
Abstract
Background
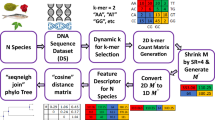
It has been a challenging task to build a genome-wide phylogenetic tree for a large group of species containing a large number of genes with long nucleotides sequences. The most popular method, called feature frequency profile (FFP-k), finds the frequency distribution for all words of certain length k over the whole genome sequence using (overlapping) windows of the same length. For a satisfactory result, the recommended word length (k) ranges from 6 to 15 and it may not be a multiple of 3 (codon length). The total number of possible words needed for FFP-k can range from 46=4096 to 415.
Results
We propose a simple improvement over the popular FFP method using only a typical word length of 3. A new method, called Trinucleotide Usage Profile (TUP), is proposed based only on the (relative) frequency distribution using non-overlapping windows of length 3. The total number of possible words needed for TUP is 43=64, which is much less than the total count for the recommended optimal “resolution” for FFP. To build a phylogenetic tree, we propose first representing each of the species by a TUP vector and then using an appropriate distance measure between pairs of the TUP vectors for the tree construction. In particular, we propose summarizing a DNA sequence by a matrix of three rows corresponding to three reading frames, recording the frequency distribution of the non-overlapping words of length 3 in each of the reading frame. We also provide a numerical measure for comparing trees constructed with various methods.
Conclusions
Compared to the FFP method, our empirical study showed that the proposed TUP method is more capable of building phylogenetic trees with a stronger biological support. We further provide some justifications on this from the information theory viewpoint. Unlike the FFP method, the TUP method takes the advantage that the starting of the first reading frame is (usually) known. Without this information, the FFP method could only rely on the frequency distribution of overlapping words, which is the average (or mixture) of the frequency distributions of three possible reading frames. Consequently, we show (from the entropy viewpoint) that the FFP procedure could dilute important gene information and therefore provides less accurate classification.
Similar content being viewed by others


Introduction
The construction of phylogenetic trees, based on the whole-genome information, is one of the challenging problems in computational biology. The difficulty is how to best utilize genome-wide DNA information. Each species has many genes and each gene can have a long DNA sequence. To capture the essential whole-genome DNA information, many different methods have been proposed. To quantify the closeness between two species, one can consider various distance functions to measure the closeness between two DNA sequences. We review some popular methods as follows.
Traditional methods were based on the classical sequence alignment methodology; see, for example, [1]. For each potential alignment, a score of similairity/dissimilarity is assigned to each base pair and an alignment score of the two sequences is obtained by summing the scores across all pairs in the sequences. The alignment with the highest score is outputted as the final aligning result. The evolutionary distance measure between two organisms is the similarity/dissimilarity of their proteinic or genomic/genic sequences. In general, such alignment-based methods would have a huge computational cost and are infeasible for entire proteomic/genomic sequence comparison. One common practice is using some selected gene(s) to represent the whole genome information. However, there is typically no general agreement about the choice of one or multiple representative genes. Additionally and most importantly, it can be hard to find common genes in all organisms under study, especially when the organisms are phylogenetically distant from one another.
To overcome the difficulties of the alignment-based methods, various alignment-free methods for phylogenetic tree construction have been proposed in the literature. One popular method is word-based, which involves counting the frequency of words of a specific length in the whole genome DNA sequence. See, for example, [2–4, 6, 7]. Most of the word-based research works have been focused on two directions: (i) choice of an optimal word size [4–6, 8] and/or (ii) choice of a proper distance measure between two word frequency distributions [2, 3, 9–11]. As pointed out in [4], some of these methods were variations of known techniques for comparing two text strings, also known as Latent Semantic Analysis (LSA). LSA is a popular technique in natural language processing used to analyze the similarity/dissimilarity between a set of documents [12]. In [4], a feature frequency profile (FFP) of length k, denoted by FFP-k, was obtained by scanning the DNA sequence with overlapping windows of size k to find the k-tuple frequency distribution (with 4k possible values) over the DNA sequence. [4] proposed estimating the optimal length or resolution of the features by using the delimiter-stripped text from some popular English books. They then used Jensen-Shannon Divergence measure as a distance between two FFPs. There are several obvious problems with this approach: (i) The optimal length could depend on the character strings considered and there is a wide range of possible lengths, say, between 6 to 15. (ii) The obtained optimal length has little, if any, biological support. (iii) If the optimal word size is large, the vector size of the corresponding FFP would grow exponentially.
For a DNA sequence, the most natural (and biologically sensible) word length is 3, which is clearly outside the optimal range of 6 to 15 for the word length as found in [4]. Denote the feature frequency profile for words of length 3 by FFP-3. The FFP-3 (or other word lengths) for a DNA sequence may fail to retain its essential information about the higher order (dimensional) structure between successive nucleotides. Keeping the word length at 3, we propose a simple modification on the counting of the word frequencies for trinucleotides (word of 3 nucleotides). The basic idea of our approach is to record the separate information from three reading frames (RFs), where the second and the third RFs are constructed from the first (original) RF by shifting one and two nucleotides, respectively. Strictly speaking, the word “codon” is generally restricted to the description of the trinucleotides on the first reading frame. In this paper, we will use the term “translation-triplet”, or simply TT, to denote either the codon in the first reading frame, or the trinucleotide in the second and third reading frames. Specifically, the proposed summary statistic is a matrix of three vectors of size 64(= 43) each: the first vector is the frequency distribution of the codons (of length 3, non-overlapping) corresponding to the first reading frame; the second and third vectors are constructed similarly from the corresponding second and third reading frames, respectively.
The rest of the paper is organized as follows. First, we describe the data under study, including the data source and format. In total, there are 56 species in this study. These species have potentially different numbers of genes and the genes have a large variation in length. Next, we discuss the general framework for alignment-free tree construction methods. We propose a summary measure function that retains the vital information associated with each species. We show in our study later that this summary measure function, called the vector-extracting function, yields a matrix based on three reading frames that can retain key information even with additional data reduction. While several methods have been proposed by researchers [13–15], they are not as intuitive as ours and often are computationally time-consuming. We also propose a simple and heuristic numerical measure for making a formal comparison among various trees. Finally, various vector-extracting functions are shown to yield consistent phylogenetic construction whereas the popular FFP-3 vector does not yield a tree that is consistent with other known species classifications. Using the trees constructed, we show the usefulness of our proposed distance measure between trees.
Description of data
Species included in the study
In this paper, we select a broad range of bacteria from several well-studied clones of eight different genera from three distinct subphyla of the Proteobacteria. To prevent bias due to variations of individual genomes, multiple genomes from different strains of a species were selected. The genera Orientia (1 species), Rickettsia (9 species/strains), and Wolbachia (2 strains) are members of a monophyletic class ([16]). These bacteria were used to represent the α-Proteobacteria subphylum. The 5 species/strains from the monophyletic genus Neisseriae [17] were used to represent the β-Proteobacteria subphylum. The monophyletic family of Escherichia (22 species/strains), Shigella (4 species), Salmonella (4 strains), and a separate monophyletic genus of Yersinia (9 species/stains) were selected to represent the γ- Proteobacteria. It should be noted that the Escherichia and Shigella are now considered as the same genus [18]. Escherichia and Salmonella are diverse from each other about 150 million years ago [19]. Most experts agree that the β- and γ-Proteobacteria are more closely related to each other than the α-Proteobacteria [20]. In total, 56 species are selected.
Source of data and processing methods
The FASTA.ffn files of 56 bacterial genomes were downloaded from the Comprehensive Microbial Research website (lbrinkac@jcvi.org). Each data file is in FASTA format and it contains the coding sequences for mRNAs in the genome, excluding the regulatory sequences and the sequences for tRNA and rRNA. Each data file has a various number of segments (or genes), depending on the genome size. In this paper, we use “segment” and “gene” interchangeably because each segment represents the coding sequence for a gene. A segment has two parts in its structure. The first part is a text paragraph describing the information about the gene such as name, location in chromosome, etc. The second part is a letter sequence of “A”, “T”, “C”, and “G”, which is the nucleotide sequence in the DNA strand. The following example is a gene segment from E coli K12 DH10B:

One can extract the nucleotide sequence from the data file using a downloadable R package “seqinr” with its function “read.fasta()”. We perform additional post-processing procedures on the nucleotide sequence as described next.
The genetic code of 64 codons, represented by three nucleotides, is reduced to 20 distinct amino acids, which are the functional building blocks of proteins. Some small percentage (less than one percent) of nucleotide sequences extracted from the data was excluded as non-informative. The gene count and a gene length summary (including minimum, average, and maximum) for each of the 56 bacterial species are listed in Table 1.
Phylogenetic tree construction methods
Alignment-free tree construction
We let S i denote the i-th strain in the study and use the notation S i ∼S j to denote that the strains S i and S j are closely related to each other. To measure the closeness of two strains S i and S j , we first find a summary function f() to produce a general summary measure for each strain S i :
and then find a distance function d() satisfying the following condition:
That is, if two strains, S i and S j , are closely related to each other, then their summary measures, M i =f(S i ) and M j =f(S j ), are expected to be close to each other as well.
The success (or failure) of the tree construction depends heavily on the choice of an appropriate summary function, f(), to represent and characterize the long whole-genome DNA sequence of the species. Generally speaking, there is a trade-off between the compactness and completeness of the chosen summary function. Clearly, the most complete statistic is the whole-genome DNA sequence itself, but it is too big to be practical for a meaningful genome-wide comparison between two species. On the other hand, choosing a simple summary function may fail to retain the vital information for a proper comparison or tree construction. We will consider some possible summary functions later.
If the summary measure M i is a vector, then we can choose d() to be any distance function. For example, the usual Euclidean distance
or the city block distance (Manhattan distance)
where x=(x 1,x 2,…,x n ) and y=(y 1,y 2,…,y n ). In our experience, there is not much difference between these two choices of the distance measure. In this paper, we choose the city block distance (Manhattan distance).
In our proposed method, there is a slight complication for phylogenetic tree construction—our proposed summary measure M i is a matrix instead of a vector. There is no standard way to define the distance between two matrices. One possible solution is to extract rows and/or columns from the summary matrix and convert them into a vector. Denote this vector extracting function by v(). Then, given two summary matrices, M i and M j , we can define the distance between them by d(v(M i ),(v M j )). Several reasonable choices of the vector extracting function v() will be discussed later.
For a proper choice of the summary function f(), vector extracting function v(), and distance function d(), one would expect
Having chosen these functions, we then perform hierarchical clustering with complete linkage. An open source software “Cluster 3.0” developed by Michael Eisen from Stanford University was used to generate the clustering results. In addition, we use GNU GPL v2 software “Java TreeView 1.1.6r2” by Alok Saldanha to display the hierarchical dendrograms. Both programs can be downloaded at http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm .
In the following, we first discuss the proposed choice of the summary function f() and then we consider various choices of the vector extracting function v().
Trinucleotide usage profile (TUP)
Given a gene with a sequence of nucleotides (“A”, “C”, “G”, “T”), there are several reasonable ways to summarize the nucleotide sequence. For example, we can group the nucleotides in the sequence in non-overlapping triplets and then count the frequency for each of the 64 possible triplets. Another popular summary measure is the frequencies of the 64 triplets in the set of the successive overlapping triplets of the sequence. The latter is a special case of the aforementioned feature frequency profile FFP-k with k=3. The vector of 4k frequency counts is commonly referred to as the FFP-k vector [4]. As mentioned earlier, the recommended word length k for the FFP-k vector is in the range of 6 to 15 depending on the sequence under study [4]. For k = 3, a natural codon length, the obtained FFP-3 vector may fail to retain vital information contained in the whole DNA sequence, as evidenced later with an example as well as by information theory.
In this paper, we propose a simple but essential modification on the FFP-3 method. For each strain (species), we find the frequency distribution of 64 TTs in each of the three reading frames and create a summary matrix of 3 rows and 64 columns as follows.
For each gene of a strain, we count the frequencies of the 64 TTs (non-overlapping) in each of its three reading frames to create a genic 3 × 64 TT count matrix. A genomic (genome-wide) TT count matrix of a species is simply the sum of all its genic TT count matrices. Specifically, let G i denote the number of genes in the i-th genome and c ig denote the genic TT count matrix of the g-th gene in the i-th genome for g=1,2,…,G i and i=1,2,…,56. Summing over all genes, we have \(\mathbf {C}_{i} = \sum _{g=1}^{G_{i}} \mathbf {c}_{ig}\) as the TT count matrix of the i-th genome.
For strain S i , we scale its count matrix C i by dividing each row element by the corresponding row total and denote the normalized matrix (of size 3x64) by M i . Let T i be the total TT counts of the first row of C i . Then the total row counts of the second and the third rows of C i are T i −1 when we omit the nucleotides that can not be in triplet due to frame shift in the second and third reading frame of a gene segment. To illustrate this, we take the aforementioned gene (E coli K12 DH10B) as an example. In the first reading frame, all the nucleotide triplets are “GTG”, “AAA”, “AAG”, …, “TAA”. However, when we shift the frame one nucleotide to the right to get the second reading frame, the triplet sequence starts with “TGA” and ends with “GCT”. So the first nucleotide (“G”) and the last two nucleotides (“AA”) can not be in triplet. These three nucleotides are excluded from the calculation. Similarly, in the third reading frame, the first two nucleotides (“GT”) and the last nucleotide (“A”) are omitted. Therefore, the total TT count for the first reading frame is one more than that for the second or the third reading frame. In practice, T i is a very large number, hence we can obtain the normalized matrix simply by
For the remainder of this paper, we refer to the summary matrix M i as the Trinucleotide Usage Profile (TUP) matrix.
Vector extracting functions
We now let a strain/bacterium be represented by a TUP matrix of size 3x64 containing the genome-wide proportions of all the 64 types of TTs corresponding to the three reading frames. To find the distance between two TUP matrices, M i and M j , we need to choose a proper vector extracting function, v(), and compute d(v(M i ),v(M j )). The following are some examples.
-
1.
Extract any of the three rows from the TUP matrix. The vectors corresponding to the first, second, and third RFs are designated as the TUP-R1 vector, TUP-R2 vector, and TUP-R3 vector, respectively.
-
2.
Extract all of the three rows from the summary matrix and concatenate them into a vector of 192 elements. The value of each element is the proportion of the combined TTs from the three RFs (3x64) of that bacterium. This vector is designated as the TUP-All vector.
-
3.
Extract the columns from the TUP matrix corresponding to a specific amino acid or stop codons. For example, we can extract the three columns from the summary matrix corresponding to the three stop codons (“TAA”, “TAG”, and “TGA”) and convert them into a vector of 9 elements. This approach was used successfully in [21] for a phylogenetic tree construction. It is interesting to observe that extracting columns corresponding to any specific amino acid, in general, has slightly inferior phylogenetic tree construction than those using stop codons. According to [21], the stop codons serve a vital role in gene expression and avoidance of transcriptional mistakes and it could offer a shortcut for whole genome analysis.
-
4.
Choose the output vector to be the sum of the three rows in the TUP matrix. This in fact gives the FFP-3 vector in [4] (see also [22]). Recall that the FFP-3 vector counts the occurrences of each of the 64 TTs by scanning the reading frame with moving window of three nucleotides to form a count vector of length 64. Therefore, this count vector is mathematically equivalent to the sum of 3 rows of our 3x64 TT count matrix. So the FFP-3 method can be viewed as performing a vector extracting function on the TUP matrix. However, the study showed (later) that the tree formed by the FFP-3 method yields a biologically inconsistent phylogenetic tree.
While choosing a simpler vector extracting function can provide more compact statistics, it may not retain or characterize certain key information contained in the summary matrix (and the original sequence). Consequently, the constructed phylogenetic tree may not be close to those trees with stronger biological support.
Results and discussion
Four phylogenic trees were constructed using vectors with (1) TUP-R1 (2), TUP-R2, (3) TUP-R3, and (4) TUP-All, respectively. Hierarchical correlation (city block, complete linkage) was used for clustering.
Constructed trees using various TUP vectors
The phylogenetic trees constructed using the four forms of vector extracting functions are shown in Figs. 1, 2, 3 and 4 respectively.

Phylogenic tree based on the TUP-R1 vector

Phylogenic tree based on the TUP-R2 vector

Phylogenic tree based on the TUP-R3 vector

Phylogenic tree based on TUP-All vector
All four trees show consistent and similar patterns. The lab strain E. coli K12-MG1655 and its clones BL21(DE3), W3110, and K12 (DH10B) are always grouped together. However, some wild-type strains, such as the Enterophathogeic strains O127-H6 and the commensal IAI1 strains, are also found to be closely associated with these lab-strains. This finding should not be surprising as the genes of most escherichial strains were the result of lifestyle adaptations [27]. Despite the genome reduction of these lab-strains, their overall genomic vectors might still be comparable to their wild-type strains. The four trees are all in accordance with current knowledge of evolution from the species taxa level. Before giving additional biological interpretations, we first explain why the phylogenetic signals in the vectors TUP-R1, TUP-R2, TUP-R3, and TUP-All are strong, despite the great variation in their numerical values.
The TUP-R1 vector is the distribution of the 64 non-overlapping codons, starting at its first reading frame of each gene, on the genome-wide DNA sequence. While TUP-R1 is a reasonable summary statistic for the DNA sequence, it cannot detect TT permutations because TT permutations do not change the distribution of the 64 codons. Likewise, the vectors of TUP-R2 and TUP-R3 are the distributions of the 64 TTs obtained from scanning the second and the third RFs, respectively. Note that the resulting count vectors are quite different due to the shift. Because the three RFs are essentially the same (long) DNA sequences, we would expect similar trees to be drawn even with three quite different vectors. On the other hand, the TUP-All vector contains more complete information and it can even detect TT permutations in the whole genome DNA sequence.
Biological interpretation of the constructed trees
As mentioned earlier, all four tress (Figs. 1, 2, 3 and 4 based on TUP-R1, TUP-R2, TUP-R3, and TUP-All, respectively) constructed are very similar to each other. Therefore, for biological interpretations of the constructed trees, we only discuss in the following the tree constructed by the TUP-R1 vectors as shown in Fig. 1. This tree correctively organizes the bacteria from the three subphyla according to their natural histories. Among the γ-Proteobacteria, all the Escherichia/Shigella species are grouped into one tight clade, which is in perfect agreement with the current views on these two genera [18]. E. fergusonii is the most remote member of this clade. The 4 strains of Salmonella are grouped into one tight clade and are closely associated with Escherichia. The correlation between the Escherichia/Shigella group and the Salmonella group is in line with the current view of their natural classification [19]. The 9 species of Yersinia form a tight group, with Y. enterocolitica being the most remote member of this group. This Yersinia clade is distinctly separated from the Escherichia/Salmonella group.
The 5 species of the Neisseriae are members of the β-Proteobacteria. They form a distinct branch but are more closely related to the γ-Proteobacteria. Although N. gonorrhoeae and N. meningitidis are often difficult to distinguish [23], the codon distributions of these two species are clearly distinguishable.
Within the α-Proteobacteria branch, all the Rickettsia species are grouped together. The placing of the Orientia as an extended family of the Rickettsia is in perfect agreement with the literature [24]. The placing of the two parasitic Wolbachia near the Rickettsia/Orientia branch is also in good agreement with the current phylogenetic assignment of this group of bacteria [16, 25, 26].
Comparison with the FFP-3 method
For the purpose of comparison, we also perform the grouping of bacteria based on the FFP-3 vector, a special case in [4]. Figure 5 is the tree constructed by the 56 FFP-3 vectors. This tree shows that the phylogenetic signals in the genome are much weaker than the phylogenetic signals in the protein-coding genes. Although the three subphyla could be distinguished by the nucleotide-triples ratios, their resolutions in separating bacterial groups are poor. Furthermore, it could not separate organisms at the lower taxa. For example, the Shigella strains are less similar to the Escherichia strains.

Phylogenic tree based on the FFP method with length 3
Unlike Figs. 1, 2, 3 and 4, Fig. 5 has the strain “E fergusonii ATCC 35469” (marked with a red dot) wrongly clustered within “E coli strains” in the constructed tree. As the FFP-3 vector is (essentially) the sum of three TUP vectors, it may dilute “key information” in DNA sequences. Thus it is very likely that the cause of the mis-classification could be attributed to the vector extracting function used in constructing the tree.
On the other hand, a statistic (e.g., TUP-R1, TUP-R2, TUP-R3, TUP-ALL, or FFP-3 vector) is more effective in classification if it is “less random” across the genes within the same species. Entropy is a popular measure for the randomness, hence it is suitable for comparing the performance of various classification variables. Next, we show theoretically and empirically that the FFP-3 method indeed has a higher entropy (more random) than all the TUP methods.
Comparing entropy among various methods
Let X be a random variable taking m possible values, t 1,t 2,…,t m , with P(X=t i )=p i for i=1,…,m. In this paper, m=64 and X represents the summary vector using TUP or FFP procedure.
The entropy associated with probability vector p=(p 1,p 2,…,p m )\(\left (\sum _{i=1}^{m} p_{i}=1\right)\) is
It is straightforward to show that \(\left (- \frac {\partial ^{2} H}{\partial p_{i} \partial p_{j}} \right)\) is a positive definite matrix, implying that H(p) is a concave function of p. Consequently, for any two probability vectors p and q and for 0<w<1, we have
for the mixture distribution of X (with probability vector p) and Y (with probability vector q) given by Z X+(1−Z)Y with P(Z=1)=w=1−P(Z=0).
Note that FFP-3 can be considered as the mixture distribution with equal weights of TUP-R1, TUP-R2, and TUP-R3. Based on this characterization, we have the following observations.
-
1.
The sample entropies calculated for TUP-R1, TUP-R2, and TUP-R3 are of similar magnitudes, which may explain their similar classification power and similar constructed trees.
-
2.
Since FFP-3 is the mixture distribution with equal weights of TUP-R1, TUP-R2, and TUP-R3, the entropy for FFP-3 is larger than the average entropy of the three TUPs. Thus FFP-3 has a higher entropy than at least one of the TUPs. Since all three TUPs have similar entropies, FFP-3 is expected to have a higher entropy than all of them. As mentioned earlier, using a more “random” statistic to represent a species is less likely to be a good characterization/classification of the given species. This may help to explain why the tree constructed by FFP-3 has less biological support than the tree constructed by using TUP-R1, TUP-R2, or TUP-R3.
-
3.
For the purpose of illustration, we consider two examples below. The first one is a real data example and the second one is a simple artificial example with an extreme case.
-
(a)
For the E coli K12 DH10B example shown earlier, the entropy for three reading frames, R1, R2, and R3, are 3.750678, 3.71317, and 3.859847, respectively. The entropy for FFP-3 is 3.995315, which is larger than the entropies of all three reading frames.
-
(b)
For an artificial example, we consider the DNA sequence of “ACTACTACTACTACTACTACT...”. The TUP-R1 will produce a probability vector with probability 1 concentrating at “ACT” and hence the entropy is 0. Similarly, TUP-R2 and TUP-R3 also have zero entropy with concentration values at “CTA” and “TAC”, respectively. On the other hand, FFP-3 will produce a probability vector with probability 1/3 concentrating at each of three possible values, “ACT”, “CTA”, and “TAC”; hence the entropy is log(3), obviously larger than the zero entropy of the three TUPs.
-
(a)
Proposed method for measuring “closeness between trees”
When the number of strains under study is large, it could be tedious to “visualize” the closeness of many variously constructed phylogenetic trees. We propose a numeric measure for the closeness between two trees. Let M i be the TUP matrix for strain S i ,d() be the distance function, and v() be the vector extracting function for the construction of the phylogenetic tree. Define a large vector (of size \({56 \choose 2}=1540\)) of pairwise distances between any two strains, say, S i and S j , as
For two different vector extracting functions, say, v 1() and v 2(), we can compute two vectors \(\mathbf {T}(_{v_{1}})\) and \(\mathbf {T}(_{v_{2}})\). If the resulting phylogenetic trees are similar to each other, the “distance” (again, in Euclidean distance or city block distance) between \(\mathbf {T}(_{v_{1}})\) and \(\mathbf {T}(_{v_{2}})\), \(d(\mathbf {T}(_{v_{1}}), \mathbf {T}(_{v_{2}}))\), should be small (and vice versa). Next, we use this proposed measure to compute the distance between each pair of the trees constructed.
Numeric results for “closeness between trees”
To evaluate the “closeness” among the trees, we use the current study as an example. Let v all ,v 1,v 2,v 3, and v FFP-3 be the vector extracting functions corresponding to TUP-All, TUP-R1, TUP-R3, TUP-R3, and FFP-3, respectively. Table 2 summarizes all the pairwise distances among the five trees constructed.
The distances \(d(\mathbf {T}(_{v_{all}}), \mathbf {T}(_{v_{i}}))\) for i=1,2,3 are 12.08,7.97,11.89, respectively, which are much smaller than the distance \(d(\mathbf {T}(_{v(_{all}})), \mathbf {T}(_{v_{\text {FFP-3}}})) (=108.36)\). The distances between \(\mathbf {T}(_{v_{\text {FFP-3}}})\) and the other three trees, \(\mathbf {T}(_{v_{1}})\), \(\mathbf {T}(_{v_{2}})\), and \(\mathbf {T}(_{v_{3}})\), are 117.82,110.49, and 96.74, respectively, which are also large. This is consistent with previous observation that the tree in Fig. 5, constructed using \(\mathbf {T}(_{v_{\text {FFP-3}}})\), is far different from the trees in Figs. 1, 2, 3 and 4, which have a stronger biological support.
Summary and extension
In this paper, we proposed a new alignment-free method for constructing a phylogenetic tree based only on the TUPs, the Trinucleotides Usage Profiles, of the genome-wide DNA sequences under study; and each TUP vector represents the (relative) frequency distribution of the 64 trinucleotides obtained by scanning over each of the DNA sequences using non-overlapping windows of length 3. Clearly, the TUP method is slightly more efficient computationally than the popular feature frequency profile FFP-k method with k=3 because the latter counts the frequency distribution for the overlapping windows of the same length. Computing efficiency, however, needs not be a key comparison criterion between these two methods because both are already very efficient when compared to alignment-based methods. Most importantly, we showed empirically and theoretically that the TUP method outperforms the FFP-3 method. In addition, the FFP method does not use the information about the starting of the reading frame, which is usually known. We also provided a numerical measure for comparing various trees constructed.
As pointed out by a reviewer, the dataset under study contains only prokaryotic genomes, which have much simpler structures compared to eukaryotic genomes. Because eukaryotic genomes are complicated by their introns and exons, the proposed method might not be suitable for eukaryotic genomes.
For a better classification result with FFP-k, a much larger value of k than 3 was recommended in [4] but with the tradeoff of the much larger number of possible categories, i.e., 4k. For example, the number of possible categories is 4096 for k=6 or 262144 for k=9. The FFP-6 or FFP-9 method is expected to provide a better classifier than the classifier based on the FFP-3 method. For a fair comparison, method FFP-6 or FFP-9 should be compared to its TUP counterpart, the “extended TUP” method (say, TUP-6 or TUP-9), that uses multiple consecutive trinucleotides of the same length. The “extended TUP” method could be useful when the number of species to be classified is huge. Based on the entropy theory provided in this paper, we expect that the classifier based on this multiple-TUP method would be superior to the classifier based on the corresponding FFP-k method.
References
Needleman S, Wunsch C. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970; 48(3):443–53.
Blaisdell B. A measure of the similarity of sets of sequences not requiring sequence alignment. PNAS. 1986; 83(14):5155–9.
Blaisdell B. Average values of a dissimilarity measure not requiring sequence alignment are twice the average of conventional mismatch counts requiring sequence alignment for a computer-generated model system. J Mol Evol. 1989; 29(6):538–47.
Sims G, Jun S, Wu G, Kim S. Alignment-free genome comparison with feature frequency profiles(FFP) and optimal resolutions. PNAS. 2009; 106(8):2677–82.
Jun S, Sims G, Wu G, Kim S. Whole-proteome phylogeny of prokaryotes by feature frequency profiles: an alignment-free method with optimal feature resolution. PNAS. 2010; 107(1):133–8.
Wu G, Jun S, Sims G, Kim S. Whole-proteome phylogeny of large dsDNA virus families by an alignment-free method. PNAS. 2009; 106(31):12826–31.
Hao B, Qi J. Prokaryote phylogeny without sequence alignment: from avoidance signature to composition distance. J Bioinforma Comput Biol. 2004; 2(1):1–19.
Wu T, Huang Y, Li L. Optimal word sizes for dissimilarity measures and estimation of the degree of dissimilarity between DNA sequences. Bioinformatics. 2005; 21(22):4125–32.
Wu T, Burke J, Davison D. A measure of DNA sequence dissimilarity based on the Mahalanobis distance between frequencies of words. Biometrics. 1997; 53(4):1431–9.
Edgar R. Local homology recognition and distance measures in linear time using compressed amino acid alphabets. Bioinformatics. 2004; 32(1):380–5.
Van Helden J. Metrics for comparing regulatory sequences on the basis of pattern counts. Bioinformatics. 2004; 20(3):399–406.
Deerwester S, Dumais S, Furnas W, Landauer T, Harshman R. Indexing by Latent Semantic Analysis. J Am Soc Inf Sci. 1990; 41(6):391–407.
Robinson D, Foulds L. Comparison of phylogenetic trees. Math Biosci. 1981; 53(1–2):131–47.
Sokal R, Rohlf F. The comparison of dendrograms by objective methods. Taxon. 1962; 11(2):33–40.
Nielsen J, Kristensen A, Mailund T, Pedersen C. A sub-cubic time algorithm for computing the quartet distance between two general trees. Algoritm Mol Biol. 2011; 6:15.
Hanage W, Fraser C, Spratt B. Fuzzy species among recombinogenic bacteria. BMC Biol. 2005; 3:6.
Escobar-Paramo P, Giudicelli C, Parsot C, Denamur E. The evolutionary history of Shigella and enteroinvasive Escherichia coli revised. J Mol Evol. 2003; 57(2):140–8.
Ochman H, Elwyn S, Moran N. Calibrating bacterial evolution. PNAS. 1999; 96(22):12638–43.
Yarza P, Ludwig W, Euzeby J, Amann R, Schleifer K-H, Glockner F, Rossell-Mra R. Update of the All-Species Living Tree Project based on 16S and 23S rRNA sequence analyses. Syst Appl Microbiol. 2010; 33(6):291–9.
Knapp J. Historical perspectives and identification of Neisseria and related species. Clin Microbiol Rev. 1988; 1(4):415–31.
Xu L, Kuo J, Liu J, Wong T. Bacterial phylogenetic tree construction based on genomic translation stop signals. Microb Inf Experimentation. 2012; 2:6.
Vinga S, Almeida J. Alignment-free sequence comparison - a review. 19. 2003; 4:513–23.
Tamura A, Ohashi N, Urakami H, Miyanura S. Classification of Rickettsia tsutsugamushi in a New Genus, Orientia gen. nov., as Orientia tsutsugamushi comb. nov. Int J Syst Bacteriol. 1995; 45(3):589–91.
Pfarr K, Foster J, Slatko B, Hoerauf A, Eisen J. On the taxonomic status of the intracellular bacterium Wolbachia pipientis: should this species name include the intracellular bacteria of filarial nematodes?Int J Syst Evol Microbiol. 2007; 57(8):1677–8.
Garzon M, Wong T. DNA chips for species identification and biological phylogenies. Nat Comput. 2011; 10(1):375–89.
Ibrahim A, Goebel B, Liesack W, Griffiths M, Stackebrandt E. The phylogeny of the genus Yersinia based on 16S rDNA sequences. FEMS Microbiol Lett. 1993; 114(2):173–7.
White A, Sibley K, Sibley C, Wasmuth J, Schaefer R, Surette M, Edge T, Neumann N. Intergenic sequence comparison of Escherichia coli isolates reveals lifestyle adaptation but not host specificity. Appl Environ Microbiol. 2011; 77(21):7620–32.
Acknowledgement
We acknowledge the usage of high performance computing facility (HPC) provided by the University of Memphis in USA for this research.
Declarations
This article has been published as part of BMC Bioinformatics Volume 17 Supplement 13, 2016: Proceedings of the 13th Annual MCBIOS conference. The full contents of the supplement are available online at http://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-17-supplement-13.
Funding
This work and its publication were partially supported by the Ministry of Science and Technology of Taiwan, ROC, Grant No. MOST-105-2811-M-009-002, MOST-105-2118-M-009-003-MY2, MOST-105-2627-M-009-005, and MOST-105-2118-M-009-005.
Availability of data and material
The links to the data sets and the software used to support the results of this work are included in the article.
Authors’ contributions
Study Design: SC, LYD, TYW. Model Development: SC, LYD, TYW. Analysis: SC, LYD, DB, JJHS. Manuscript Preparation: SC, LYD, JJHS, DB, BM, HHSL, TYW. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver(http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, S., Deng, LY., Bowman, D. et al. Phylogenetic tree construction using trinucleotide usage profile (TUP). BMC Bioinformatics 17 (Suppl 13), 381 (2016). https://doi.org/10.1186/s12859-016-1222-3
Published:
DOI: https://doi.org/10.1186/s12859-016-1222-3