
Abstract
Background
The well-known Genome-Wide Association Studies (GWAS) had led to many scientific discoveries using SNP data. Even so, they were not able to explain the full heritability of complex diseases. Now, other structural variants like copy number variants or DNA inversions, either germ-line or in mosaicism events, are being studies. We present the R package affy2sv to pre-process Affymetrix CytoScan HD/750k array (also for Genome-Wide SNP 5.0/6.0 and Axiom) in structural variant studies.
Results
We illustrate the capabilities of affy2sv using two different complete pipelines on real data. The first one performing a GWAS and a mosaic alterations detection study, and the other detecting CNVs and performing an inversion calling.
Conclusion
Both examples presented in the article show up how affy2sv can be used as part of more complex pipelines aimed to analyze Affymetrix SNP arrays data in genetic association studies, where different types of structural variants are considered.
Similar content being viewed by others


Background
Genome-Wide Association Studies (GWAS) interrogate a large number of genetic variants with high-throughput technologies using single nucleotide polymorphisms (SNPs). Up to now, GWAS have led to many scientific discoveries including genes and gene variants related to cancer [1–4], asthma [5–7] or obesity [8, 9] among others. Nonetheless, SNPs have explained relatively little of the total heritability of complex diseases [10, 11]. In order to overcome this difficulty, researchers are also analyzing other structural genomic variants (SVs) such as copy number variants (CNVs) [12–14], inversions [15, 16] or chromosomal rearrangements present in mosaicism [17–19]. This has been possible due to the efforts made by scientific community in developing new tools to detect SV using existing SNP array data [20–22].
Over the last few years, commercial enterprises such as Affymetrix and Illumina, have produced high-density SNP arrays that made possible to genotype many markers in a single assay. These arrays are excellent tools to perform GWAS not only with SNPs but also with common and rare SVs. An example of it is Affymetrix CytoScan family, that includes a high-density array (CytoScan HD) and a light version array (CytoScan 750K) [23, 24]. This family of arrays was designed to provide a genome-wide overview of the whole genome since they include markers for constitutional and cancer genes and OMIM and RefSeq genes.
Affymetrix provides a wide range of software to analyze the data obtained from their arrays. The most common software to analyze CytoScan data is called Chromosome Analysis Suite (ChAS) [25]. Despite the benefits, the usage of ad hoc software from Affymetrix has two main limitations. On one hand, while the raw data can be processed in a high throughput way, the analysis of the results is recommended to be performed by groups of three subjects. On the other hand, the set of available analysis is reduced to the algorithms included in the software, so no other custom-functionality can be added to help researchers to perform downstream analyses.
In order to overcome these drawbacks an R package called affy2sv has been created. This R package improves the advantages provided by ChAS incorporating new functionalities that make possible the analysis of CytoScan data using other existing R packages (MAD [26], R-GADA [27], snpStats [28], invClust [29, 30]) and external software (PLINK [31], PennCNV [32–34]), as well as data visualization. Therefore, affy2sv will facilitate the analysis of CytoScan data in SNPs, CNVs, mosaicism or inversion association studies using pipelines under R environment.
In this article, we illustrate affy2sv's performance by analyzing two different sets of SNP array generated with CytoScan platform. The first set includes population of two different locations: 429 subjects from general population of Toronto and 198 subjects from Nijmegen (Dataset A). The second set includes 315 subjects diagnosed with intellectual disability (Dataset B). Dataset A is used to illustrate how to compare genetic variants between two general populations under GWAS framework and how to detect mosaicism events. Dataset B is used to illustrate how to detect potentially pathogenic CNVs and how to perform inversion calling. The result obtained from the inversion analysis is the genotype of a well-known inversion located at chromosome 8p23.1 [35].
Implementation
affy2sv is implemented as a R package freely available from its web page [36] and through CREAL-installer [37]. affy2sv is based on standard CRAN and Bioconductor classes allowing for full flexibility, modularity and integration with other R packages.
Input data
affy2sv is compatible with the newest Affymetrix SNP array CytoScan HD/750k, but it also accepts Genome-Wide SNP 5.0/6.0 and Axiom arrays. It works with the raw data files, known as .CEL files. Internally, affy2sv uses the package CRLMM [38–41] to extract some measures [genotype, Log R Ratio (LRR) and B Allele Frequency (BAF)] from Genome-Wide SNP 5.0/6.0 raw data. To deal with Axiom and CytoScan arrays and to extract the homologous measures (genotype, allele peaks, allele intensities, LRR and BAF), affy2sv uses the Affymetrix Power Tools (APT) [42].
Output data
affy2sv can be used to process .CEL files and to generate R objects and files compatibles with snpStats, MAD, R-GADA, PLINK, and PennCNV. These R packages and programs are specifically designed to perform GWAS, analyze mosaicism and CNVs, respectively.
The R object generated for snpStats is called SnpMatrix Container. This object contains a MAP and a SnpMatrix. The MAP is a data.frame that includes an annotation for each SNP (SNP's name, chromosome, cM, position and alleles). The genotypes are stored in a SnpMatrix object. The file compatible with MAD and R-GADA is a tabular file for each subject containing the BAF, the LRR and the genotype of each SNP (SNP's name, chromosome, position, LRR, BAF and genotype). The compatibility with PLINK is reached creating a TPED file (transposed format), which contains the chromosome, SNP's name, genetic distance and position, followed by all the genotype-pairs. To work with PennCNV several files are required. The tools manual, available on its web page [43], explains its composition and how to generate them. affy2sv creates the a file that contains the LRR, BAF and genotype, called signal intensity file.
Method
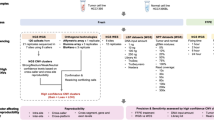
affy2sv is a set of R functions used to process a certain type of raw data and generate a specific output file. There are two steps to process the data from Affymetrix CytoScan arrays with affy2sv: 1) read raw data and calculate measures (genotype, LRR and BAF) 2) generate a specific output. This two-step process is illustrated in Fig. 1a. The figure shows that intermediate files are created with the first step. Also that these intermediate files are used as input for the second step and used to generate a specific output.

Schema of the application of affy2sv to analyze CytoScan data. Part A of the figure shows the work-flows available in the R package affy2sv. These work-flows are composed by two steps: generate intermediate files and generate specific output. First step us done using the function Cyto2APT. The seconds step is done using the functions Cyto2Mad and Cyto2SnpMatrix. Part B of the figure shows the pipelines used to perform the two studies detailed in the article. The two CytoScan HD populations were pre-process using affy2sv and then analyzed using different tools
Step 1: Process raw data and get BAF, LRR and genotype
This step is performed using the function Cyto2APT. Cyto2APT is in charge to call the APT. These tools require a series of library and annotation files depending on the array-technology used. These files can be downloaded from the Affymetrix Library [44] and from the Affymetrix annotation [45] web pages. The user needs to download the files corresponding to their own data's technology. Later, the function APTparam creates a required object that indicates the correct system call to deal with apt-copynumber-cyto from APT. The following code illustrates the use of a standard call:

This code indicates that the raw .CEL files are located at /home/cydata. The argument output.path indicates where the intermediate files will be saved. In analysis.path is indicated the path where all the library and annotation files are stored. All the other arguments refer to the library and annotation files required by the function. These argument define the technology used in the array, the distribution of the probes, the name of each probe (and the related SNP) and others.
We thought these technical arguments could be hidden, but leaving them unmasked would allow the user to have more than one library (for example, one for CytoScan HD and another one for CytoScan 750K) or more than one version of a single library. The term intermediate files is used to refer to the files generated by Cyto2APT. These files are, in fact, the plain text version of the common .cychp files generated by apt-copynumber-cyto. So, at the end of this step, the intermediate files generated by Cyto2APT are the same files that could be obtained by using ChAS. This is because the system call to apt-copynumber-cyto generated by affy2sv is the recommended by Affymetrix in the tool's manual [46, 47].
In order to increase the versatility of the package affy2sv, we also make possible to create a personalized system call to apt-copynumber-cyto through APTparam. This can be done by setting the argument type from standard to custom . Then, it is needed to fill the argument param with a string containing all the arguments for apt-copynumber-cyto (arguments like cel.list, output.path… must not to be set on APTparam but in the string to param). An example of how to do it is available in the supplementary material (Additional file 1).
Once APTparam set up the arguments, Cyto2APT will manage with apt-copynumber-cyto to create the intermediate files. The following code is an example of how to use Cyto2APT:

Step 2: Generate a specific output
The R package affy2sv can create objects or files compatible with MAD, R-GADA, snpStats, PLINK and PennCNV. This is done using Cyto2Mad or Cyto2SnpMatrix depending on the desired output.
The function Cyto2Mad creates the files compatible with MAD, R-GADA and with PennCNV. The following code shows how to create the files compatible with MAD:

The first argument, cychp.files, indicates where the intermediate files are stored (in this case it takes the value /home/tmp). The second one, output.name, indicates where the files compatible with MAD will be saved (they will be saved into /home/mad). The third argument specifies the output's format (MAD). The last argument, annotation.file is filled with the path to the annotation file (in CSV format), provided by Affymetrix.
To create the files compatible compatible with PennCNV only the value of output.type needs to be changed from mad to penncnv:

The function Cyto2SnpMatrix is in charge of creating a SnpMatrix Container, an object compatible with the R package snpStats. An example of how this function is used:

The argument cychp.files (/home/tmp) takes the path where the intermediate files generated with Cyto2APT are stored. annotation.file is filled with the path to the annotation file (in CSV format), provided by Affymetrix. The output.type is set to snpmatrix to generate the SnpMatrix Container.
Setting the value of output.type to plink, and adding and filling the argument output.name with a valid directory, Cyto2SnpMatrix creates a file compatible with PLINK:

Visualization
affy2sv can create a series of plots to help to perform a quality control process on CytoScan populations. The function Cyto2QCView allows to create three type of plots: 1) a plot to see how a single probe was genotyped for all the population 2) a plot, for a single individual, where the intensities of all its probes are shown 3) a plot, for a single individual, that displays the strength and the contrasts of all its probes. The following code shows how Cyto2QCView can be used:

Results and discussion
To show how affy2sv can be integrated in pipelines developed in R, two different datasets have been analyzed. Figure 1b shows a schema of these two analysis. Dataset A is used to illustrate how to perform a GWAS using CytoScan data. The same data is used to show how to detect genetic mosaicisms. Dataset B is used to describe how to analyze large CNVs and how to genetoype the well-known 8p23.1 inversion.
Dataset A includes a set of two populations. 429 subjects corresponding to Toronto general population that comes from The Ontario Population Genomics Platform between the ages of 20 and 79 [48]. Dataset A also includes 198 samples from Nijmegen coming from a full set of 1000 subjects of a previous study [49]. The GWAS consisted in comparing the genotypes between the two populations. We aimed to find the top 10 SNPs that best differentiate the two poplulations. Table 1 shows the results found by using combined efforts of affy2sv and snpStats. Figure 2 shows the Manhattan plot result of this analysis. The complete code to perform this GWAS, including the quality control performed over the SNPs, can be found in supplementary material (Additional file 1). We observe that there is an SNP that passes genome-wide significance level of p-value < 10 −8.

Manhattan plot result of comparing the two populations in Dataset A. Manhattan plot result of the GWAS study comparing general population from Nijmegen versus Toronto (Dataset A), performed with affy2sv and snpStats. It shows the log10 of the p-value given to each SNP in chromosome 1 to X
The mosaicism study in Dataset A was done by using the R package MAD (Additional file 1). Table 2 shows the three unique events found in the entire dataset. Figure 3 shows two of the three events found in the Toronto population. They correspond to a mosaic terminal deletion of 22 Mb at chromosome 8p and a mosaic of 35 Mb gain at terminal of chromosome 18q of the same individual.

Two of the three mosaic events detected by MAD of Dataset A after being pre-processed by affy2sv. The plots show two mosaic events found by MAD after the pre-process Dataset A with affy2sv Each plot represents the whole chromosome where the mosaic event is located. The black dots show the value of the LRR for each single SNP while the red points show the value of the BAF; placing at the top the ones corresponding to AA allele (with a value close to 1), at the middle the ones corresponding to AB allele (with a value around 0.5) and at the bottom the ones corresponding to BB allele (with a value close to 0). Part A shows a 33 Mb mosaic deletion at terminal 8p. Part B shows a 25 Mb mosaic duplication at terminal 18q. The presence of both events in the same sample (from Toronto general population) indicated that the individual carries an unbalanced chromosomal translocation (8p; 18q) in a proportion of cells
Dataset B includes 315 subjects with intellectual disability from the Biobank of the Galician Foundation of Genomics Medicine (the use of the samples for this purpose was authorized by the Ethical Committee of the institution). For the CNV study the R package R-GADA was used to detect regions with copy gains and losses (Additional file 1). The detected CNVs can be seen in Table 3. Two of these events are represented in Fig. 4a and Fig. 4b. The plots show an interstitial gain in chromosome 7q and an interstitial loss in chromosome 8p in two subjects diagnosed with intellectual disability. Finally, Dataset B is used to genotype 8p23.1 inversion. This was performed using the R package invClust (Additional file 1). The classification of each individual of the population, according to the inversion haplotypes, can be seen in Fig. 4c. Genotype frequencies were: 87 for the allele I/I, 197 to the NI/I and 61 to NI/NI (being I the inverted allele and NI the non-inverted). The inversion appears with a frequency of 46 % in Dataset B population, similar than in general population.

Two CNV events found in the Dataset B population (diagnosed with intellectual disability) with R-GADA and calling of 8p23.1 inversion in the same population with invClust. Part A shows a subject from Dataset B having an interstitial gain on chromosome 7q. The black dots show the value of the LRR for each single SNP while the red points show the value of the BAF; placing at the top the ones corresponding to AA allele (with a value close to 1), at the middle the ones corresponding to AB allele (with a value around 0.5) and at the bottom the ones corresponding to BB allele (with a value close to 0). Part B shows another subject from the same Dataset B having a interstitial loss on chromosome 8p. Part C Shows the calling of the inversion 8p23.1 in the entry dataset. The three groups correspond to each genotype; being the blue points the individuals that contain the inversion in both alleles, the green corresponds to the heterozygous individuals and the red ones are the individuals without the inversion. The plot is obtained after performing a MDS reduction over the population. 2-D density curves indicate the probability of belonging to each genotype being the more closed circles the highest probability
The R package affy2sv includes a function to perform a simple but visual quality control on CytoScan samples. A plot where a single SNP is displayed for all samples can be created (Fig. 5a). Another plot for allele intensities (Fig. 5b) or a plot that shows the strength versus the contrast of each probe (Fig. 5c). See Additional file 1.

The three type of plots affy2sv can draw on CytoScan samples to perform a visual QC. Plot A shows the log2 of the intensities of both alleles for a single SNP across all the population. In the case, a random probe (A-4DTYM) was selected and drawn across the population diagnosed with intellectual disability (Dataset B). Plot B shows the values corresponding to the log2 of the intensity of both alleles for all the probes in a random subject (3C136, from Dataset B). Plot C draws the strength and the contrast of all the probes for a random individual (3C136), being the strength log(A + B) and contrast (A-B)/(A + B)
Conclusion
affy2sv is an R package to pre-process raw .CEL files from Affymetrix CytoScan HD and 750k arrays (also the old SNP arrays called Genome-Wide SNP 5.0/6.0 and Axiom). The package can be used to create a wide range of output files and object compatibles with other R packages, like snpStats or MAD, and external software, like PLINK and PennCNV, used in genetic structural variants studies.
Availability & requirements
-
Package's name: affy2sv
-
Package's state: affy2sv 1.0.12 with APT 1.16.1
-
Package's web page: affy2sv is available at Bioinformatic Research Group in Epidemiology (BRGE - CREAL) software page http://www.creal.cat/brge.htm. Also at its own page on bitbucket https://bitbucket.org/brge/affy2sv.
-
Package's manual: The package comes with its standard R documentation. A web page manual is available at the packages own page on bitbucket https://bitbucket.org/brge/affy2sv/wiki.
-
Package's requirements:
-
○ operating systems: Multiplatform (Windows, GNU/Linux and MAC OS)
-
○ r dependence: R (> = 3.0.0), snpStats, crlmm, oligo, oligoClasses, VanillaICE, SNPchip, genomewidesnp6Crlmm, genomewidesnp5Crlmm, ff, pd.genomewidesnp.6, pd.genomewidesnp.5, stringr, biomaRt, ggplot2, gtable, grid, data.table, Biobase, parallel, methods
-
○ external dependences: python 2.7, numpy (> = 1.7), pandas
-
-
Programming language: R, Python and C/C++
-
License: GPL-2
-
Any restrictions to use by non-academics: No restrictions to use affy2sv, check the license for APT at its own web page.
References
Chih-yu Chen, I-Shou C, Chao AH and Wyeth WW On the identification of potential regulatory variants within genome wide association candidate SNP sets. BMC Medical Genomics. 2014; doi:10.1186/1755-8794-7-34.
Barrdahl M, Canzian F, Joshi AD, Travis RC, Chang-Claude J, Auer PL, et al. Post-GWAS gene-environment interplay in breast cancer: results from the Breast and Prostate Cancer Cohort Consortium and a meta-analysis on 79 000 women. Hum Mol Genet. 2014. doi:10.1093/hmg/ddu223.
Na L , Ping Z, Jian Z, Jieqiong D, Hongchun W, Wei L, et al. A Polymorphism rs12325489C>T in the LincRNA-ENST00000515084 Exon Was Found to Modulate Breast Cancer Risk via GWAS-Based Association Analyses. PLoS One. 2014. doi:10.1371/journal.pone.0098251.
Johnson ME, Schug J, Wells AD, Kaestner KH, Grant SF. Genome-Wide Analyses of ChIP-Seq Derived FOXA2 DNA Occupancy in Liver Points to Genetic Networks Underpinning Multiple Complex Traits. J Clin Endocrinol Metab. 2014. doi:10.1210/jc.2013-4503.
Melén E, Granell R, Kogevinas M, Strachan D, Gonzalez JR, Wjst M, et al. Genome-wide association study of body mass index in 23 000 individuals with and without asthma. Clin Exp Allergy. 2013. doi:10.1111/cea.12054.
Myers RA, Scott NM, Gauderman WJ, Qiu W, Mathias RA, Romieu I, et al. Genome-wide interaction studies reveal sex-specific asthma risk alleles. Hum Mol Genet. 2014. doi:10.1093/hmg/ddu222.
Castro-Giner F, Kogevinas M, Imboden M, de Cid R, Jarvis D, Mächler M, et al. Joint effect of obesity and TNFA variability on asthma: two international cohort studies. Eur Respir J. 2009. doi:10.1183/09031936.00140608.
González JR, Estévez MN, Giralt PS, Cáceres A, Pérez LM, González-Carpio M, et al. Genetic risk profiles for a childhood with severely overweight. Pediatr Obes. 2013. doi:10.1111/j.2047-6310.2013.00166.x.
González JR, Cáceres A, Esko T, Cuscó I, Puig M, Esnaola M, et al. A common 16p11.2 inversion underlies the joint susceptibility to asthma and obesity. Am J Hum Genet. 2014. doi:10.1016/j.ajhg.2014.01.015.
Maher B. Personal genomes: The case of the missing heritability. Nature. 2008. doi:10.1038/456018a.
Gusev A, Bhatia G, Zaitlen N, Vilhjalmsson BJ, Diogo D, Stahl EA, et al. Quantifying Missing Heritability at Known GWAS Loci. PLoS Genetics. 2013. doi:10.1371/journal.pgen.1003993.
Harrison SM, Granberg CF, Keays M, Hill M, Grimsby GM, Baker LA. DNA Copy-Number Variations in 46,XY Disorders of Sex Development. J Urol. 2014. doi:10.1016/j.juro.2014.06.040.
Sehn JK, Abel HJ, Duncavage EJ. Copy number variants in clinical next-generation sequencing data can define the relationship between simultaneous tumors in an individual patient. Exp Mol Pathol. 2014. doi:10.1016/j.yexmp.2014.05.008.
Lee HW, Seol HJ, Choi YL, Ju HJ, Joo KM, Ko YH, et al. Genomic copy number alterations associated with the early brain metastasis of non-small cell lung cancer. Int J Oncol. 2012. doi:10.3892/ijo.2012.1663.
Cartwright IM, Genet MD, Fujimori A, Kato TA. Role of LET and chromatin structure on chromosomal inversion in CHO10B2 cells. Genome Integrity. 2014. doi:10.1186/2041-9414-5-1.
Fouet C, Gray E, Besansky NJ, Costantini C. Adaptation to aridity in the malaria mosquito Anopheles gambiae: chromosomal inversion polymorphism and body size influence resistance to desiccation. PloS One. 2012. doi:10.1371/journal.pone.0034841.
Frank SA. Somatic Mosaicism and Disease. Curr Biol. 2014. doi:10.1016/j.cub.2014.05.021.
Machiela MJ, Chanock SJ. Detectable clonal mosaicism in the human genome. Semin Hematol. 2013. doi:10.1053/j.seminhematol.2013.09.001.
Valind A, Pal N, Asmundsson J, Gisselsson D, Mengelbier LH. Confined trisomy 8 mosaicism of meiotic origin: a rare cause of aneuploidy in childhood cancer. Genes Chromosomes Cancer. 2014. doi:10.1002/gcc.22173.
Pique-Regi R, Cáceres A, González JR. R-Gada: a fast and flexible pipeline for copy number analysis in association studies. BMC Bioinformatics. 2010. doi:10.1186/1471-2105-11-380.
González JT, Rodríguez-Santiago B, Cáceres A, Pique-Regi R, Rothman N, Chanock SJ, et al. A fast and accurate method to detect allelic genomic imbalances underlying mosaic rearrangements using SNP array data. BMC Bioinformatics. 2011. doi:10.1186/1471-2105-12-166.
Cáceres A, Sindi SS, Raphael BJ, Cáceres M, González JR. Identification of polymorphic inversions from genotypes. BMC Bioinformatics. 2012. doi:10.1186/1471-2105-13-28.
Affymetrix “Data Sheet: The CytoScan® HD Cytogenetics Solution” http://media.affymetrix.com/support/technical/datasheets/cytoscan_hd_datasheet.pdf. Accessed April 7, 2015.
Affymetrix “Data Sheet: The CytoScan® 750K Cytogenetics Solution” http://media.affymetrix.com/support/technical/datasheets/cytoscan750k_datasheet.pdf. Accessed April 7, 2015.
Affymetrix, “Chromosome Analysis Suite (ChAS)” [Computer Software] http://www.affymetrix.com/support/learning/training_tutorials/chromosome_analysis/chas.affx. Accessed May 1, 2015.
Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012. doi:10.1038/ng.2270.
Gonzalez JR et al. “gada: Genome Alteration Detection Algorithm (GADA)” [Computer Software] http://R-Forge.R-project.org/projects/gada. Accessed April 7, 2015.
David Clayton “snpStats: SnpMatrix and XSnpMatrix classes and methods” [Computer Software] http://www.bioconductor.org/packages/release/bioc/html/snpStats.html. Accessed April 7, 2015.
Cáceres A, González JR. Following the footprints of polymorphic inversions on SNP data: from detection to association tests. NAR. 2015. doi:10.1093/nar/gkv073.
Cáceres A et al. “invClust R package” [Computer Software] http://www.creal.cat/jrgonzalez/software.htm#ancla-invClust. Accessed April 7, 2015.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. American Journal of Human Genetics. 2007. doi:10.1086/519795.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Research. 2007. doi:10.1101/gr.6861907.
Diskin SJ, Li M, Hou C, Yang S, Glessner J, Hakonarson H, et al. Adjustment of genomic waves in signal intensities from whole-genome SNP genotyping platforms. NAR. 2008. doi:10.1093/nar/gkn556l
Wang K, Chen Z, Tadesse MG, Glessner J, Grant SFA, Hakonarson H, et al. Modeling genetic inheritance of copy number variations. NAR. 2008. doi:10.1093/nar/gkn641.
Maximilian PA, Horswell SD, Hutchison CE, Speedy HE, Yang X, Liang L, et al. The origin, global distribution, and functional impact of the human 8p23 inversion polymorphism. Genome Res. 2012. doi:10.1101/gr.126037.111.
Hernandez-Ferrer C et al. “affy2sv: A tool for pre-processing Affymetrix SNP array data” [Computer Software] https://bitbucket.org/brge/affy2sv/wiki/Home. Accessed April 7, 2015.
BRGE (CREAL) “Software Development – BRGE (CREAL)” http://www.creal.cat/jrgonzalez/software.htm. Accessed April 7, 2015.
Carvalho BS, Louis TA, Irizarry RA. Quantifying uncertainty in genotype calls. Bioinformatics. 2010. doi:10.1093/bioinformatics/btp624.
Ritchie ME, Carvalho BS, Hetrick KN, Tavaré S, Irizarry RA. R/Bioconductor software for Illumina's Infinium whole-genome genotyping BeadChips. Bioinformatics. 2009. doi:10.1093/bioinformatics/btp470.
Scharpf RB, Ruczinski I, Carvalho B, Doan B, Chakravarti A, Irizarry RA. A multilevel model to address batch effects in copy number estimation using SNP arrays. Bioinformatics. 2011. doi:10.1093/biostatistics/kxq043.
Scharpf RB, Irizarry RA, Ritchie ME, Carvalho B, Ruczinski I. Using the R Package crlmm for Genotyping and Copy Number Estimation. Journal of Statistical Software. 2011;40(12):1–32.
Afymetrix “Affymetrix Power Tools” [Computer Software] http://www.affymetrix.com/estore/partners_programs/programs/developer/tools/powertools.affx. Accessed April 7, 2015.
Wang K “PennCNV Input File Formats” [Computer Software] http://www.openbioinformatics.org/penncnv/penncnv_input.html.
Affymetrix “Affymetrix Library Files” [webpage] http://www.affymetrix.com/support/technical/libraryfilesmain.affx. Accessed April 7, 2015.
Affymetrix “Affymetrix Annotation Files” [webpage] http://www.affymetrix.com/support/technical/annotationfilesmain.affx. Accessed April 7, 2015.
Affymetrix “MANUAL: apt-copynumber-cyto (1.16.1)” [webpage] http://media.affymetrix.com/support/developer/powertools/changelog/apt-copynumber-cyto.html. Accessed April 7, 2015.
Affymetrix “Affymetrix Power Tools (APT) -- Release 1.16.1” [webpage] http://media.affymetrix.com/support/developer/powertools/changelog/index.html. Accessed April 7, 2015.
Uddin M, Thiruvahindrapuram B, Walker S, Wang Z, Hu P, Lamoureux S, et al.. A high-resolution copy-number variation resource for clinical and population genetics. Genet Med. 2014. doi:10.1038/gim.2014.178.
Stevens-Kroef MJ, van den Berg E, Olde Weghuis D, Geurts van Kessel A, Pfundt R, Linssen-Wiersma M, et al. “Identification of prognostic relevant chromosomal abnormalities in chronic lymphocytic leukemia using microarray-based genomic profiling. Mol Cytogenet. 2014. doi:10.1186/1755-8166-7-3.
Acknowledgements
This work was partly supported by the Spanish Ministry of Science and Innovation (MTM2011-26515), FIS PI1002512 and a predoctoral fellowship of the Universitat Pompeu Fabra (to CH-F).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
LAP-J is a founder and member of the scientific advisory board of the company qGenomics. KD is actually working for Affymetrix UK Ltd.
Author's contributions
CH-F created the package. LAP-J, JRG and CH-F analyzed the data, interpreted the results and wrote the manuscript draft. IQ, AC and KD designed the resource population and led collection of phenotypic data. KD come up with the tool's parameters. JRG conceived and supervised the project. All authors read and approved the final manuscript.
Additional file
Below is the link to the electronic supplementary material.
Additional file 1:
It includes a plot for mosaic events detected in Dataset A and for the CNV events detected in Dataset B. It also includes the full code used in both analysis of both Dataset A and Dataset B.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Hernandez-Ferrer, C., Quintela Garcia, I., Danielski, K. et al. affy2sv: an R package to pre-process Affymetrix CytoScan HD and 750K arrays for SNP, CNV, inversion and mosaicism calling. BMC Bioinformatics 16, 167 (2015). https://doi.org/10.1186/s12859-015-0608-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12859-015-0608-y