
Abstract
Background
DNA methylation profiling of pediatric brain tumors offers a new way of diagnosing and subgrouping these tumors which improves current clinical diagnostics based on histopathology. We have therefore developed the MethPed classifier, which is a multiclass random forest algorithm, based on DNA methylation profiles from many subgroups of pediatric brain tumors.
Results
We developed an R package that implements the MethPed classifier, making it easily available and accessible. The package can be used for estimating the probability that an unknown sample belongs to each of nine pediatric brain tumor diagnoses/subgroups.
Conclusions
The MethPed R package efficiently classifies pediatric brain tumors using the developed MethPed classifier. MethPed is available via Bioconductor: http://bioconductor.org/packages/MethPed/
Similar content being viewed by others


Background
Carcinogenesis involves changes in gene expression that results in tumor specific gene and protein signatures. Such signatures have been used to classify different subtypes of cancers. Gene expression is partly regulated by the methylation state of CpG islands. Cancer tissue is characterized by an increased variability in DNA methylation patterns. DNA methylation profiling has been reported as a robust method to classify and subgroup tumors of different origin [1]. For most pediatric brain tumor diagnoses, methylation profiling can divide the tumors into clinically relevant subgroups reflecting the diverse biology of the different subtypes which further highlights the need for specific therapeutic strategies to target different subgroups. With the increased knowledge about specific brain tumor subgroups and the development of targeted therapy for different entities, it is essential to quickly and accurately determine the correct diagnosis for pediatric brain tumor patients. The most popular and commonly used platform for genome-wide methylation profiling is the Illumina Infinium Human Methylation 450 BeadChip arrays. These arrays profile ~485,000 CpG sites and have been used by the Cancer Genome Atlas Project (TCGA) and in numerous studies of pediatric brain tumors. A correct diagnosis is vital for determining the appropriate treatment protocol for a specific patient, to select the right patients for clinical trials investigating novel therapy for specific diagnoses and subgroups and for basic researchers to be able to draw correct conclusions from experiments. We therefore developed the MethPed classifier [2], which is a multiclass random forest algorithm [3], based on DNA methylation profiles from many subgroups of pediatric brain tumors. We have now developed an R package that uses this method, making it easily available and accessible.
Implementation
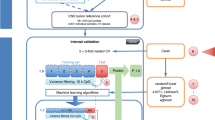
The MethPed classifier was developed using the Random Forest (RF) algorithm [3] for robust classification of unknown brain tumor samples into subtypes as described in Danielsson et al. [2]. Briefly, the RF algorithm was applied on beta values which are the estimate of methylation levels (between 0 and 1 with 0 being unmethylated and 1 fully methylated) using the ratio of intensities between methylated and unmethylated alleles generated by the Illumina Infinium HumanMethylation 450 BeadChip array. A training probe pool of 900 methylation sites that showed the highest predictive power (AUC values) in a large number of regression analyses was selected from 472 clinically diagnosed brain tumor cases available on GEO after necessary data cleaning and KNN imputation of missing values. The RF algorithm was then applied to classify unknown samples based on the selected training probe pool. See Fig. 1 for a summary of the workflow for the MethPed classifier and R package.

Implementation and workflow of the MethPed classifier and package
The MethPed classifier can be accessed through the ‘MethPed’ package that can be downloaded from Bioconductor, a repository for bioinformatics related applications. The ‘MethPed’ package includes the MethPed classifier and an example data set of two tumors. The example data can be read into the R computing environment with the help of the data function after installing the package.
Data for running MethPed is generated by the Illumina Infinium HumanMethylation 450 BeadChip arrays. Beta values for two samples (Tumor A and Tumor B) are provided with the ‘MethPed’ package as an example [2]. This data has no missing values. If missing values exist in a data set, the impute package can be used for missing value imputation, according to the MethPed vignette in Bioconductor.
Results and discussion
The MethPed analysis starts with loading the data, checking for missing values in the data file and thereafter runs through the classification. Error rate of the prediction is estimated and the probability that a sample belongs to one of nine tumor diagnoses/subgroups is given. In the current version of MethPed the following groups are included; glioblastoma (GBM), pilocytic astrocytoma, medulloblastoma (Wnt, Shh, group 3 and group 4), diffuse intrinsic pontine glioma (DIPG), ependymoma and embryonal tumor with multilayered rosettes (ETMR). These include the most common diagnoses and subgroups but not all. The robustness of a classifier is highly dependent on the accuracy of the training data and therefore we choose not to build in groups with limited data avaliable. With the MethPed classifier, the probability that a tumor sample belongs to a specific tumor group is presented (Fig. 2a), or if preferred, the group with the maximum probability leaving zero to the other tumor groups by supplying an extra parameter ‘prob = FALSE’ in the classifier (Fig. 2b). It should be note that for tumors belonging to diagnoses that are not included in the MethPed classification, these will be classified as inconclusive (with low probabilities of belonging to any of the groups) or to the most similar tumor form that is present in the classifier. For more information, see Danielsson et al. [2].

Bar plots on the diagnosis prediction of the two test samples. a Classification probability of a sample belonging to each of the pediatric brain tumor diagnoses currently included in MethPed and (b) Maximum classification probability of a specific tumor group for each sample
The conditional probability matrix of the classification from the MethPed output can be observed by the ‘summary’ command in R. For visualization of the prediction, bar plots can be generated by using the ‘plot’ command (Fig. 2). If missing probes from the sample compared with the training data included with the package exist, these can be observed by the ‘probeMis’ command.
MethPed is currently the only publically available tool for classification of pediatric brain tumors. The use of methylation profiling for classification of these tumors adds new and important knowledge in the clinical setting for choosing the optimal care and treatment of these patients and will therefore likely complement histopathological diagnoses in the near future [1, 2].
Conclusions
The MethPed R package can be used to efficiently classify pediatric brain tumors using DNA methylation profiles generated by the Illumina 450 K methylation arrays.
Abbreviations
DIPG, diffuse intrinsic pontine glioma; ETMR, embryonal tumor with multilayered rosettes; GBM, glioblastoma; TCGA, the Cancer Genome Atlas Project.
References
Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DT, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I, et al. New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell. 2016;164:1060–72.
Danielsson A, Nemes S, Tisell M, Lannering B, Nordborg C, Sabel M, Carén H. MethPed: a DNA methylation classifier tool for the identification of pediatric brain tumor subtypes. Clin Epigenetics. 2015;7:62.
Breiman L. Random forests. Mach Learn. 2001;45:5–32.
Funding
This work was supported by the Swedish Cancer Society; the Swedish Children’s Cancer Society; the Swedish Research Council; the Swedish Society for Medical Research; a Marie Curie CIG from the EU’s Seventh Framework Programme (FP7) and the Wenner-Gren foundation.
Availability of data and materials
Data used for building MethPed is available at GEO (accession numbers: GSE50022, GSE55712, GSE36278, GSE52556, GSE54880, GSE45353 and GSE44684).
Project home page: http://bioconductor.org/packages/MethPed/
Operating systems: any operating system supporting R
Programming language: R
Other requirements: working R installation, with Bioconductor version 3.3
Any restriction to use by non-academics: none.
Authors’ contributions
AD and HC initiated the study. MTA developed the R package under guidance from SN, AD and HC. MTA and HC wrote the manuscript. All authors read, contributed to and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The inclusion of the two test samples were approved by the Regional Ethics Committee in Gothenburg, EPN, Dnr 604–12. Samples were obtained after signed informed consent from the parents of children who underwent surgery at the Sahlgrenska University Hospital.
Author information
Authors and Affiliations
Corresponding author
Additional information
Mohammad Tanvir Ahamed and Anna Danielsson are joint first authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ahamed, M.T., Danielsson, A., Nemes, S. et al. MethPed: an R package for the identification of pediatric brain tumor subtypes. BMC Bioinformatics 17, 262 (2016). https://doi.org/10.1186/s12859-016-1144-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-016-1144-0