] to extract sequences with non-redundant proteins (Sequence identity cut-off 0.7). Finally, we obtained 873 DSBs and 183 SSBs (Additional file 2), which is called Uniprot1065. To deal with the unbalanced datasets, a larger number of samples were selected by down-sampling methods during the training process. We obtained a “Negative sample” dataset by randomly selecting subsequences which has the equal size of the SSBs dataset from DSBs dataset.
Independent datasets
Further, an independent dataset was obtained from PDB (www.rcsb.org/pdb/) to evaluate the performance in predicting novel proteins. PISCES is used (http://dunbrack.fccc.edu/Guoli/PISCES.php) to obtain the non-redundant PDB401 dataset, in which every structure is determined by X-ray or NMR, and resolution better than 3 Å. The sequence similarity is lower than 30%, and the sequence length is higher than 40 residues. In addition, we checked the similarity between the training and independent test sets. We also used the CD_HIT toolkit to extract the non-redundant proteins in the independent dataset. As a result, we obtained the non-redundant independent set of 125 DSBs and 41 SSBs (Additional file 3).
Protein features
Sequence-derived features can reflect the characteristics of the protein sequences. Here, we consider four types of sequence-derived features, including overall amino acid composition (OAAC), dipeptide composition, PSSM profiles and physicochemical properties. The overall amino acid composition expresses the global descriptors of proteins. Dipeptide composition is the detailed descriptors of sequences and the other two kinds of properties are transformed with the split amino acid composition for describing local features of sequence. The details of features are described as follows.
Overall amino acid composition (OAAC)’
The OAAC method is a 20-dimensional descriptor of a protein sequence, which describes the frequencies of amino acids in the sequence. It is defined as the follow:
Where p i is the occurrence frequency of the i-th amino acids occurrence, L is the total sequence length, and n i is the sum of the i-th amino acids in the sequence.
Researches have shown that a better result can be reached by computing the square root of p i [38]. Therefore, f i is used for the OAAC features.
Dipeptide composition
Dipeptide component is an important representation of a protein sequence, and has been widely used in the secondary structure prediction [39], subcellular localization and fold recognition [24]. Dipeptide composition contains two consecutive residues information of each sequence, which has 400 patterns [40]. In this work, three types of dipeptide compositions were calculated for every two residues in case of 0, 1 and 2 of intervals respectively, as illustrated in Fig. 2. The dipeptide composition is defined as:

Schematic representation for three kinds of dipeptide composition. The dipeptide compositions are calculated for every two residues in case of 0, 1 and 2 of intervals respectively
Where D s (i, j) represents the total of each type of i and j dipeptides with s of intervals where s = 0, 1, 2, and N is the sequence length of protein. f s (i, j) is the occurrence frequency of every dipeptides. Finally, we got a total of 1200 dimensional vectors with dipeptides of varying intervals together.
Physicochemical properties
Physicochemical properties play a major role in analyzing DNA-binding mechanism. AAindex is widely used in many studies of physicochemical properties of amino acids. A great number of algorithms for predicting protein functions had been developed by using physicochemical properties from AAindex. Here, we used 28 AAindex properties (Table 1) which are selected by the Auto-IDPCPs methods [41]. Each protein is represented by a set of 28*L matrix array along with the L-residue number.
PSSM profiles
The PSSM is an important tool to predict protein function, and the PSSM profiles represent the evolution information, which has been widely used in protein function prediction [42]. Here, PSSM profiles are obtained by using PSI-BLAST [43]. The PSSM was calculated by three iterations of PSI-BLAST to search the non-redundant NCBI database based on the substitution matrix of BLOSUM62. The parameter of e-value was set to 0.001. This PSSM scoring matrix has L rows and 20 columns, and L rows are the sequence length of a protein, and 20 columns represent the occurrence of each kind of 20 amino acids.
Split amino acid (SAA) transformation
SAA transformation was used to describe the local composition of protein sequences [44]. SAA transformation partitions each sequence into three regions: the parts of the N-terminal, middle and C-terminal. The composition of each region is shown in Fig. 3. The variable length sequences were partitioned with a fixed length pattern of the 6 dimensional vectors. The sequences are defined as N-terminal regions, middle regions and C-terminal regions based on their position. For the sequences with varied lengths, we used three definitions to represent the local composition (Fig. 3).

The SAA method defining the N-terminal, middle, and C-terminal regions based on the sequence length L. If L is no less than 4d N + 20 + d c , the N-terminal of each part d N is represented 25 residues in the start region of sequence. The middle regions have more than 20 residues, and the length of the C-terminal regions d c is set as 10 residues (a). If L is between 4d N + d c and 4d N + 20 + d c , the N-terminal of each part d N is represented by 20 residues in the start part. Thus, the middle part has less than 20 residues (b). For short sequence, if L is no less than 4d N + d c , the N-terminal part is not divided and the lengths of the middle and N-terminal regions are set as (L-d c )/2 (c)
Classification model and evaluation method
The classification models are built by using SVM and random forest with above mentioned features. SVM models are implemented by the SVM package in Matlab 2012a. The default parameters of SVM are adopted in the experiments. The random forest models are implemented by using Andy Liaw’s Matlab package. The number of trees is set to 3000. Two classifiers are used to build prediction models and then compared. The performances of classification models were evaluated by AUC (area under the ROC curve), F1 (F-measure), Acc (accuracy), Spe (specificity), Sen (sensitivity) and MCC (Matthew’s correlation coefficient).
The 10-fold cross-validation is usually adopted to evaluation performances of classification models. In the 10-fold cross-validation, data is randomly divided into ten equal parts. In each fold, one part is kept for the testing dataset and nine parts are used as the training dataset. In each training dataset, classification models were constructed based on different features and predictions for testing set is given by combining outputs of classification models by majority voting strategy. The ensemble learning is a strategy to improve the performances of classification, and has lots if successful applications [45,46,47,48,49,50,51,52,53,54]. The majority voting strategy is a popular way of the ensemble learning, and can combine various sequence-derived to predict single-stranded and double-stranded DNA binding proteins.
Results and discussion
OAAC results
To evaluate the OAAC method, we detected the sequence composition of two kinds of proteins, and the comparisons of the two types of proteins are shown in Fig. 4. DSBs residues have only slightly higher frequency than SSBs, including Arg (R), Lys (K), Glu (E), Pro (P), Ser (S), Leu (L) and His (H). Clearly, the positive charge residues (Arg, His and Lys) in DSBs have a higher level than these of SSBs, and it coincides with the fact that dsDNA strand has higher negative charge than ssDNA strand, and dsDNA has a stabilized double-helix structure while ssDNA presents unwound and irregular helix. Therefore, the positive charges of sequence residues are more enriched to DSBs than SSBs. Asn (N), Gly (G), Phe (F), Tyr (Y) and Val (V) in the SSBs sequences have a higher frequency than those in the DSBs. It is believed that the differences of sequences can be used to distinguish DSBs and SSBs. The OAAC values (20 × N dimension matrices, N is the number of proteins) of each protein are used as features.

The frequency distributions of 20 kinds of amino acids in DSBs and SSBs
Dipeptide composition analysis
In the study, dipeptide compositions are used to obtain the global sequence information. The dipeptide compositions is analyzed statistically by computing pairs of amino acids conditions with 0, 1 and 2 of intervals respectively [55]. The dipeptide frequency of 0, 1 and 2 of intervals is shown in Fig. 5. The differences for 16 kinds of dipeptide of frequency are more than 0.003 (AL, RA, EE, EL, GN, GQ, GG, LE, LS, KA, KE, KG, KT, PK, VA and VE) and 2 kinds of dipeptide (ES, EB) are less than 0.005 in Fig. 5a. The frequency differences of 25 kinds of dipeptide are more than 0.003, which are shown in Fig. 5b (AR, AN, AK, RR, RP, RT, NN, NG, NV, DV, QG, EI, EL, EV, GA, GG, GL, LD, LE, KE, KL, PG, PT, SG and SL) and those 6 kinds of dipeptide are less than 0.005 (KL, LE, GG, GL, EI and NG). The frequency difference of 19 kinds of dipeptide are more than 0.003, which are shown in Fig. 5c (RR, RL, NN, NG, DG, DP, QL, EE, EK, GA, GQ, GG, LL, LK, KR, KK, FF, SL and TE) and the 5 kinds of dipeptide are less than 0.005 (GG, GQ, LK, EE and RR). The results show the effectiveness of the dipeptide compositions feature.

The frequency distributions of paired amino acids in the condition with 0, 1 and 2 of intervals. a The dipeptide frequency in case of 0 of intervals. b The dipeptide frequency in case of 1 of intervals. c The dipeptide frequency in case of 2 of intervals
The studies on protein-DNA binding have found some related physicochemical properties of amino acid, which were regarded as critical factors of protein-DNA binding mechanism. There are several typical physicochemical properties which are discovered to be associated with protein-DNA binding including charge, hydrophobicity, flexibility, solvent accessibility, polarity, volume and pK, etc. However, it is still unknown that whether those physicochemical properties are associated with the proteins specific binding to dsDNA or ssDNA. It is difficult to screen out related physicochemical properties for predicting SSBs and DSBs specific to biological methods. Therefore, 28 kinds of typical physicochemical properties, which are significantly different in 6 parts of SAA, are selected to analyze DSBs and SSBs. The physicochemical properties were evaluated for revealing protein-DNA interactive mechanism by computational methods. Here, we present a feature analytical method of the physicochemical features. The method is defined as follows.
Where X is the physicochemical properties difference in rate for DSBs and SSBs. \( \overline{K_1} \) and \( \overline{K_2} \) are the average for all physicochemical properties in every part of SAA.
The differences of physicochemical properties are shown in Fig. 6. According to statistical results, DSBs and SSBs show significant difference in some properties, and the AAindex IDs of physicochemical properties are shown in Table 2. Obviously, five parts share some properties, including RADA880108 (Mean polarity), NADH010104 (Hydropathy scale, 20% accessibility), NADH010106 (Hydropathy scale, 36% accessibility), and four parts share KLEP840101 properties (Net charge), CIDH920103 (Standardized hydrophobicity measures), MIYS990104 (Self-consistent estimation of inter-residue protein) [56]. The N-terminal (N1 and N3) and C-terminal share GUYH850105 (Amino acid side-chain partition energies). There are significant differences with polarity, hydropathy, net charge and protein contact energies between DSBs and SSBs. Therefore, these properties are playing critical roles in selecting the specific binding of dsDNA and ssDNA.

The physicochemical properties different rate in 6 parts of SAA between DSBs and SSBs. SAA transformation partitions each sequence into 6 parts: the parts of the N-terminal (N1, N2, N3 and N4), middle (M) and C-terminal (C)
Prediction performance of the classifiers
In order to identify whether the selected features can be employed to classify DSBs and SSBs, we constructed the SVM and random forest classifier with different features by 10-fold cross validation and leave-one-out cross validation in Uniprot1065 set. Here, we constructed six predicting models with individual features. ROC plots the summarizing results of the SVM and random forest testing in the dataset using the different features described in Figs. 7 and 8, and the ROC shows that all features dramatically improved the predicted performance. In addition, the results of leave-one-out cross validation are shown in Additional file 4. The Gini importance of each feature type is an importance characteristic parameter in random forest. We tested the Gini importance of each feature, and obtained the average value of Gini values in 10-fold cross-validation, and found out some significant difference in the features. The figures of Gini importance are provided in Additional file 5. For example, we can observe that Leu (L), Gly (G), Phe (F), Asn (N) and Trp (W) are hydrophobic, and have comparatively high Gini importance in Additional file 5: Figure S1. The results show that hydrophobicity may be one of the most significant characteristics between DSBs and SSBs.

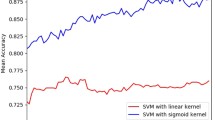
The ROC curve of the SVM model. The ROC plots the summarizing results of the SVM testing in the dataset using the different features. The red curve represents the result of independent test on PDB dataset, and others represent the results of 10-fold cross validation on Uniprot1065 set

The ROC curve of random forest models. The ROC plots the summarizing results of random forest testing in the dataset using the different features. The red curve represents the result of independent test on PDB dataset, and others represent the results of 10-fold cross validation on Uniprot1065 set
From the results of Tables 3 and 4, we can find that all single features can predict SSBs and DSBs with good performance. In experimentations, we found that the best precision is obtained by using multiple features with the highest prediction accuracy of 0.887, SN of 0.908, SP of 0.788 and AUC of 0.919 based on random forest model. The results suggest that the OAAC, PSSM, Dipeptide and AAindex features are important features to predict SSBs from DSBs sequences. Moreover, comparing Table 3 with Table 4, the random forest models also show better classification performance than SVM models. The results show that the random forest may be advantageous to deal with problems with high dimensions and unbalanced samples. Based on the results of individual modules, the predicting models based on Dipeptide = 1 feature obtained better performance than any other features, which illustrate that the Dipeptide = 1 feature of the sequences is more crucial to predicting DSBs from SSBs.
Independent test on PDB set
Furthermore, we had done an independent test using PDB401 dataset to validate our method. The results listed in Table 5 demonstrate that SVM classifier obtains a better performance. In SVM model, the combination of all features achieves the good performance with accuracy of 0.642, sensitivity of 0.613, specificity of 0.732, Matthew correlation coefficient of 0.298, and AUC of 0.672, respectively. The random forest method achieves a higher accuracy of 0.727, but the results of random forest methods show some of the biases in sensitivity and specificity. The random forest method achieves a higher sensitivity but a lower specificity, and the SVM model performs better than that of random forest in specificity and AUC. As we know that there is no computational works to identify SSBs and DSBs sequences, therefore we had train a baseline model by permuting the labels of SSBs and DSBs in the training data, and applied this model to predict the independent dataset. The results are shown in Table 5. In addition, we extracted the structures of 727 un-annotated DNA binding proteins from PDB. Then, we used CD-HIT to get the non-redundant set. We finally got 568 un-annotated proteins. The un-annotated proteins are predicted by using the prediction method, and the results are shown in Additional file 6. In general, the result indicates that our method has good generalization abilities in classifying DNA-binding proteins in novel proteins.
Conclusions
In this study, we compile a non-redundant sequence dataset consisting of 873 DSBs and 183 SSBs, and build four kinds of typical features underlying DNA binding proteins sequences. Using the features, we developed SVM-based model and RF-based model to predict SSBs from DSBs sequences. The results confirmed the distinguishing abilities of the features. Interestingly, OAAC, dipeptide compositions and physicochemical properties presents remarkable difference between DSBs and SSBs. The independent test confirms the effectiveness of the model. Based on the sequence-derived features, RF model has a prediction accuracy of 88.7% and AUC of 0.919, and SVM performs better in independent data set. In general, our results indicate that the method can effectively predict DSBs and SSBs sequence to investigate DNA binding protein sequences, and these amino acid properties may be critical to describe the specific binding of a protein for ssDNA or dsDNA molecule.
Abbreviations
- Acc:
-
Accuracy
- AUC:
-
Area under the curve
- DSBs:
-
Double-stranded DNA binding proteins
- dsDNA:
-
Double-stranded DNA
- F1:
-
F-measure
- MCC:
-
Mathews Correlation Coefficient
- MCC:
-
Matthew’s correlation coefficient
- OAAC:
-
Overall amino acid composition
- PSSM:
-
Position-specific scoring matrix
- RF:
-
Random forest
- ROC:
-
Receiver operating characteristics
- SAA:
-
Split amino acid
- Sen:
-
Sensitivity
- Spe:
-
Specificity
- SSBs:
-
Single-stranded DNA binding proteins,
- ssDNA:
-
Single-stranded DNA
- SVM:
-
Support vector machine
References
Edsö JR, Gustafsson C, Cohn M. Single- and double-stranded DNA binding proteins act in concert to conserve a telomeric DNA core sequence. Genome Integrity. 2011;2(1):1–9.
Attaiech L, Olivier A, Mortier-Barrière I, Soulet AL, Granadel C, Martin B, et al. Role of the single-stranded DNA-binding protein SsbB in pneumococcal transformation: maintenance of a reservoir for genetic plasticity. PLoS Genet. 2011;7(6):1–12.
Shlyakhtenko LS, Lushnikov AY, Miyagi A, Lyubchenko YL. Specificity of binding of single-stranded DNA-binding protein to its target. Biochemistry-US. 2012;51(7):1500–9.
Richard DJ, Bolderson E, Cubeddu L, Richard DJ, Bolderson E, Cubeddu L, et al. Single-stranded DNA-binding protein hssb1 is critical for genomic stability. Nature. 2008;453(5):677–81.
Delagoutte E, Heneman-Masurel A, Baldacci G. Single-stranded DNA binding proteins unwind the newly synthesized double-stranded DNA of model miniforks. Biochemistry. 2011;50(6):932–44.
Kur J, Olszewski M, Długołecka A, Filipkowski P. Single-stranded DNA-binding proteins (SSBs)-sources and applications in molecular biology. ACTA Biochimica Polonica-English Edition. 2005;52(3):569–74.
Shi H, Zhang Y, Zhang G, Guo J, Zhang X, Song H, et al. Systematic functional comparative analysis of four single-stranded DNA-binding proteins and their affection on viral RNA metabolism. PLoS One. 2013;8(1):e55076.
Morgan HP, Estibeiro P, Wear MA, Max KE, Heinemann U, Cubeddu L, et al. Sequence specificity of single-stranded DNA-binding proteins: a novel DNA microarray approach. Nucleic Acids Res. 2007;35(10):e75.
Kresten LL, Best RB, Depristo MA, Dobson CM, Michele V. Simultaneous determination of protein structure and dynamics. Nature. 2005;433(7022):128–32.
Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461(7268):1248–53.
Dickey TH, Altschuler SE, Wuttke DS. Single-stranded DNA-binding proteins: multiple domains for multiple functions. Structure. 2013;21(7):1074–84.
Kerr ID, Wadsworth RIM, Cubeddu L, Blankenfeldt W, Naismith JH, White MF. Insights into ssDNA recognition by the OB fold from a structural and thermodynamic study of Sulfolobus SSB protein. EMBO J. 2003;22(11):2561–70.
Marceau AH, Bahng S, Massoni SC, George NP, Sandler SJ, Marians KJ, et al. Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J. 2011;30(20):4236–47.
Pretto DI, Tsutakawa S, Brosey CA, Castillo A, Chagot ME, Smith JA, et al. Structural dynamics and ssDNA binding activity of the three n-terminal domains of the large subunit of replication protein a from small angle X-ray scattering. Biochemistry-US. 2010;13(49):2880–9.
Wakamatsu T, Kitamura Y, Kotera Y, Nakagawa N, Kuramitsu S, Masui R. Structure of RecJ exonuclease defines its specificity for single-stranded DNA. J Biol Chem. 2010;285(13):9762–9.
Dey S, Pal A, Guharoy M, Sonavane S, Chakrabarti P. Characterization and prediction of the binding site in DNA-binding proteins improvement of accuracy by combining residue composition, evolutionary conservation and structural parameters. Nucleic Acids Res. 2012;40(15):7150–61.
Xiong Y, Liu J, Wei DQ. An accurate feature-based method for identifying DNA-binding residues on protein surfaces. Proteins Struct Funct Bioinf. 2011;79(2):509–17.
Xiong Y, Xia J, Zhang W, Liu J. Exploiting a reduced set of weighted average features to improve prediction of DNA-binding residues from 3D structures. PLoS One. 2011;6(12):e28440.
Qian ZL, Cai YD, Li YX. A novel computational method to predict transcription factor DNA binding preference. Biochem Biophys Res Commun. 2006;348(3):1034–7.
Zhu X, Ericksen SS, Mitchell JC. DBSI: DNA-binding site identifier. Nucleic Acids Res. 2013;41(16):e160.
Kuznetsov IB, Gou Z, Li R, Hwang S. Using evolutionary and structural information to predict DNA binding sites on DNA-binding proteins. Proteins Struct Funct Bioinf. 2006;64(1):19–27.
Wei W, Juan L, Yi X. Analysis and classification of DNA-binding sites in single-stranded and double-stranded DNA-binding proteins using protein information. IET Syst Biol. 2014;4(8):176–83.
Nimrod G, Szilágyi A, Leslie C, Ben-Tal N. Identification of DNA-binding proteins using structural, electrostatic and evolutionary features. J Mol Biol. 2009;387(4):1040–53.
Lin WZ, Fang JA, Xiao X, Chou KC. IDNA-prot: identification of DNA binding proteins using random forest with grey model. PLoS One. 2011;6(9):e24756.
Szabóová A, Kuželka O, Sergio ME, Železn F, Tolar J. Prediction of DNA-binding propensity of proteins by the ball-histogram method using automatic template search. BMC Bioinformatics. 2012;13(Suppl 10):S3.
Yan C, Terribilini M, Wu F, Jernigan R, Dobbs D, Honavar V. Predicting DNA-binding sites of proteins from amino acid sequence. BMC Bioinformatics. 2006;7(1):262.
Zhou W, Yan H. Prediction of DNA-binding protein based on statistical and geometric features and supportvector machines. Proteome Sci. 2011;9(12):1–6.
Shazman S, Elber G, Mandel-Gutfreund Y. From face to interface recognition: a differential geometric approach to distinguish DNA from RNA binding surfaces. Nucleic Acids Res. 2011;39(17):7390–9.
Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, et al. DNA-binding specificities of human transcription factors. Cell. 2013;152(1-2):327–39.
Wang W, Liu J, Zhou X. Identification of single-stranded and double-stranded DNA binding proteins based on protein structure. BMC Bioinformatics. 2014;12(15):12.
Cai YD, Doig AJ. Prediction of Saccharomyces cerevisiae protein functional class from functional domain composition. Bioinformatics. 2004;20(8):1292–300.
Brameier M, Haan J, Krings A, Maccallum RM. Automatic discovery of cross-family sequence features associated with protein function. BMC Bioinformatics. 2006;7(1):16.
Yu EY, Wang F, Lei M, Lue N. A proposed OB-fold with a protein-interaction surfacein Candida albicans telomerase protein Est3. Nat Struct Mol Biol. 2008;15(9):985–9.
Nanni L, Brahnam S, Lumini A. High performance set of PseAAC and sequence based descriptors for protein classification. J Theor Biol. 2010;266(1):1–10.
Song J, Tan H, Takemoto K, Akutsu T. HSEpred: predict half-sphere exposure from protein sequences. Bioinformatics. 2008;24(13):1489–97.
Zhang Z, Kochhar S, Grigorov MG. Descriptor-based protein remote homology identification. Protein Sci. 2005;14(2):431–44.
Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26(5):680–2.
Feng ZP, Zhang CT. Prediction of the subcellular location of prokaryotic proteins based on a new representation of the amino acid composition. Int J Biol Macromol. 2001;28(3):255–61.
Lin H, Li QZ. Using pseudo amino acid composition to predict protein structural class: approached by incorporating 400 dipeptide components. J Comput Chem. 2007;28(9):1463–6.
Garg A, Raghava GP. ESLpred2. Improved method for predicting subcellular localization of eukaryotic proteins. BMC Bioinformatics. 2008;9(1):503.
Huang HL, Lin IC, Liou YF, Tsai CT, Hsu KT, Huang WL, et al. Predicting and analyzing DNA-binding domains using a systematic approach to identifying a set of informative physicochemical and biochemical properties. BMC Bioinformatics. 2011;12(Suppl 1):S47.
Ahmad S, Sarai A. PSSM-based prediction of DNA binding sites in proteins. BMC Bioinformatics. 2005;6(1):33.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402.
Afridi TH, Khan A, Lee YS. Mito-GSAAC: mitochondria prediction using genetic ensemble classifier and split amino acid composition. Amino Acids. 2012;42(4):1443–54.
Zhang W, Chen Y, Tu S, Liu F, Qu Q. Drug side effect prediction through linear neighborhoods and multiple data source integration, IEEE International Conference on Bioinformatics and Biomedicine; 2016. p. 427–34.
Zhang W, Chen Y, Liu F, Luo F, Tian G, Li X. Predicting potential drug-drug interactions by integrating chemical, biological, phenotypic and network data. BMC Bioinformatics. 2017;18(1):18.
Zhang W, Zhu X, Fu Y, Tsuji J, Weng Z. The prediction of human splicing branchpoints by multi-label learning, IEEE International Conference on Bioinformatics and Biomedicine; 2016. p. 254–9.
Li D, Luo L, Zhang W, Liu F, Luo F. A genetic algorithm-based weighted ensemble method for predicting transposon-derived piRNAs. BMC Bioinformatics. 2016;17(1):329.
Luo L, Li D, Zhang W, Tu S, Zhu X, Tian G. Accurate prediction of transposon-derived piRNAs by integrating various sequential and physicochemical features. PLoS One. 2016;11(4):e0153268.
Zhang W, Liu F, Luo L, Zhang J. Predicting drug side effects by multi-label learning and ensemble learning. BMC Bioinformatics. 2015;16:365.
Zhang W, Zou H, Luo L, Liu Q, Wu W. Wenyi Xiao. Predicting potential side effects of drugs by recommender methods and ensemble learning. Neurocomputing. 2015;173(3):979–87.
Zhang W, Niu Y, Zou H, Luo L, Liu Q, Wu W. Accurate prediction of immunogenic T-cell epitopes from epitope sequences using the genetic algorithm-based ensemble learning. PLoS One. 2015;10(5):e0128194.
Zhang W, Niu Y, Xiong Y, Zhao M, Rongwei Yu, Juan Liu. Computational prediction of conformational B-cell epitopes from antigen primary structures by ensemble learning. PLoS One. 2012; 7(8): e43575.
Zhang W, Liu J, Zhao M, Li Q. Predicting linear B-cell epitopes by using sequence-derived structural and physicochemical features. Int J Data Mining Bioinformatics. 2012;6(5):557–69.
Govindan G, Nair AS. New feature vector for apoptosis protein subcellular localization prediction. In: Advances in Computing and Communications Communications, vol. 190; 2011. p. 294–301.
Naderi-Manesh H, Sadeghi M, Arab S, Moosavi Movahedi AA. Prediction of protein surface accessibility with information theory. Proteins Struct Funct Bioinf. 2001;42(4):452–9.
Acknowledgements
The authors thank editorial staff for their help in editing this manuscript and we would like to thank the anonymous reviewers for their suggestions and comments to improve the manuscript.
Funding
This work was supported by the Science and Technology Research Key Project of Educational Department of Henan Province (No. 16A520016, 17B520002, 17B520036), Key Project of Science and Technology Department of Henan Province (No. 142102210056), National Natural Science Foundation of China (No. 61402153), Project funded by China Postdoctoral Science Foundation, and Ph.D. Research Startup Foundation of Henan Normal University (Nos. qd15130, qd15132, qd15129). None the funding bodies played any role in the design of this project, conclusion of this study, or collection of data.
Availability of data and materials
Original data of this article can be available at UniProtKB/Swiss-Prot database (http://www.uniprot.org) and PDB database (http://www.rcsb.org/pdb/).
Authors’ contributions
WW developed the methodology. WW, LS and SZ executed the experiments. HJ, TJ, KL, and JS. performed additional analysis of the results. WW wrote this paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
This file contains the complete list of UniProt codes for the whole DNA-binding protein sets from UniProtKB/Swiss-Prot (www.uniprot.org). (DOCX 26 kb)
Additional file 2:
This file contains the list of UniProt codes for non-redundant DNA-binding protein sets from UniProtKB/Swiss-Prot (www.uniprot.org). (DOCX 19 kb)
Additional file 3:
This file contains the list of PDB codes for non-redundant DNA-binding protein independent sets from PDB (www.rcsb.org/pdb/). (DOCX 16 kb)
Additional file 4:
This file contains the results of leave-one-out cross validation. (DOCX 24 kb)
Additional file 5:
This file contains Gini importance of each feature type in random forest. (XLSX 86 kb)
Additional file 6:
This file contains the prediction results for 568 un-annotated proteins. (XLSX 22 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, W., Sun, L., Zhang, S. et al. Analysis and prediction of single-stranded and double-stranded DNA binding proteins based on protein sequences. BMC Bioinformatics 18, 300 (2017). https://doi.org/10.1186/s12859-017-1715-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-017-1715-8