
Abstract
Although liposomes have improved patient safety and the pharmacokinetic profile of free drugs, their therapeutic efficacy has only shown marginal improvement. The incorporation of active-targeted ligands to enhance cellular uptake has shown promise in preclinical studies. However, no active-targeted liposomes have successfully translated into clinical use thus far. This study aimed to evaluate the targeting ability and antitumor efficiency of A6, a specific short peptide (KPSSPPEE) when incorporated into PEGylated liposomal doxorubicin (PLD). The results revealed significantly enhanced cellular uptake. The cytotoxicity of the formulations was determined by 3 h and 6 h incubation of formulations with cells, followed by 48 h incubation to evaluate the targeted ability of the formulations and the results indicated the higher cytotoxicity of A6-PLD (IC50 of 7.52 µg/mL after 6 h incubation) in the CD44 overexpressing C26 cell line compared to non-targeted PLD (IC50 of 15.02 µg/mL after 6 h incubation). However, CD44-negative NIH-3T3 cells exhibited similar uptake and in vitro cytotoxicity for both A6-PLD (IC50 of 38.05 µg/mL) and PLD (IC50 of 34.87 µg/mL). In animal studies, A6-PLD demonstrated significantly higher tumor localization of doxorubicin (Dox) (~ 8 and 15 µg Dox/g tumor for 24 and 48 after injection) compared to PLD (~ 6 and 8 µg Dox/g tumor for 24 and 48 after injection), resulting in effective inhibition of tumor growth. The median survival time (MST) for Dextrose 5% was 10, PLD was 14 and A6-PLD was 22 days. In conclusion, A6-PLD, a simple and effective targeted liposome formulation, exhibits high potential for clinical translation. Its improved targetability and antitumor efficacy make it a promising candidate for future clinical applications.
Similar content being viewed by others
Introduction
Targeted drug delivery technology in tumor therapy research provides a promising approach to enhance therapeutic efficacy and mitigate the adverse effects of anticancer drugs on normal tissues. Various targeting ligands, including antibodies, aptamers, and peptides, have been extensively explored for specifically targeting cancer cells (Hashemi et al. 2020a). In recent years, short peptides with distinct biological functions have garnered significant attention for their potential applications in diagnostic and therapeutic biology (Apostolopoulos et al. 2021). Furthermore, diverse targeted nanocarriers such as liposomes, PLGA, and gold nanoparticles have been investigated to improve the therapeutic effect, enhance the biodistribution of anticancer drugs, leverage the enhanced permeability and retention (EPR) effect, and mitigate their toxic side effects (Yazdian-Robati et al. 2019; Hashemi et al. 2020a; Arabi et al. 2015). CD44, a multifunctional cell surface glycoprotein, plays a crucial role in cancer progression, with elevated levels observed in various cancers including gallbladder, breast, colon, prostate, and ovarian cancers. Its presence correlates with aggressive biological behavior, metastasis, and poor prognosis (Hassn Mesrati et al. 2021). Consequently, targeting CD44 in therapeutic strategies holds great potential for inhibiting tumor cell invasion and metastasis (Chen et al. 2018). Among the molecules targeting CD44, the human urokinase plasminogen activator (uPA) stands out (Finlayson 2015). A6 (KPSSPPEE), a small peptide derived from the connecting peptide domain of uPA, exhibits high binding affinity to CD44 ligands. This peptide possesses anti-invasive, anti-migratory, and anti-angiogenic properties, making it a promising candidate for enhancing the efficiency and effectiveness of cancer treatment (Franco et al. 2006).
Stealth PEGylated liposomal doxorubicin (PLD, commercially available as Caelyx®, Doxil®) is the first FDA-approved nano-drug, extended circulation time of free doxorubicin (Dox) and significantly reduced toxicity compared to free Dox (Barenholz 2012). However, this nanoformulation is non-cell-selective, which limits its site-specific bioavailability (Blanco et al. 2015). Decoration of PLD with specific peptides, aptamers, or antibodies, to improve cell selectivity offers an efficient therapeutic strategy against various cancers (Moosavian et al. 2018; Amin et al. 2013). Numerous studies have been conducted on the functionalization of PLD (Mashreghi et al. 2021a, 2020, 2021b; Zamani et al. 2020). To our knowledge, there is no report on employing A6 as a targeting ligand in liposomal delivery systems. Moreover, clinical trials of the A6 peptide have demonstrated its excellent safety (Gu et al. 2019).
In this study, our objective was to investigate the potential of utilizing the A6 peptide as a targeting agent for CD44-positive cancer cells by conjugating it to PLD (A6-PLD). Building upon our prior research, we hypothesized that incorporating A6 as a targeting ligand would enhance the therapeutic effectiveness of PLD. To achieve this, we conducted a comprehensive characterization of the liposomes. Subsequently, both in vitro and in vivo studies were conducted to assess and compare the therapeutic efficacy of A6-PLD with that of PLD.
Materials and methods
Materials
The commercially available PLD (Caelyx®) was procured from Behestan Darou Company located in Tehran, Iran. The thiolated A6 peptide-amine (SH-KPSSPPEEC-NH2) was acquired from ChinaPeptides Co. Ltd based in Shanghai, China. The 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-mPEG2000-mal) was obtained from Avanti polar lipids in Alabaster, AL. Tetrazolium dye (MTT) was purchased from Merck in Darmstadt, Germany. RPMI 1640 and DMEM culture media, as well as fetal bovine serum (FBS), were sourced from Gibco, a division of Thermo Fisher Scientific based in the USA.
Experimental methods
Conjugation of A6 peptide to DSPE-mPEG2000-mal
The A6 peptide, which was modified with a thiol group at the lysine residue to facilitate its linkage to the liposomes, was synthesized by ChinaPeptide Company with a purity of 95%. To create DSPE-mPEG2000-A6, activated DSPE-mPEG2000-mal was utilized to conjugate A6 to DSPE-PEG2000 through a thioether bond formed between the thiol group of a cysteine residue in the A6 peptide and the pyrrole group of maleimide (Yazdian-Robati et al. 2022).
The peptide, dissolved in DMSO at a concentration of 10 mg/ml, and the lipid, dissolved in chloroform at a concentration of 10 mg/ml, were mixed at a molar ratio of 1.2:1 (peptide to lipid) and incubated overnight at room temperature in the dark under an argon atmosphere with continuous agitation. The effectiveness of the linkage was assessed using silica thin-layer chromatography (TLC) with a developing solvent composed of chloroform/methanol/water (90/10/2) and exposure to iodine vapor. Subsequently, the solvents were evaporated using a heated nitrogen stream, and the content of the tube was freeze-dried overnight. The resulting white powder was then appropriately resuspended in deionized water (Darban et al. 2017; Nik et al. 2019). The lipid–peptide conjugation was confirmed by high-performance liquid chromatography (HPLC) analysis.
Preparation and characterization of A6-targeted liposomal doxorubicin by post-insertion methods
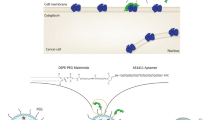
The activated A6-DSPE-PEG (2000) micelles were incubated with PLD formulation for one hour at 60 ℃ to DSPE-mPEG-2000-A6 incorporated into the liposomes (Fig. 1) (Moosavian et al. 2016) The targeted liposomes were characterized by measuring their size, polydispersity index, and zeta potential using a particle size analyzer called Nano-ZS, manufactured by Malvern in the UK. The concentration of encapsulated Dox (doxorubicin) before and after the post-insertion step was determined using fluorimetry. To do this, small portions of the samples were dissolved in acidified isopropyl alcohol, ensuring that the concentration of Dox was below the level where self-quenching occurs. The concentration of Dox was then measured using a spectrofluorometer (Shimadzu RF5000U, Japan) with an excitation wavelength of 480 nm and an emission wavelength of 580 nm. The measured concentrations were compared to a reference standard curve, which was constructed using serial dilutions of a standard PLD (phospholipid) solution (Arabi et al. 2015). The size of the liposomes were examined using a FESEM. Formulations were diluted in DW and then sprayed onto aluminum foil and dried. The nanoparticles were sprinkled with gold film and examined under a microscope.

Schematic representation of thiolated-A6-peptide coupled to the distal end of DSPE-mPEG2000-mal in the lipid bilayer of the liposome
Cell culture
C26 cell line (murine colon carcinoma cells) and NIH-3T3 cell line (mouse embryonic fibroblast cells) were acquired from the Pasteur Institute of Iran located in Tehran, Iran. The cells were cultured at a temperature of 37 ℃ in an environment of 5% CO2 and 95% air with humidity. The culture medium used for maintaining the cells was RPMI 1640, supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 100 IU/ml penicillin, and 100 mg/ml streptomycin (provided by Gibco, UK).
Cytotoxicity assay
The antiproliferative effect of A6-targeted liposomes on cancer and normal cells was determined using the MTT test. C26 and NIH-3T3 cells were seeded in 96-well plates at a concentration of 2500 cells per well in 100 μl of culture medium. The cells were then treated with liposomal formulations, including PLD (standard liposomes) and A6-PLD (A6-targeted liposomes), as well as free Dox (doxorubicin), and incubated for either 3 or 6 h at a temperature of 37 ℃
Subsequently, the culture medium in the wells was replaced with a complete culture medium and allowed to incubate at 37 ℃ for 48 h. The untreated cells were used as a control group for comparison. Following the incubation period, the contents of all wells were substituted with a freshly prepared MTT solution (5 mg/ml) in a medium without fetal bovine serum (FBS), and further incubated for 4 h in the dark. Afterward, 200 μl of dimethyl sulfoxide (DMSO) was added to all wells and thoroughly mixed to dissolve the formazan crystals formed by the MTT reaction. The optical densities (ODs) of the wells were determined by measuring the spectrometric absorbance at 570 nm, with a background measurement at 630 nm, using a Stat-Fax 2100 microplate reader manufactured by Awareness Technology Inc. in the USA. Subsequently, the IC50 values were calculated using Calcusyn version 2.0 software.
Cellular uptake
Flourimetry assay
A total of 200,000 C26 or NIH-3T3 cells were seeded in each well of a 24-well plate and incubated overnight. The cells were then treated with free Dox, PLD, and A6-PLD at a concentration of 20 µg/ml, using serum-free media, and were exposed to these treatments at a temperature of 37 ℃ for 1, 3, and 6 h. Alternatively, another set of cells was exposed to the treatments at 4 ℃ for 1 h. Following the designated incubation periods, the medium in each well was removed, and the cells were washed three times with PBS (pH 7.4). Subsequently, 0.9 ml of acidified isopropanol was added to each well and incubated overnight at 4 ℃. Cell debris was eliminated through centrifugation, and the extracted Dox associated with the cells was measured in the supernatant.
Flow cytometry assay
A population of 1 × 106 cells was suspended in PBS (0.1 M) containing 0.1% BSA and subsequently incubated with PLD or A6-PLD at a concentration of 20 μg/ml. The incubation was carried out at 37 ℃ for a duration of 1 h. To assess the fluorescence signal, flow cytometry (FACSCalibur, BD, USA) was utilized, specifically reading the signal in the FL2 channel.
For the competition assay, the cells were pre-treated with an excessive amount of free A6 peptide for a period of 30 min. Following the pre-treatment, the cells were incubated with A6-PLD as described above.
Animal study
Female BALB/c mice with a weight range of 18–20 g were selected for in vivo experiments. All animal procedures were conducted following the guidelines and approval of the Research Ethics Committees of the National Institute for Medical Research Development (approval code: IR.NIMAD.REC.1399.122).
For the antitumor test, a total of 5 mice were randomly divided into groups. The mice were anesthetized using ketamine (100 mg/kg) and xylazine (10 mg/kg) and then injected subcutaneously with 3 × 105 C26 cells per mouse at the right flank. After 1 week of inoculation, the drugs (Dox, PLD, and A6-PLD) at a dosage of 10 mg/kg were administered intravenously. During the experiment, the mice were weighed daily, and their survival rates were monitored for a period of 30 days.
To study biodistribution and pharmacokinetics, a separate group of mice (n = 3) received different formulations (Dox, PLD, and A6-PLD) at a dosage of 15 mg/kg via intravenous injection 2 weeks after inoculation. At specific time intervals (3, 12, 24, 48, and 72 h) post-injection, blood samples were collected. At 24 and 48 h post-injection, mice were killed, and tissue samples including spleen, liver, kidney, lung, heart, and tumor were dissected. The concentration of Dox in each sample was measured using a spectrofluorometer with excitation and emission wavelengths set at 485 nm and 590 nm, respectively, using a device such as the Perkin-Elmer from the UK, as previously described in the literature (Moosavian et al. 2018; Shahraki et al. 2021; Huang et al. 2009).
Statistical analysis
GraphPad Prism 6.0 was used to analyze the data (GraphPad Software, Inc., San Diego, CA, USA). Data were presented as mean ± SEM of three independent experiments. The Kaplan–Meier method used to calculate the median survival time (MST). P < 0.05 was considered statistically significant.
Results and discussion
In our study, we employed the A6 peptide as a targeting agent to attach to the surface of PLD (PEGylated liposomal doxorubicin) to target CD44 in C26 cells. To accomplish this, we utilized the reaction between the thiol group (-SH) of the A6 peptide and the maleimide functional group of DSPE-mPEG2000-mal (see Fig. 1). This resulted in the formation of nanomicelles that could decorate the outer surface of PLD using a post-insertion technique (Mashreghi et al. 2020). The efficiency of the coupling process was assessed through TLC (thin-layer chromatography) and HPLC (high-performance liquid chromatography) analyses. These techniques provided a means to evaluate the success of the coupling reaction. The post-insertion technique utilized in this study is a straightforward and effective method for incorporating peptide ligands onto preformed stealth liposomes (Moreira et al. 2002).
Physicochemical characterization of liposomes
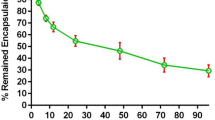
According to the data presented in Table 1, the average particle size of Calyx® before the insertion of the A6 peptide was approximately 94 nm, with a polydispersity index (PDI) of 0.061. It exhibited a zeta potential of −14.4 ± 0.1. Following the conjugation of the A6 peptide, the average particle size of A6-PLD slightly increased to 96 nm, with a PDI of 0.065. This indicates a successful conjugation of DSPE-mPEG2000-A6 peptide to the surface of PLD. The FESEM analysis also indicated the size of the liposomes were consistent with the results of DLS (see Fig. 2). However, this increase was not statistically significant (p > 0.05). These findings suggest that A6 has minimal impact on the pharmacokinetic profile of PLD (Moosavian and Sahebkar 2019). This is consistent with our previous studies, which demonstrated that the binding of a small targeting agent to the outer surface of PLD results in only a slight increase in particle size (Moosavian et al. 2018; Wei et al. 2013). The mean diameter of the liposomes, which remained below 100 nm before and after A6 attachment, confirms that A6-PLD maintains a suitable size for tumor targeting (Darban et al. 2017). To assess the stability of the liposomes after the post-insertion process, the amount of encapsulated Dox was measured before and after the post-insertion step. The results indicated no significant difference in the amount of encapsulated Dox before and after the post-insertion (p > 0.005).

Characterization. FESEM micrograph of the A PLD and B A6-PLD
In vitro studies
Cellular uptake
The targeted formulation was tested for its ability to bind to and uptake target cell lines through binding and uptake assays. At 4 °C, all energy-dependent endocytic pathways were halted, while at 37 °C, endocytic pathways were normal (Thorley et al. 2014). In accordance with previous studies (Arabi et al. 2015), C26 cells were utilized as CD44-positive cell lines, while NIH-3T3 cells were used as CD44-negative cell lines. As depicted in Fig. 3A, B, the percentage of associated Dox in C26 cells treated with A6-PLD was significantly higher compared to C26 cells treated with PLD at a temperature of 37 ℃. This indicates the contribution of A6-CD44 interaction in the cellular uptake of liposomes. Furthermore, the percentage of Dox–cell association increased proportionally with the incubation time. Conversely, there was no significant difference in the amount of Dox observed from different formulations when incubated at 4 ℃. This can be attributed to the rigidity of the cellular membrane, which hampers energy-dependent uptake and passive diffusion at lower temperatures (Allen et al. 2002). Interestingly, the percentage of associated Dox in NIH-3T3 cells incubated with PLD and A6-PLD did not show a significant difference. These results suggest that the presence of A6 on the surface of liposomes facilitates the internalization process of liposomal Dox specifically in C26 cells. In both cell lines, free Dox exhibited higher cellular association, indicating its ability to freely pass through the cell membrane.

In vitro cellular association of PLD, A6- PLD and Dox at A NIH3 cell, B C26 cell
To investigate the cell uptake further, flow cytometry analysis was conducted. As illustrated in Fig. 4A, B, A6-PLD demonstrated a significantly higher affinity (p < 0.01) for C26 cells compared to PLD alone. In contrast, the uptake of PLD and A6-PLD by NIH-3T3 cells did not show a significant difference.

In vitro cellular uptake A6-PLD, PLD, and C26 cells (A) and NIH-3T3 cells (B) at 37 ℃. Green line: control cells, black line: cells incubated with PLD for one hour at 37 ℃, Blue line: cells incubated with A6-PLD for one hour at 37 ℃, red line: cells pre-treated with free A6 and then incubated with A6-PLD for one hour at 37 ℃
Notably, the fluorescence intensity of C26 cells decreased significantly when they were pre-treated with free A6 peptides before being exposed to A6-PLD (p < 0.01). This indicates that the excess amount of free anti-CD44 peptides competitively inhibits the specific binding of A6-PLD to C26 cells.
Cytotoxicity study
To assess the cytotoxicity of A6-PLD, free DOX, and PLD, two cell lines, C-26 and NIH-3T3, were selected. Various concentrations of both liposomal formulations and free DOX were used to treat the cells for 3 and 6 h. The cell viability was then evaluated using the MTT assay for the following 72 h.
Table 2 presents the results, indicating that A6-PLD exhibited significant cytotoxicity compared to PLD (p < 0.001) in the C26 cell line. Conversely, this significant difference was not observed in NIH-3T3 cells. In contrast to C26 cells, NIH-3T3 cells are CD44-negative and displayed a lower response to A6-PLD. As a result, the A6 peptide delivered PLD more efficiently to the targeted cells, resulting in a difference in response. It is consistent with previous studies demonstrating receptor-mediated endocytosis of nanoparticles that these findings are in agreement with the concept of ligand-mediated targeting (Ahmad and Allen 1992; Chang et al. 2013).
Animal studies
Biodistribution study
To assess the impact of the A6 peptide on the biodistribution pattern of PLD, we administered A6-PLD and PLD (at a dose of 10 mg/kg) via intravenous injection into mice with subcutaneous C26 colon cancer tumors. We performed analyses by measuring the intrinsic autofluorescence signal of Dox in various tissues, including serum, liver, spleen, tumor, kidneys, heart, lung, and muscle.
Figure 5A demonstrates the concentration of Dox in plasma at 4, 12, 24, and 48 h after injection with Dox, PLD, and A6-PLD. The results reveal that both PLD and A6-PLD exhibited an extended blood circulation time compared to free Dox. Numerous studies have confirmed that encapsulating Dox into liposomes significantly improves the pharmacokinetic profile and prolongs blood circulation time compared to free Dox (Munster et al. 2018; Sugarbaker and Stuart 2019). Figure 5B indicates that A6-PLD notably increased the tumor accumulation of Dox, highlighting the efficiency of the active-targeting strategy (Mashreghi et al. 2021a). Furthermore, we investigated and compared the concentrations of Dox in major organs among groups receiving free Dox, PLD, and A6-PLD. As illustrated in Fig. 5C, the accumulation of Dox in the liver and spleen was significantly higher for A6-PLD compared to PLD (p < 0.01). At 24 h post-injection, the concentration of Dox in the liver and spleen for mice receiving A6-PLD was 1.37 times greater than those receiving PLD. However, there were no significant differences in the concentration of doxorubicin in lung and kidney tissues, and it followed the same pattern in both liposomal groups.

Biodistribution of PLD and A6-PLD at different time points (24 and 48 h) in organs including A serum, B liver, spleen, heart, kidney, and lung and C tumor in BALB/c mice bearing C26 tumor after a single dose of 15 mg/kg liposomal doxorubicin administered i.v. 2 weeks after the tumor inoculation. D represents a ratio of doxorubicin concentration in the liver, spleen, heart, and tumor to serum, at 48 h. Data expressed as mean ± SEM. (n = 3)
An important observation in major organs was the lower concentration of Dox in the heart of mice that received PLD and A6-PLD formulations compared to the Dox-injected group. One of the major limitations of Dox usage is its cardiotoxicity, which can be mitigated by liposomes, reducing the risk of this adverse effect (Xing et al. 2015). Figure 5D also indicates that, after 48 h post-injection, the tumor/serum ratio of Dox concentration in the A6-PLD group was higher than that in the PLD group. This suggests that A6-PLD effectively delivered Dox to the tumor site, and due to the higher expression of CD44 in C26 tumors, there was a higher accumulation (Nik et al. 2019). It has been shown that long-circulating liposomes can preferentially accumulate in solid tumor sites through the enhanced permeability and retention (EPR) effect. Both targeted and non-targeted liposomes follow the same mechanism of distribution during this step. Previous studies have shown that functionalization can alter the size and surface charge of liposomes, consequently affecting their pharmacokinetic profile. Therefore, a targeting ligand with minimal interference in the pharmacokinetic profile is desirable. After tumor localization, ligand-targeted liposomes can enter cells via receptor-mediated endocytosis, improving targeting efficiency and retention at the target site (Behzadi et al. 2017; Rosenblum et al. 2018; Moosavian et al. 2021). It has been reported that A6 peptide can enhance tumor penetration in tumor cells (Gu et al. 2019).
In vivo antitumor study
The therapeutic efficacy of the A6-PLD formulation was investigated in C26 tumor models and compared with PLD. During the course of 1 month, various parameters including body weight, tumor growth rate (measured in mm3), and survival rate were monitored (Fig. 6A–C). Body weight reduction is commonly used as a standard method to evaluate the toxicity of chemotherapeutic drugs (Tuscano et al. 2010). As shown, there was no significant weight change observed in the group treated with A6-PLD compared to the PLD and dextrose 5% control groups. The difference in weight changes was not statistically significant (p > 0.05), suggesting that the single treatments with the targeted formulation did not cause any obvious toxicity and were well tolerated. The MST for dextrose 5% was 10, PLD was 14 and A6-PLD was 22 days.

In vivo therapeutic efficacy of PLD and A6- PLD in mice bearing C-26 colon carcinoma tumor after i.v. administration of a single dose of 10 mg/kg liposomal doxorubicin or dextrose 5% on day 8 after tumor inoculation. A Tumor volume, B percentage change in animal body weight, C survival curve. Data represented as mean ± SE (n = 5)
After the intravenous injection of A6-PLD in the C26 tumor model, the tumor growth rate was more effectively inhibited compared to mice receiving non-targeted PLD at equal doses (p < 0.0001). Survival data, depicted in a Kaplan–Meier plot, further supported the significant differences in therapeutic efficacy between the targeted and non-targeted liposomal formulations (Arabi et al. 2015). The survival results demonstrated that treatment with A6-PLD extended mouse survival compared to PLD and Dextrose 5% control groups. These findings align with the results observed in the biodistribution studies, which showed high levels of Dox accumulation in the tumor tissue of the A6-PLD-treated group. Thus, it can be concluded that A6-PLD improves the overall therapeutic efficacy compared to free Dox and PLD. These findings are consistent with previous studies that have reported enhanced therapeutic efficacy of polymerosomal epirubicin following modification with the A6 peptide (Gu et al. 2019). Other studies have also demonstrated that modification with targeting ligands enhances the therapeutic efficacy of liposomal Dox (Makwana et al. 2021; Lin et al. 2021). This is the first study to evaluate the therapeutic efficacy, pharmacokinetics, and biodistribution of PLD using the A6 peptide in a C26 colon carcinoma tumor model. As reported previously, this study confirms that liposomes modified with a targeting ligand, such as a peptide, can improve therapeutic outcomes of non-targeted liposome formulations (Shahin et al. 2013; Shmeeda et al. 2009).
Conclusion
PLD serves as the standard treatment for ovarian cancer, multiple myeloma, and AIDS-related Kaposi's sarcoma, as approved by the FDA. However, this drug has certain limitations, including inadequate cellular uptake and a low release rate of Dox at the tumor site. To address these limitations, the present study successfully developed a surface-functionalized PLD by incorporating the urokinase-derived peptide (A6) through post-insertion techniques. The characterization of this formulation confirmed its suitability in terms of size for effective delivery to the tumor site through the enhanced permeability and retention (EPR) effect. In vitro studies on cell cytotoxicity and uptake demonstrated that the functionalized A6-PLD system facilitates the active delivery of Dox into tumor cells. Furthermore, in vivo results revealed the efficacy of A6-PLD in reducing tumor volume and improving the survival of animals compared to treatment with PLD alone. Nevertheless, further investigations are warranted to fully evaluate the efficacy of A6-PLD before its translation into clinical settings. These studies should include histological evaluations and the use of nude mice tumor models to provide a comprehensive assessment of A6-PLD's potential benefits and effectiveness.
Availability of data and materials
Data will be made available on request.
References
Ahmad I, Allen TM (1992) Antibody-mediated specific binding and cytotoxicity of liposome-entrapped doxorubicin to lung cancer cells in vitro. Can Res 52(17):4817–4820
Allen TM, Sapra P, Moase E, Moreira J, Iden D (2002) Adventures in targeting. J Liposome Res 12(1–2):5–12
Amin M, Badiee A, Jaafari MR (2013) Improvement of pharmacokinetic and antitumor activity of PEGylated liposomal doxorubicin by targeting with N-methylated cyclic RGD peptide in mice bearing C-26 colon carcinomas. Int J Pharm 458(2):324–333
Apostolopoulos V, Bojarska J, Chai T-T, Elnagdy S, Kaczmarek K, Matsoukas J, New R, Parang K, Lopez OP, Parhiz H (2021) A global review on short peptides: frontiers and perspectives. Molecules 26(2):430
Arabi L, Badiee A, Mosaffa F, Jaafari MR (2015) Targeting CD44 expressing cancer cells with anti-CD44 monoclonal antibody improves cellular uptake and antitumor efficacy of liposomal doxorubicin. J Control Release 220:275–286
Barenholz YC (2012) Doxil®—the first FDA-approved nano-drug: lessons learned. J Control Release 160(2):117–134
Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC, Brown D, Alkilany AM, Farokhzad OC, Mahmoudi M (2017) Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev 46(14):4218–4244
Blanco E, Shen H, Ferrari M (2015) Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol 33(9):941–951
Chang D-K, Li P-C, Lu R-M, Jane W-N, Wu H-C (2013) Peptide-mediated liposomal Doxorubicin enhances drug delivery efficiency and therapeutic efficacy in animal models. PLoS ONE 8(12):e83239
Chen C, Zhao S, Karnad A, Freeman JW (2018) The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol 11(1):64
Darban SA, Badiee A, Jaafari MR (2017) PNC27 anticancer peptide as targeting ligand significantly improved antitumor efficacy of Doxil in HDM2-expressing cells. Nanomedicine 12(12):1475–1490
Finlayson M (2015) Modulation of CD44 activity by A6-Peptide. Front Immunol. https://doi.org/10.3389/fimmu.2015.00135
Franco P, Vocca I, Carriero MV, Alfano D, Cito L, Longanesi-Cattani I, Grieco P, Ossowski L, Stoppelli MP (2006) Activation of urokinase receptor by a novel interaction between the connecting peptide region of urokinase and αvβ5 integrin. J Cell Sci 119(16):3424–3434
Gu W, An J, Meng H, Yu N, Zhong Y, Meng F, Xu Y, Cornelissen JJLM, Zhong Z (2019) CD44-Specific A6 short peptide boosts targetability and anticancer efficacy of polymersomal epirubicin to orthotopic human multiple myeloma. Adv Mater 31(46):1904742
Hashemi M, Shamshiri A, Saeedi M, Tayebi L, Yazdian-Robati R (2020a) Aptamer-conjugated PLGA nanoparticles for delivery and imaging of cancer therapeutic drugs. Arch Biochem Biophys 691:108485
Hassn Mesrati M, Syafruddin SE, Mohtar MA, Syahir A (2021) CD44: a multifunctional mediator of cancer progression. Biomolecules 11(12):1850
Huang Z, Jaafari MR, Szoka FC Jr (2009) Disterolphospholipids: nonexchangeable lipids and their application to liposomal drug delivery. Angew Chem Int Ed Engl 48(23):4146–4149
Lin W-W, Cheng Y-A, Li C-C, Ho K-W, Chen H-J, Chen I-J, Huang B-C, Liu H-J, Lu Y-C, Cheng C-M (2021) Enhancement of tumor tropism of mPEGylated nanoparticles by anti-mPEG bispecific antibody for ovarian cancer therapy. Sci Rep 11(1):1–12
Makwana V, Karanjia J, Haselhorst T, Anoopkumar-Dukie S, Rudrawar S (2021) Liposomal doxorubicin as targeted delivery platform: current trends in surface functionalization. Int J Pharm 593:120117
Mashreghi M, Zamani P, Moosavian SA, Jaafari MR (2020) Anti-Epcam aptamer (Syl3c)-functionalized liposome for targeted delivery of doxorubicin: in vitro and in vivo antitumor studies in mice bearing C26 colon carcinoma. Nanoscale Res Lett 15(1):1–13
Mashreghi M, Faal Maleki M, Karimi M, Kalalinia F, Badiee A, Jaafari MR (2021a) Improving anti-tumour efficacy of PEGylated liposomal doxorubicin by dual targeting of tumour cells and tumour endothelial cells using anti-p32 CGKRK peptide. J Drug Target 29(6):617–630
Mashreghi M, Zamani P, Karimi M, Mehrabian A, Arabsalmani M, Zarqi J, Moosavian SA, Jaafari MR (2021b) Anti-epithelial cell adhesion molecule RNA aptamer-conjugated liposomal doxorubicin as an efficient targeted therapy in mice bearing colon carcinoma tumor model. Biotechnol Prog 37(3):e3116
Moosavian SA, Sahebkar A (2019) Aptamer-functionalized liposomes for targeted cancer therapy. Cancer Lett 448:144–154
Moosavian SA, Abnous K, Badiee A, Jaafari MR (2016) Improvement in the drug delivery and anti-tumor efficacy of PEGylated liposomal doxorubicin by targeting RNA aptamers in mice bearing breast tumor model. Colloids Surf, B 139:228–236
Moosavian SA, Abnous K, Akhtari J, Arabi L, Gholamzade Dewin A, Jafari M (2018) 5TR1 aptamer-PEGylated liposomal doxorubicin enhances cellular uptake and suppresses tumour growth by targeting MUC1 on the surface of cancer cells. Artif Cells Nanomed Biotechnol 46(8):2054–2065
Moosavian SA, Bianconi V, Pirro M, Sahebkar A (2021) Challenges and pitfalls in the development of liposomal delivery systems for cancer therapy. Semin Cancer Biol 69:337–348
Moreira JN, Ishida T, Gaspar R, Allen TM (2002) Use of the post-insertion technique to insert peptide ligands into pre-formed stealth liposomes with retention of binding activity and cytotoxicity. Pharm Res 19(3):265–269
Munster P, Krop IE, LoRusso P, Ma C, Siegel BA, Shields AF, Molnár I, Wickham TJ, Reynolds J, Campbell K (2018) Safety and pharmacokinetics of MM-302, a HER2-targeted antibody–liposomal doxorubicin conjugate, in patients with advanced HER2-positive breast cancer: a phase 1 dose-escalation study. Br J Cancer 119(9):1086–1093
Nik ME, Malaekeh-Nikouei B, Amin M, Hatamipour M, Teymouri M, Sadeghnia HR, Iranshahi M, Jaafari MR (2019) Liposomal formulation of Galbanic acid improved therapeutic efficacy of pegylated liposomal Doxorubicin in mouse colon carcinoma. Sci Rep 9(1):9527
Rosenblum D, Joshi N, Tao W, Karp JM, Peer D (2018) Progress and challenges towards targeted delivery of cancer therapeutics. Nat Commun 9(1):1410
Shahin M, Soudy R, Aliabadi HM, Kneteman N, Kaur K, Lavasanifar A (2013) Engineered breast tumor targeting peptide ligand modified liposomal doxorubicin and the effect of peptide density on anticancer activity. Biomaterials 34(16):4089–4097
Shahraki N, Mehrabian A, Amiri-Darban S, Moosavian SA, Jaafari MR (2021) Preparation and characterization of PEGylated liposomal Doxorubicin targeted with leptin-derived peptide and evaluation of their anti-tumor effects, in vitro and in vivo in mice bearing C26 colon carcinoma. Colloids Surf, B 200:111589
Shmeeda H, Tzemach D, Mak L, Gabizon A (2009) Her2-targeted pegylated liposomal doxorubicin: retention of target-specific binding and cytotoxicity after in vivo passage. J Control Release 136(2):155–160
Sugarbaker PH, Stuart OA (2019) Pharmacokinetics of the intraperitoneal nanoparticle pegylated liposomal doxorubicin in patients with peritoneal metastases. Eur J Surg Oncol. https://doi.org/10.1016/j.ejso.2019.03.035
Thorley AJ, Ruenraroengsak P, Potter TE, Tetley TD (2014) Critical determinants of uptake and translocation of nanoparticles by the human pulmonary alveolar epithelium. ACS Nano 8(11):11778–11789
Tuscano JM, Martin SM, Ma Y, Zamboni W, O’Donnell RT (2010) Efficacy, biodistribution, and pharmacokinetics of CD22-targeted pegylated liposomal doxorubicin in a B-cell non–Hodgkin’s lymphoma xenograft mouse model. Clin Cancer Res 16(10):2760–2768
Wei T, Liu J, Ma H, Cheng Q, Huang Y, Zhao J, Huo S, Xue X, Liang Z, Liang X-J (2013) Functionalized nanoscale micelles improve drug delivery for cancer therapy in vitro and in vivo. Nano Lett 13(6):2528–2534
Xing M, Yan F, Yu S, Shen P (2015) Efficacy and cardiotoxicity of liposomal doxorubicin-based chemotherapy in advanced breast cancer: a meta-analysis of ten randomized controlled trials. PLoS ONE 10(7):e0133569
Yazdian-Robati R, Arab A, Ramezani M, Rafatpanah H, Bahreyni A, Nabavinia MS, Abnous K, Taghdisi SM (2019) Smart aptamer-modified calcium carbonate nanoparticles for controlled release and targeted delivery of epirubicin and melittin into cancer cells in vitro and in vivo. Drug Dev Ind Pharm 45(4):603–610
Yazdian-Robati R, Bayat P, Dehestani S, Hashemi M, Taghdisi SM, Abnous K (2022) Smart delivery of epirubicin to cancer cells using aptamer-modified ferritin nanoparticles. J Drug Target. https://doi.org/10.1080/1061186X.2022.2025600
Zamani P, Navashenaq JG, Teymouri M, Karimi M, Mashreghi M, Jaafari MR (2020) Combination therapy with liposomal doxorubicin and liposomal vaccine containing E75, an HER-2/neu-derived peptide, reduces myeloid-derived suppressor cells and improved tumor therapy. Life Sci 252:117646
Acknowledgements
This study was financially supported by the National Institute for Medical Research Development (NIMAD), Tehran, Iran (project number: 990019). We also thank Mashhad University of Medical Sciences (MUMS) which provided facilities and equipment for this research.
Funding
This study was financially supported by National Institute for Medical Research Development (NIMAD), Tehran, Iran (Project Number: 990019).
Author information
Authors and Affiliations
Contributions
RYR and EM performed methodology, investigation, and writing-original draft. HK, AKH and SMT performed supervision, data curation, conceptualization, and writing—reviewing and editing. MRJ and MM performed formal analysis, software, supervision, and writing and editing of the manuscript and SAM secured the grant and performed supervision.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal work was approved by the Institutional Ethical Committee and Research Advisory Committee of Mashhad University of Medical Sciences (Ethical number: 990019). All animal experiments and methods complied with the relevant guidelines and regulations approved by the ethical committee and the Animal Research: Reporting of In vivo Experiments (ARRIVE) and performed in accordance with the UK Animals (Scientific Procedures) Act, 1986.
Consent for publication
All the authors contributed to the article and approved the submitted version.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yazdian-Robati, R., Amiri, E., Kamali, H. et al. CD44-specific short peptide A6 boosts cellular uptake and anticancer efficacy of PEGylated liposomal doxorubicin in vitro and in vivo. Cancer Nano 14, 84 (2023). https://doi.org/10.1186/s12645-023-00236-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12645-023-00236-0