
Abstract
Purpose
Owing to the complexity of cancer, a synergistic combination of chemotherapy and gene therapy can be a promising therapeutic strategy. This study aimed to use stem cell membrane (SCM)-camouflaged polydopamine nanoparticles for simultaneous delivery of curcumin (CUR) and siRNA-targeting STAT3 (CPDA/siSTAT3@SCM NPs) for osteosarcoma (OS).
Methods
Transmission electron microscopy, UV–Vis absorbance spectra, zeta potential, cell co-localization, and Coomassie bright blue staining were used to characterize CPDA/siSTAT3@SCM NPs constructed by the self-assembly method. Drug release, cellular uptake, cell proliferation, apoptosis, wound healing, and transwell assays were evaluated in vitro. The expression levels of epithelial–mesenchymal transition (EMT)- and apoptosis-related proteins were measured by western blotting. Furthermore, the biodistribution, antitumor efficacy, and biosafety of CPDA/siSTAT3@SCM NPs in an MG63 xenograft mouse model were evaluated.
Results
CPDA/siSTAT3@SCM NPs were successfully synthesized to deliver CUR and siRNA simultaneously, and they showed osteosarcoma-targeting ability. Furthermore, it showed high cellular uptake and excellent synergistic antitumor effects in vitro. CPDA/siSTAT3@SCM NPs suppressed OS cell proliferation, migration, invasion, and EMT progression, and promoted the apoptotic process. In tumor-bearing mice, the treatment with CPDA/siSTAT3@SCM NPs showed an excellent antitumor effect with no side effects in major organs.
Conclusion
This study revealed that CPDA/siSTAT3@SCM NPs can target drug delivery by biomimetic multifunctional nanoparticles to treat OS through chemo-gene combined therapy.
Similar content being viewed by others


Introduction
Osteosarcoma (OS) is the most commonly diagnosed primary malignant bone tumor, which is highly metastatic and locally aggressive, and frequently involves the long diaphysis metaphysis (Corre, et al. 2020). Various therapeutic advancements and strategies such as surgical techniques, neoadjuvant chemotherapy, radiofrequency ablation, and cryotherapy have been widely developed for OS (Liu et al. 2022); however, the 5-year survival rate of patients with OS remains less than 30% (Moukengue 2022). Furthermore, there has been no significant progress in the chemotherapy treatment effect over the last decades because of poor chemoresistance and selectivity in OS (Panagi et al. 2022). A new consensus on the key genes involved in the development and progression of OS has shifted the treatment strategy to developing novel therapies for effective molecular targets (Li et al. 2022). The combination of chemotherapy with gene therapy has received increased research attention and offers a promising treatment option for OS.
The signal transducer and activator of transcription (STAT) family of proteins are latent cytoplasmic transcription factors involved in regulating cellular growth, differentiation, and survival (Zou et al. 2020; Wang et al. 2022). STAT3 is a crucial regulator of osteosarcoma progression and has been linked to poor clinical prognosis (Yang et al. 2022). STAT3 mutations play a crucial role in OS formation, metastasis, and drug resistance and it has been identified as a potential therapeutic target (Liu et al. 2021). SiRNA effectually inhibited carcinoma proliferation and metastasis in the MG63 OS model by selectively suppressing STAT3 (Jin et al. 2020). Furthermore, Kong et al. found that STAT3 regulates OS cell migration and invasion by altering the content of epithelial–mesenchymal transition (EMT)-related proteins, such as E-cadherin, N-cadherin, vimentin, and fibronectin (Kong et al. 2017). Therefore, RNA interference-mediated gene silencing appears to be a promising therapeutic strategy for various cancers, including OS.
Curcumin (CUR) is derived from a perennial herb and has been reported to have powerful antioxidant and anti-inflammatory properties; it has been used for the treatment of tumors, such as OS, colorectal cancer, prostate cancer, and breast cancer (Xu et al. 2021; Termini et al. 2020; Pricci et al. 2020; Wang et al. 2019). In addition, CUR has been reported to inhibit cancer development, metastasis, and progression via diverse signaling molecular pathways (Xu et al. 2021). CUR suppresses EMT progression in pancreatic and colorectal cancer by regulating related genes (Jia et al. 2021; Cao et al. 2016). Moreover, curcumin effectively regulated Bcl-2 and BAX, resulting in OS cancer cell apoptosis, which is associated with the sensitivity of OS to chemotherapy (Xu et al. 2021). Notably, CUR and its derivatives can specifically target STAT3, inhibiting OS cell proliferation and invasion (Fossey et al. 2011; Sun et al. 2019; Lu, et al. 2020). However, the antitumor effectiveness of CUR is severely limited due to its low hydrophobicity and bioavailability, which limits its clinical applications (Tan et al. 2021). Therefore, there is an urgent need to improve CUR bioavailability and combine it with gene therapy to provide broad therapeutic strategies for OS.
Combinational therapy of chemotherapeutics and genes using nanoparticles as carriers has been extensively developed in tumor-targeting therapy over the years, providing a feasible solution to issues regarding CUR mentioned and siRNA (Wu et al. 2022). Currently, various nanoplatform drug delivery systems, such as liposomes (Chi et al. 2017), polymeric nanoparticles (Yin et al. 2020), nanomicelles (Li et al. 2020), and dendrimers(Gonçalves et al. 2014), have been studied and validated in OS treatment. These systems have the characteristics of controllable drug release, remarkable biocompatibility, tumor targeting, and prolonged blood circulation half-life of drugs (Wu et al. 2022). Polydopamine (PDA) is a highly adhesive, hydrophilic, stable, and biocompatible nanoparticle that spontaneously polymerizes by dopamine under alkaline conditions (Su et al. 2021). Furthermore, PDA can carry various functional molecules, such as chemotherapy drugs, gene drugs, and proteins, to the surface because of its variable morphology and multiple catechols and amino functional groups (Zhang et al. 2020). PDA can condense polyanionic and unstable siRNA, thereby helping it enter solid tumor cells and escape intracellular lysosomes. It can also reduce the side effects of chemotherapeutics, prolong the half-life of drugs, and greatly improve the anticancer effect (Mu et al. 2021). NPs is easily identified and eliminated by the reticuloendothelial system, resulting in immune rejection because they have low diffusion efficiency at the tumor site. Researchers are attempting to modify the surface of NPs with membranes to improve their biocompatibility and targeting efficiency (Zhang and Huang 2022). Human umbilical cord mesenchymal stem cells (Huc-MSCs) appear to have the lowest immunogenicity, longest circulation time, targeting ability, and receptor integration of all the cells (Zhang and Huang 2022; Thanuja et al. 2018). The role of MSC in carcinomas is controversial; however, they have been reported to migrate to the tumor site by rolling, capture, adhesion, and extravasation across epithelial layers (Hass et al. 2011). The main membrane proteins CXCR4, CXCR2, and CD74 mediate tumor homing, and the receptors that recognize tumor secretion, including platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF), are retained during MSC cell membrane extraction (Wu et al. 2019). Stem cell membrane (SCM) coated with NPs with targeting and immune camouflage properties is a promising biomedical application that combines the advantages and properties of SCM and NPs.
The present study aimed to design and synthesize a PDA nanoparticle (CPDA/siSTAT3@SCM) that is disguised and modified by SCM to co-load CUR and siSTAT3, to achieve chemical-gene combined therapy of OS and induce apoptosis of osteosarcoma cells. Inhibiting the progression of OS epithelial–mesenchymal transition provides a more effective treatment strategy for Patients with OS.
Materials and methods
Materials
Dopamine hydrochloride and CUR were obtained from Sigma–Aldrich (St. Louis, MO, USA). Hoechst stain, DiO, and DiR iodide were purchased from Yeasen. The 0.8 μm polycarbonate membrane was purchased from MilliporeSigma (Billerica, MA, USA). Tris (hydroxymethyl) aminoethane was purchased from Adamas Reagent Co. Ltd. (Shanghai, China). Western blotting antibodies were purchased from Bioss USA and Proteintech Technology (Rosemount, IL, USA). siRNAs targeting STAT3 (5′-CCCGGAAAUUUAACAUUCUTT-3′) and siRNA negative control (siRNA-NC, 5′-GUUGAAAUCAAAGUCGUCC-3′) labeled with CY3 were synthesized by Shanghai GenePharma (Chona). MG63, Hfob1.19, Lo2, RAW246.7, and Huc-MSCs were obtained from the Chinese Academy of Sciences (Shanghai, China). The cells were cultured according to the guidelines recommended by American Type Culture Collection (ATCC).
Preparation of the MSC membrane
MSC membrane extraction was performed following a previously published protocol, with minor modifications (Mu et al. 2021). Briefly, MSCs were transferred to a hypotonic lysis buffer at 4 °C overnight and sonicated for 5 min on ice. Cells were then centrifuged at 2000×g for 10 min at 4 °C; the resulting supernatant was centrifuged again at 22,000×g for 30 min at 4 °C to obtain a cell membrane precipitate. Finally, the cell membrane precipitate was resuspended in phosphate-buffered saline (PBS) before use.
Synthesis and characterization of CPDA/siSTAT3@SCM NPs
First, 32 mL of ethanol and 72 mL of ultrapure water were magnetically stirred and mixed at room temperature, and 120 mg of tris was dissolved and stirred for 30 min. The pH of the mixed solution was adjusted to 8.5 with 1.6 mL of ammonia water. Dopamine hydrochloride (200 mg) was then added and magnetically stirred for 12 h until the solution turned dark brown. The mixture was centrifuged at 22,000×g for 30 min at room temperature, washed with ultrapure water, centrifuged three times, powdered using a low-temperature lyophilizer, and then weighed with electronic precision to determine its concentration.
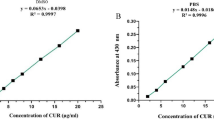
In addition, 1 mg PDA NPs, 2 mg CUR solution, and 10 mL ultrapure water were magnetically stirred for 24 h, and the undissolved CUR precipitate was removed by low-speed centrifugation (1000×g, 10 min). The PDA-CUR precipitate was obtained by high-speed centrifugation and purification. After washing with ultrapure water three times, the supernatant was collected, the UV–visible absorption spectrum absorbance of curcumin at 425 nm wavelength was obtained using a UV-5200 PC spectrophotometer, and CUR quality was analyzed. The drug-loading efficiency of CUR was calculated as follows: drug-loading rate% = mass of loaded CUR/(mass of PDA NPs + mass of loaded CUR) × 100%.
Cy3-labeled STAT3 siRNA (100 nM) to the PDA NP solution (0, 0.05, 0.1, 0.2, 0.4, and 0.8 mg/mL) dissolved in RNase-free ultrapure water. The mixture was incubated at room temperature for 40 min, centrifuged at 20,000×g for 20 min, and washed 3 times with RNase-free ultrapure water. The supernatant was collected, the fluorescence spectrum of the supernatant was recorded using a fluorescence spectrophotometer (Tecan Infinite® 200 Pro; Tecan, Männedorf, Switzerland), and the optimal ratio of STAT3 siRNA to PDA NPs was calculated.
The obtained cell membrane precipitation solution was repeatedly extruded 14 times through a porous polycarbonate membrane with a pore size of 200 nm using a nano-miniature extruder. The extruded stem cell membrane vesicles and CPDA/siSTAT3@SCM NPs were sonicated on an ice bath for 5 min and then extruded 14 times through a 200 nm polycarbonate membrane using a nano-miniature extruder to prepare CPDA/siSTAT3 @SCM NPs.
Transition electron microscopy (TEM, 2100F; JEOL Ltd., Tokyo, Japan) was used to image the NPs after they were stained with 1% uranyl acetate. The particle size and zeta potential were measured using a Zeta-sizer Nano ZS90 (Malvern Instruments Ltd., Malvern, UK).
Sodium dodecyl sulfate and polyacrylamide gel electrophoresis (SDS-PAGE) analysis of membrane proteins
SDS-PAGE was used to examine proteins in SCM vesicles and PDA@SCM NPs. The prepared NPs and MSC membrane were lysed with RIPA lysis buffer, and the concentration of SCM proteins was determined using a BCA protein kit. The proteins (50 μg/well) were separated by 10% SDS-PAGE, stained with Coomassie Brilliant Blue, and imaged.
Drug-loading content
The concentration of CUR was determined using UV–Vis spectroscopy (UV-3600plus; Shimadzu Co., Kyoto, Japan). Acetonitrile was used to extract the CUR entrapped in the NPs. The following formula was used to calculate the loading efficiency of CUR:
In addition, siSTAT3 (100 nM) was dissolved with different PDA concentrations (0, 0.05, 0.10, 0.20, 0.40, and 0.80 μg/μL) to measure the encapsulation rate of Cy3-labeled siSTAT3. The prepared CPDA/siSTAT3 NPs were centrifuged at 20,000×g for 10 min, and the supernatant fluorescence spectra were recorded using a spectrophotometer (Tecan Infinite ®200 Pro; Tecan).
In vitro drug release
The dialysis bag served as the intermediate infiltration material for the release study, and the recipient bag was filled with CPDA/siSTAT3 NPs and CPDA/siSTAT3 NPs @SCM NPs, which were immersed in a PBS solution at a pH of 5.0 or 7.4 and moderately shaken at 37 °C. Analyses were performed on 1 mL of medium using a UV–visible spectrometer at 4, 8, 12, 16, 20, and 24 h to evaluate CUR release. The release medium was replaced with the same amount of liquid for each measurement to maintain a constant volume.
Cellular uptake and internalization of CPDA/siSTAT3 NPs @SCM NPs
Co-localization assays were used to verify membranes and core co-localization. CUR was co-loaded into the PDA core, and Dil was used to stain the MSC membranes. The dual-labeled CPDA/siSTAT3 @SCM NPs were incubated with MG63s for 4 h. Cells were rinsed, fixed with 4% formaldehyde, and stained with hoechst33342 before imaging under a CLSM (LSM 510 Metanlo; Zeiss, Jena, Germany) at excitation wavelengths of 405, 488, and 594 nm. Cy3-labeled siRNA and CUR were loaded into the NPs. CUR was co-loaded into the PDA core to confirm membrane and core co-localization, and Dil was used to stain the MSC membranes. The MG63s were incubated with dual-labeled CPDA/siSTAT3 NPs or CPDA/siSTAT3 @SCM NPs for 24 h. The subsequent steps were the same as those previously described. Furthermore, MG63, hFOB1.19, LO2, and RAW264.7 cells were incubated for 4 h with Cy3-labeled CPDA/siSTAT3 NPs or CPDA/siSTAT3 NPs @SCM.Ps, stained with hoechst33342, and photographed using a fluorescence microscope (Nikon, Tokyo, Japan).
Gene silencing efficiency of siSTAT3 via CPDA/siSTAT3 NPs
In 6-well plates (2 × 105 cells/well), MG63 cells were seeded and incubated for 24 h with naked siSTAT3 or CPDA/siSTAT3@SCM. The positive control was siSTAT3 (100 nM) encapsulated in Lipofectamine 3000 (Lipo3k). Total RNA obtained from cells was reverse transcribed using a kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. PCR amplification was performed using an ABI Step One Plus system and an SYBR Green Master Mix kit (Thermo Fisher Scientific, Waltham, MA, USA) to measure STAT3 expression.
Cell proliferation assays
The cell-counting kit (CCK)-8 assays and colony formation assay was used to assess cell proliferation. First, MG63 cells were plated at a density of 5 × 104 in a 96-well plate and co-incubated for 24 h with a different PBS formulation, free CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3, CPDA/siSTAT3@SCM. CCK-8 solution (10 μL) was added to each well and incubated at 37 °C for 1 h, and the absorbance of the samples was measured using a microplate reader (ELx-800; BioTek Instruments, Winooski, VT, USA) at 450 nm. MG63 cells (2 × 105 cells/well) were incubated in 6-well plates for plate colony assay with different groups of nanocomposites, as described above. The cells were resuspended and inoculated at 2 × 103 cells into 6-well plates for 24 h and then cultured for 14 days. Finally, cells were fixed with 4% paraformaldehyde, stained with crystal violet, and photographed.
Apoptosis assay
MG63 cells (2 × 105/per well) were incubated in 6-well plates and treated with PBS, free CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3, or CPDA/siSTAT3@SCM NPs to quantitatively analyze cell apoptosis. After incubation for 48 h, the cells were treated with Annexin V-FITC/PI, following the instructions of the manufacturer. All samples were scanned using FCM.
Wound healing assay
MG63 cells were seeded in 6-well plates (2 × 105 cells/well) with different groups of nanocomposites for the wound healing assay. After incubation for 24 h, a linear scratch was made with a 100 μL pipette tip, and the cells were washed twice. Subsequently, the cells were photographed with an inverted microscope (Olympus, Tokyo, Japan) at 0 and 48 h and assessed using ImageJ software.
In vitro migration and invasion
Transwells were seeded with MG63 cells (2 × 104 cells/well) for the migration assay, with or without Matrigel (B.D. Biosciences, Franklin Lakes, NJ, USA), to assess invasion and migration. The upper transwell chambers in both assays were filled with 300 μL of serum-free medium whereas the lower chambers were filled with a medium containing 10% FBS as a chemoattractant. Cells were fixed in 4% paraformaldehyde and stained with crystal violet after 24 h of culture at 37 °C, then examined under an inverted microscope (Olympus, Japan).
RNA isolation and quantitative real-time PCR (qRT-PCR)
The total RNA from the cells was extracted using RNAios Plus according to the manufacturer's protocol. To measure the expression of STAT3, the Hairpin-it™ miRNA RT-PCR Quantitation Kit was used for reverse transcription and qRT-PCR. The RNA was reverse-transcribed with the PrimerScriptTM reagent kit, and the expression level of STAT3 mRNA was analyzed using SYBR Green, with β-actin serving as the internal control. The primer sequences were as follows: STAT3 forward, 5′-GGAGGAGTTGCAGCAAAAAG-3′, STAT3 reverse, 5′-TGTGTTTGTGCCCAGAATGT-3′, β-actin forward, 5′-CCAAGGCCAACCGCGAGAAGATGAC-3′, β-actin reverse, 5′-AGGGTACATGGTGGTGCCGCCAG-3′.
Western blot assay
As previously described, different groups of MG63 cells (2 × 104 cells/well) were harvested and digested using RIPA lysis buffer (Beyotime, Nantong, China). Electrophoretic separation of the expectant protein was conducted using SDS-PAGE, followed by electrotransfer. The membranes were incubated at 4 °C with the primary antibodies GAPDH, STAT3, Bcl-2, Bax, E-cadherin, and N-cadherin (all 1:1000 dilutions; Bioss USA) after blocking with 5% skim milk. Membranes were then incubated with secondary antibodies (goat anti-rabbit, 1:5000 dilution; Proteintech) at room temperature for 1 h. The abundance of the target protein bands was measured and analyzed using a Bio-Rad imaging system (Bio-Rad Laboratories, Hercules, CA, USA).
In vivo antitumor study
Male BALB/c nude mice (6 weeks old) were obtained from the Vital River Company (Beijing, China) and housed in an SPF animal room. The Animal Welfare and Research Ethics Committee of Jilin University approved all experimental animal procedures. The OS mouse model was established by subcutaneously injecting 1 × 107 MG63 cells into the right flank of mice. Approximately 10 days later, spontaneous elevations of the tumor reached approximately 100 mm3 (tumor volume (V) = 0.5 × length × width2), and saline, free CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3, and CPDA/siSTAT3@SCM (CUR dose: 5 mg/kg; siSTAT3 dose: 0.31 mg/kg) were injected through the tail vein 7 times at an interval of 2 days. All mice were euthanized on day 29, and the tumor tissues or major organs were excised and collected. Paraformaldehyde 4% was used to fix tumors and organ samples, and paraffin was used as a fixative for histological analysis. In addition, hematoxylin and eosin (H&E), Ki67, and tunnel staining were performed on the tumors to further investigate the effect of the co-delivered NPs. In addition, STAT3 expression levels in the tumor tissue sections were evaluated by immunohistochemical staining using anti-STAT3 as the primary antibody.
In vivo biodistribution
To observe NP biodistribution, CPDA/siSTAT3 NPs and CPDA/siSTAT3@SCM NPs were injected using a DiR dye (Yeasen Biotech Corporation, Shanghai, China) at a concentration of 0.4 mg/kg via tail vein injection. An in vivo imagining system was used to collect whole-body fluorescence images (Xenogen Co., Alameda, CA, USA). The mice were euthanized 6, 24, and 48 h after treatment, and the major organs and tumors were excised and immediately imaged for ex vivo imaging.
Results and discussion
Construction and characterization of CPDA/siSTAT3@SCM NPs
Scheme 1 depicts a schematic representation of the co-delivery system CPDA/siSTAT3@SCM NPs for CUR and siSTAT3 and its inhibition of EMT-related and apoptosis-related OS pathways. First, we used dopamine hydrochloride to oxidize and self-polymerize into PDA NPs in a weakly alkaline aqueous phase. During the 24 h-synthesis process, the reaction system changed color from light brown to brown-black, as shown in Additional file 1: Fig. S1A. And, TEM (Additional file 1: Fig. S1B) analysis revealed the synthesized PDA NPs were regular spherical (the basic shape) with uniform particle size and good distribution. We loaded CUR onto PDA NPs through the π–π conjugated stacking interaction. The absorption peak was detected by the UV–Vis absorption spectrum because CUR has a characteristic absorption peak at a wavelength of 425 nm whereas PDA NPs have no special absorption peak. CPDA NPs showed a characteristic absorption peak at a wavelength of 425 nm, indicating that CUR was successfully loaded onto PDA (Fig. 1A). The CUR-loaded PDA NPs are hereafter referred to as CPDA NPs.

Schematic representation of the preparation process for the Cur and siSTAT3 co-delivery system, CPDA/siSTAT3@SCM NPs, and its inhibitory effects on EMT-related and apoptosis-related pathways in OS

The characterization of CPDA/siSTAT3@SCM NPs. A UV–Vis absorbance spectra of CUR, PDA NPs, and PDA–CUR NPs. B Fluorescence of 100 nM Cy3-siSTAT3 mixed with different concentrations of PDA NPs (0, 0.05, 0.10, 0.20, 0.40, and 0.80 μg/μL). C TEM images of the synthesized CPDA/siSTAT3 NP, SCM vesicle, and CPDA/siSTAT3@SCM NP. Scale bar = 50 nm. D SDS-PAGE-based protein analyses of SCM, CPDA/siSTAT3 NPs, and CPDA/siSTAT3@SCM NPs. E Intracellular co-localization of MSC membrane (red) and PDA/siSTAT3core (green). F Surface zeta potential of different formulations
As shown in Fig. 1B, when the concentration of PDA NPs reached 0.80 mg/mL, the fluorescence of 100 nM Cy3 was almost completely extinguished, indicating that almost all Cy3-STAT3 siRNA was loaded by PDA NPs, showing the highest fluorescence quenching efficiency. The average siRNA drug-loading was 41.25 ± 2.62 (μg/10 mg), confirming that PDA NPs still have high siRNA loading efficiency. In subsequent experiments of this study, the PDA NPs loaded with CUR and STAT3 siRNA are referred to as CPDA/siSTAT3 NPs. As shown in Fig. 1C, TEM analysis revealed that the CPDA/siSTAT3 NPs were solid and spherical in shape, with an average particle size of 101 ± 7 nm. After wrapping the MSC membrane, the average diameter of CPDA/siSTAT3@SCM NPs increased by approximately 20 nm, indicating that we have successfully camouflaged the umbilical cord MSC on the surface of CPDA/siSTAT3 NPs and obtained CPDA/siSTAT3@SCM NPs. The hydrated particle sizes of PDA, CPDA/siSTAT3, and CPDA/siSTAT3@SCM were 96 ± 5 nm, 101 ± 4 nm, and 117 ± 9 nm, respectively (Additional file 1: Fig. S2).
Phagocytosis is the process by which cells take up foreign matter, including bacteria, viruses, cell debris, and other foreign bodies. This process involves some special cells in the immune system, such as macrophages, neutrophils, and monocytes, which recognize and engulf these foreign substances through receptors on the cell membrane, and decompose and digest them (Feng et al. 2019). In the present study, after co-incubating the double-labeled CPDA/siSTAT3@SCM NPs with MG63 cells for 4 h, the cells were photographed and observed with a confocal microscope. The overlapping of the results of red fluorescent Bio-dil dye and green fluorescent CUR, as shown in Fig. 1E, confirmed that we successfully prepared CPDA/siSTAT3@SCM NPs and that the NPs can be recognized and phagocytized by MG63 cells. Moreover, the NP structure remains intact for a period of time after being taken up by cells.
As shown in Fig. 1F, the zeta potential of PDA increased to -21.8 ± 1.72 mV after CUR loading, whereas it was − 29.5 ± 2.13 mV after STAT3 siRNA loading because the negative charge carried by siRNA lowers the overall potential. Similarly, after further covering SCM, the zeta potential of CPDA/siSTAT3@SCM NPs decreased by approximately − 37.53 ± 2.41 mV because the MSC membrane vesicles also showed a negative charge drop, which further decreased the overall zeta potential.
Cellular uptake and safety evaluation
CUR was co-loaded into CPDA/siSTAT3 or CPDA/siSTAT3@SCM NPs with Cy3-labeled siSTAT3 to determine the tumor-homing effect of the prepared NPs in dual-agent co-delivery. As shown in Fig. 2A, compared with the CPDA/siSTAT3 NP group, the CPDA/siSTAT3@SCM NP group exhibited better drug phagocytosis accumulation in MG63 cells, indicating that CPDA/siSTAT3@SCM NPs improved the tumor-homing ability of MSC surface membrane and that the tumor-targeting ability of the NPs was enhanced. As shown in Fig. 2B, it can be observed by the naked eye that the red blood cells in distilled water (ddHaO) produced obvious hemolysis. After co-incubating particles at concentrations of 20 μg/mL, 50 μg/mL, and 100 μg/mL for 4 h, the resulting hemolysis ratios of red blood cells were 1.35%, 2.03%, and 4.51%, respectively. These values remained below the 5% threshold. This result indicates that CPDA/siSTAT3@SCM NPs have good hemocompatibility in blood. As shown in Additional file 1: Fig. S3, at a concentration within 500 μg/mL, the NPs did not affect the viability of LO2 cells, whereas, at a concentration > 500 μg/mL, the NPs reduced the viability of LO2 cells, indicating that the CPDA/siSTAT3@SCM NPs exhibit nanotoxicity at high concentrations. The qRT-PCR results shown in Fig. 3C show that the knockout efficiency of STAT3 mRNA in MG63 cells treated with PDA/siSTAT3 NPs was similar to that of the Lipo3k/siSTAT3 group.

Phagocytosis and safety evaluation of CPDA/siSTAT3@SCM NPs. A Cellular uptake of CPDA/siSTAT3 NPs and CPDA/siSTAT3@SCM NPs at 24 h analyzed by CLSM. Scale bar = 50 μm. B Hemolysis assay of multiple densities of CPDA/siSTAT3@SCM NPs. C Relative STAT3 mRNA expression in MG63 cells after transfection with naked siSTAT3, Lipo3k/siSTAT3, or CPDA/siSTAT3@SCM NPs. D Fluorescence microscopy images of MG63, hFOB1.19, LO2, and RAW264.7 cells after 4 h incubation with Cy3-labeled CPDA/siSTAT3 NPs and CPDA/siSTAT3@SCM NPs (nucleus visualized with blue Hoechst33342 dyes; NPs visualized with red Cy3 dyes). Scale bar = 100 μm
As shown in Fig. 2D, after co-culture with MG63 cells for 24 h, the number of phagocytic NPs in cells co-cultured in the Cy3-CPDA/siSTAT3@SCM NP group was significantly higher than that in the Cy3-CPDA/siSTAT3 NP group. However, after co-culture with normal bone Hfob1.19 cells and liver LO2 cells for 24 h, the number of phagocytic NPs in the Cy3-CPDA/siSTAT3@SCM NP group was reduced compared to that in the Cy3-CPDA/siSTAT3 NP group. In contrast, after co-incubation with RAW264.7 for 24 h, the fluorescence intensity in the cells co-cultured in the Cy3-CPDA/siSTAT3@SCM NP group was significantly lower than that in the Cy3-CPDA/siSTAT3 NP group, indicating that the surface proteins and immune evasion properties of mesenchymal stem cells are preserved in the SCM membrane vesicles, which may enable them to evade macrophage phagocytosis.
In vitro release of NPs
As shown in Fig. 3A, the drug release curve showed that at pH 5.0, CPDA/siSTAT3@SCM NPs released approximately 52% of CUR, which was two-fold higher than that at pH 7.4, indicating that the CUR release from the NPs is better in acidic tumor tissues. Notably, in the tumor microenvironment, the functional groups on the surface of PDA NPs can interact with the amino groups in CUR molecules to weaken the π-π stacking effect, endowing the CPDA/siSTAT3@SCM NPs with a good pH-dependent cumulative drug release capability, which can reduce dosing frequency. In addition, under the same pH environment, the CUR release of CPDA/siSTAT3@SCM NP was slightly lower than that of bare NPs without MSC membrane, indicating that MSC membrane vesicles can slow down CUR release from NPs and improve the effective circulating drug concentrations.

In vitro experiments of CPDA/siSTAT3@SCM NPs. A Release profile of CUR from CPDA/siSTAT3 NPs and CPDA/siSTAT3@SCM NPs at different pH within 24 h. B Comparison of relative viability of MG63 cells cultured with different formulations for 24 h and 48 h. C Representative images of colony formation using various formulations in MG63 cells. D Apoptosis of MG63 cells induced by PBS, free CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3 NPs, and CPDA/siSTAT3@SCM NPs analyzed by FCM. E Wound healing width of MG63 cells induced by various formulations measured and photographed at 0 h and 48 h
Combined antitumor therapeutic efficacy in vitro
As shown in Additional file 1: Fig. S4, the 50% inhibitory concentration (IC50) of MG63 cells was 54.74 ± 6.12 μg/mL. Therefore, subsequent experiments in this study used CUR, CPDA, CPDA/siSTAT3, and CPDA/siSTAT3@SCM NPs with a CUR equivalent concentration of 10 μg/mL (below IC50).
As shown in Fig. 3B, compared with the PBS group, the MG63 cells in the CUR group alone, the CPDA group, and the PDA/siSTAT3 group showed tumor cell inhibitory effects. However, compared with the above three groups, the reduction in tumor cell activity in the CPDA/siSTAT3 group was more significant, indicating that the CPDA/siSTAT3 NPs loaded with both CUR and STAT3 siRNA exerted both the anticancer effect of CUR and the gene expression silencing effect of STAT3 siRNA. In addition, compared with CPDA/siSTAT3 NPs, CPDA/siSTAT3@SCM NPs exhibited higher cytotoxicity to MG63 cells, which is likely because SCM membrane vesicles can increase the accumulation of NPs in MG63 cells and subsequently increase the number of NPs; drug accumulation can better kill tumor cells. After CPDA/siSTAT3@SCM NPs were co-incubated with OS cells for 48 h, the average relative viability of the cells was 18.23%, indicating that CPDA/siSTAT3@SCM NPs have a good antitumoral effect.
As shown in Fig. 3C, compared with the PBS group in MG63 cells, the average cell colony formation rates of the CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3, and CPDA/siSTAT3@SCM groups decreased by nearly 18.6%, 22.4%, 40.3%, 63.7%, and 80.2%, respectively. Compared with the CPDA and PDA/siSTAT3 groups, the effect of the CPDA/siSTAT3@SCM NP group in inhibiting MG63 cell proliferation was significant, indicating that the CPDA/siSTAT3@SCM NP group can better inhibit tumor cell proliferation than the single administration groups.
As shown in Additional file 1: Fig. S5, the results of live/dead staining of MG63 cells were consistent with the above results. The number of dead MG63 cells was higher in the CPDA/siSTAT3@SCM group than in the PBS, CUR, CPDA, PDA/siSTAT3, and CPDA/siSTAT3 groups. Moreover, both the CPDA/siSTAT3 and CPDA/siSTAT3@SCM groups showed good tumor cell-killing ability, with the CPDA/siSTAT3@SCM group having the best tumor-killing effect with an average rate of dead cells of 75.2%. These results indicate that the SCM membrane vesicles can increase NP accumulation in MG63 cells to enhance the tumor-killing effect.
The apoptotic rates of each MG63 cell group with PBS, CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3, and CPDA/siSTAT3@SCM were 4.74 ± 1.04%, 6.55 ± 2.12%, 9.24 ± 5.16%, 20.12 ± 7.12%, 37.65 ± 7.58%, and 48.65 ± 8.24%, respectively, indicating that the CPDA/siSTAT3@SCM NP group showed the strongest ability to induce apoptosis of MG63 cells (Fig. 3D).
Effects of CPDA/siSTAT3@SCM nanoparticles on OS cell migration and invasion
We used the cell scratch experiment to detect the effects of PBS, CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3, and CPDA/siSTAT3@SCM NPs on the migration ability of MG63 cells. As shown in Fig. 3E, after 48 h of observation, the healing speed of tumor cells in the other five experimental groups decreased compared with that of the PBS group; the cell healing speed of MG63 cells in the CPDA/siSTAT3 NP and the CPDA/siSTAT3@SCM groups was significantly lower than that of other groups (P < 0.01). Moreover, the CPDA/siSTAT3@SCM NPs had the best inhibitory effect on OS cells, and the migration rates of MG63 cells were 7.48 ± 3.06%, indicating that MSC membrane vesicles can increase the accumulation of NPs in OS cells to OS cell migration.
As shown in Fig. 4A, the transwell migration assay showed that the number of MG63 cells passing through the bottom membrane of the Transwell chamber decreased sequentially: PBS > CUR > CPDA > PDA/siSTAT3 > CPDA/siSTAT3 > CPDA/siSTAT3 groups. In the @SCM group, the CPDA/siSTAT3 NP and CPDA/siSTAT3@SCM NP groups significantly reduced the number of MG63 cells compared to other groups, indicating a synergistic antitumor therapeutic effect. Moreover, in the @SCM NP group, the number of CPDA/siSTAT3 MG63 passing through the bottom membrane of the Transwell chamber was the lowest, indicating that the migration of OS cells was significantly inhibited. Therefore, the results of the Transwell invasion experiment were consistent with the above results. In addition, the number of OS cells passing through the bottom membrane of the Transwell chamber in the CPDA/siSTAT3@SCM NP group was significantly lower than that in the other groups (P < 0.01), indicating that CPDA/siSTAT3@SCM NPs can significantly inhibit the migration and invasion of MG63 cells.

In vitro experiments of CPDA/siSTAT3@SCM NPs. A Representative images of migrated and invaded MG63 cells after incubation with PBS, free CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3 NPs, and CPDA/siSTAT3@SCM NPs for 24 h. Scale bars: 50 μm. B Relative STAT3 mRNA in different groups was detected using QRT-PCR. C Bcl-2, Bax, E-Cadherin, and N-Cadherin were analyzed using Western blotting
CPDA/siSTAT3@SCM-mediated enhanced apoptosis and migration inhibition in vitro
We further detected the expression levels of STAT3 mRNA in MG63 cells treated with CUR, CPDA, PDA/siSTAT3, CPDA/siSTAT3, and CPDA/siSTAT3@SCM NPs by qRT-PCR for 48 h. STAT3 expression was downregulated in the CUR, CPDA, PDA/siSTAT3, and CPDA/siSTAT3 groups; however, it was not significant compared with that in the PBS group. In addition, STAT3 was significantly downregulated in the CPDA/siSTAT3 and CPDA/siSTAT3@SCM NP groups, with the CPDA/siSTAT3@SCM NP group showing the most significant downregulation, further demonstrating the effect of gene-drug combination therapy and the therapeutic advantages of membrane-camouflaged encapsulated NPs (Fig. 4B).
Cancer growth represents a dysregulated imbalance between cell gain and loss, and resistance to apoptosis is an acquired characteristic of tumor cells, contributing to tumor evolution and growth, as well as treatment failure (Kulbay et al. 2022). Therefore, the efficacy of cancer therapy depends not only strictly on the cellular damage caused but also on the ability of the cell to activate its apoptotic program (Morana et al. 2022).
As shown in Fig. 4C, western blot confirmed that Bax protein expression levels were increased in the CUR, CPDA, and PDA/siSTAT3 groups; the CPDA/siSTAT3 NP group had a relatively strong ability to promote Bax protein whereas the CPDA /siSTAT3@SCM NP showed the strongest ability to promote Bax protein expression. The opposite trend was observed for the expression of Bcl-2 protein in MG6 cells with the CPDA/siSTAT3@SCM NP group having the highest Bcl-2 protein expression levels.
Furthermore, EMT, a common process in tumor cells characterized by cells losing their epithelial and acquiring mesenchymal features, is associated with various tumor functions, including tumor initiation, malignant progression, tumor stemness, tumor cell migration, blood infiltration, metastasis, and therapy resistance (Ramesh et al. 2020). As shown in Fig. 4C, western blot analysis confirmed that the expression of N-cadherin proteins was reduced in MG63 cells treated with CUR, CPDA, and PDA/siSTAT3, with the CPDA/siSTAT3 NP group having a relatively weak inhibitory effect and the CPDA/siSTAT3@SCM NP group showing the strongest inhibition effect on N-cadherin protein expression. The opposite trend was observed for E-cadherin protein expression in MG63 cells. These results suggest that CPDA/siSTAT3@SCM NPs can inhibit the expression of N-cadherin protein in OS cells and promote the expression of E-cadherin protein, indicating that they significantly inhibited EMT in MG63 cells.
Evaluation of antitumor efficacy
A subcutaneous MG63 tumor-bearing mouse model was used to evaluate CPDA/siSTAT3@SCM NPs antitumor efficacy in vivo. When the tumor volume reached approximately 100 mm3, the mice received injections of saline, NPs were injected via the tail vein, and 7 injections were administered every 2 days (Fig. 5A).

The therapeutic efficacy of CPDA/siSTAT3@SCM NPs in orthotopic MG63 tumor models. A The body weight of MG63 subcutaneous xenograft mice treated with various formulations. B Representative images of excised tumors. C Tumor weight of each group after treatment. D Tumor growth curves and E tumor weights of MG63 tumor-bearing mice. F After the antitumor study, tumor tissues were stained with H&E, TUNEL, Ki67, and STAT3 immunohistochemistry. Scale bars: H&E: 100 μm; others: 50 μm
Figure 5B depicts no weight loss in the various PDA-based groups, suggesting that the co-delivery of CUR and siSTAT3 by PDA@SCM may have almost no systemic acute toxicity compared to CUR alone. Tumor volume growth analysis revealed that the CPDA/siSTAT3 NP groups inhibited tumor growth moderately compared to other groups, whereas CPDA/siSTAT3@SCM NP groups exhibited the strongest antitumor effect in vivo because of the synergistic effect of CUR and siSTAT3 and “homing affinity” to tumor sites (Fig. 5C, D). The results of the tumor tissue weight analysis showed a similar trend as those of the tumor volume growth analysis, with the CPDA/siSTAT3@SCM NP group having the smallest average tumor weight (183 mg) compared to the other groups (Fig. 5E).
In addition, H&E staining and TUNEL immunohistochemistry revealed that the CPDA/siSTAT3@SCM NP group had the most necrotic area and the highest apoptosis levels compared to the free saline CUR, CPDA, PDA/siSTAT3, and CPDA/siSTAT3 groups (Fig. 5F). The blank areas of cells treated with the CPDA/siSTAT3@SCM NP group were the largest, and cell nuclei were shrunk, revealing the prominent antitumor effect of CPDA/siSTAT3@SCM NPs. Furthermore, Ki67 levels indicated the reduced proliferative activity of OS cells in mice treated with the prepared NPs, demonstrating the high efficacy of CPDA/siSTAT3@SCM NPs in inhibiting OS cell growth. We used immunohistochemical analyses to determine STAT3 expression levels in tumors after treatment and found that mice treated with CPDA/siSTAT3@SCM NPs had significantly decreased STAT3 expression compared with mice treated with saline, free CUR, CPDA, PDA/siSTAT3, and CPDA/siSTAT3@SCM NP (Fig. 6F).
In vivo fluorescence imaging
The in vivo biodistribution of CPDA/siSTAT3 NPs and CPDA/siSTAT3@SCM NPs was measured using a live imaging system to analyze the “homing affinity” of SCM to tumor sites (Fig. 6A). The mice were injected with DIR-CPDA/siSTAT3 NPs and DIR-CPDA/siSTAT3@SCM NPs via the tail vein and imaged 6, 24, and 48 h after injection. The findings suggested that DiR-CPDA/siSTAT3 NPs accumulated in small amounts at the tumor site, probably due to the enhanced permeability and retention effect (EPR) effect, whereas DIR-CPDA/siSTAT3@SCM NPs increased the tumor-targeting ability of NPs.

Biodistribution of CPDA/siSTAT3 NPs and CPDA/siSTAT3@SCM NPs. A Whole-body and major organ fluorescence images of MG63 tumor-bearing mice at 6, 24, and 48 h after injection of DiR-labeled CPDA/siSTAT3 NPs and CPDA/siSTAT3@SCM NPs. B Representative H&E staining images of major organs from MG63 tumor-bearing mice injected with CPDA/siSTAT3@SCM NPs. Scale bar: 50 μm
Similar to the in vitro uptake experiments, DIR-CPDA/siSTAT3 NPs and DIR-CPDA/siSTAT3@SCM NPs accumulated in a time-dependent tumor manner and were detected in other tissues. In the liver, SCM-coated NPs exhibited a lower fluorescent intensity than naked NPs, suggesting that SCM can reduce NP phagocytosis by body organs.
In vivo safety evaluation of CPDA/siSTAT3@SCM NPs
No obvious pathological changes were observed in the section of the major organs in the CPDA/siSTAT3@SCM NP group, whereas liver necrosis was observed in mice treated with free CUR (Fig. 6B). These results indicate that the synergistic effect of CSNP@SCM NPs not only increased the concentration of CUR at the tumor site but also improved the effect of siSTAT3 in vivo, leading to a more efficient synergistic treatment strategy for OS.
In addition, we collected blood from the eyeballs of OS-bearing mice in each group to analyze the commonly used clinical serum biochemical indicators (alkaline phosphatase [ALP], alanine aminotransferase [ALT], albumin [ALB], aspartic acid aminotransferase [AST], creatine kinase [CK], lactate dehydrogenase [LDH], blood urea nitrogen [BUN], and creatinine [CREA]). As shown in Additional file 1: Fig. S6, the levels of serum biochemical indicators ALP, ALT, ALB, AST, CK, LDH, BUN, and CREA in the CPDA/siSTAT3@SCM NP group were not significantly different from those in the saline group. These results indicate that in OS-bearing mice, the synthesized CPDA/siSTAT3@SCM NPs have no obvious NP toxicity and side effects, indicating that they have good biological safety in vivo.
The above experimental results prove that our synthesized product CPDA/siSTAT3@SCM NPs can carry curcumin and siSTAT3 to target OS cells, with low nanotoxicity and good biological safety. Moreover, the immune escape and tumor-targeting capabilities of SCM were not destroyed during the synthesis process. CPDA/siSTAT3@SCM NPs can effectively exert the synergistic therapeutic effect of curcumin and siSTAT3, achieve the effect of drug-gene combined therapy, promote the expression of apoptosis-related proteins, and inhibit the progress of EMT in OS, thereby promoting OS cell apoptosis process and inhibiting OS proliferation, invasion, and migration. However, although the results of the in vitro and in vivo studies are promising, more experiments are needed to validate the clinical use of our NP drug delivery system. Overall, the CPDA/siSTAT3@SCM NP system represents a novel class of nanoplatforms for effective targeted drug delivery and provides a promising strategy for treating patients with OS.
Conclusion
In the present study, we developed a multifunctional nanoplatform for combining cancer chemotherapy and gene therapy by co-loading hydrophobic chemotherapeutics and siRNA. Our findings revealed that the developed NPs have a desirable drug delivery capacity, tumor-homing, and good biostability. Pre-clinical studies revealed that CPDA/siSTAT3@SCM NPs have good inhibitory effects on OS cell growth and the EMT process. Moreover, the in vivo biodistribution study indicated that CPDA/siSTAT3@SCM NPs can target and accumulate at the tumor sites. However, although the synthesized CPDA/siSTAT3@SCM NP exhibited good antitumor effects in the OS-bearing mouse model, their effectiveness should be verified in higher-level mammalian animal models. In addition, molecular biochemistry and other methods can also be used to modify the CPDA/siSTAT3@SCM NP drug-loading system to increase its biocompatibility and self-degradation ability in vivo, thereby improving the possibility of the clinical translation of our NPs. We hope that the nano-therapeutic strategy can be further developed to meet the clinical application and provide more effective and convenient treatment for patients with OS.
Abbreviations
- OS:
-
Osteosarcoma
- SCM:
-
Stem cell membrane
- CUR:
-
Curcumin
- OS:
-
Osteosarcoma
- EMT:
-
Epithelial–mesenchymal transition
- STAT:
-
Signal transducer and activator of transcription
- PDA:
-
Polydopamine
- MSCs:
-
Mesenchymal stem cells
- VEGE:
-
Vascular endothelial growth factor
- PDGF:
-
Platelet-derived growth factor
- siSTAT3:
-
Small interfering RNA STAT3
- SDS-PAGE:
-
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- CLSM:
-
Confocal laser scanning microscope
- H&E:
-
Hematoxylin and eosin
References
Cao L et al (2016) Curcumin inhibits hypoxia-induced epithelial-mesenchymal transition in pancreatic cancer cells via suppression of the hedgehog signaling pathway. Oncol Rep 35(6):3728–3734
Chi Y et al (2017) Redox-sensitive and hyaluronic acid functionalized liposomes for cytoplasmic drug delivery to osteosarcoma in animal models. J Control Release 261:113–125
Corre I et al (2020) The osteosarcoma microenvironment: a complex but targetable ecosystem. Cells 9(4):976
Feng M et al (2019) Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 19(10):568–586
Fossey SL et al (2011) The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 11:112
Gonçalves M et al (2014) Dendrimer-assisted formation of fluorescent nanogels for drug delivery and intracellular imaging. Biomacromol 15(2):492–499
Hass R et al (2011) Different populations and sources of human mesenchymal stem cells (MSC): a comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal 9:12
Jia F et al (2021) Self-assembled fluorescent hybrid nanoparticles-mediated collaborative lncRNA CCAT1 silencing and curcumin delivery for synchronous colorectal cancer theranostics. J Nanobiotechnol 19(1):238
Jin X, Wang C, Wang L (2020) Costunolide inhibits osteosarcoma growth and metastasis via suppressing STAT3 signal pathway. Biomed Pharmacother 121:109659
Kong G et al (2017) Irisin reverses the IL-6 induced epithelial-mesenchymal transition in osteosarcoma cell migration and invasion through the STAT3/Snail signaling pathway. Oncol Rep 38(5):2647–2656
Kulbay M et al (2022) Review of cancer cell resistance mechanisms to apoptosis and actual targeted therapies. J Cell Biochem 123(11):1736–1761
Li Y et al (2020) Co-delivery of doxorubicin and paclitaxel by reduction/pH dual responsive nanocarriers for osteosarcoma therapy. Drug Deliv 27(1):1044–1053
Li ZQ et al (2022) LRIG2 regulates cell proliferation, migration and apoptosis of osteosarcoma. BMC Cancer 22(1):1029
Liu Y et al (2021) STAT3 and its targeting inhibitors in osteosarcoma. Cell Prolif 54(2):e12974
Liu X et al (2022) A conductive gelatin methacrylamide hydrogel for synergistic therapy of osteosarcoma and potential bone regeneration. Int J Biol Macromol 228:111–122
Lu KH et al (2020) Curcumin analogue L48H37 suppresses human osteosarcoma U2OS and MG-63 cells’ migration and invasion in culture by inhibition of uPA via the JAK/STAT signaling pathway. Molecules 26(1):30
Morana O, Wood W, Gregory CD (2022) The apoptosis paradox in cancer. Int J Mol Sci 23(3):1328
Moukengue B et al (2022) Origin and therapies of osteosarcoma. Cancers 14(14):3503
Mu X et al (2021) Doxorubicin and PD-L1 siRNA co-delivery with stem cell membrane-coated polydopamine nanoparticles for the targeted chemoimmunotherapy of PCa bone metastases. Nanoscale 13(19):8998–9008
Panagi M et al (2022) Immunotherapy in soft tissue and bone sarcoma: unraveling the barriers to effectiveness. Theranostics 12(14):6106–6129
Pricci M et al (2020) Curcumin and colorectal cancer: from basic to clinical evidences. Int J Mol Sci 21(7):2364
Ramesh V, Brabletz T, Ceppi P (2020) Targeting EMT in cancer with repurposed metabolic inhibitors. Trends Cancer 6(11):942–950
Su R et al (2021) Photo-enhanced antibacterial activity of polydopamine-curcumin nanocomposites with excellent photodynamic and photothermal abilities. Photodiagnosis Photodyn Ther 35:102417
Sun Y et al (2019) Curcumin inhibits the proliferation and invasion of MG-63 cells through inactivation of the p-JAK2/p-STAT3 pathway. Onco Targets Ther 12:2011–2021
Tan B et al (2021) Curcumin-microsphere/IR820 hybrid bifunctional hydrogels for in situ osteosarcoma chemo-co-thermal therapy and bone reconstruction. ACS Appl Mater Interfaces 13(27):31542–31553
Termini D et al (2020) Curcumin against prostate cancer: current evidence. Biomolecules 10(11):1536
Thanuja MY, Anupama C, Ranganath SH (2018) Bioengineered cellular and cell membrane-derived vehicles for actively targeted drug delivery: so near and yet so far. Adv Drug Deliv Rev 132:57–80
Wang L et al (2019) Curcumin derivative WZ35 inhibits tumor cell growth via ROS-YAP-JNK signaling pathway in breast cancer. J Exp Clin Cancer Res 38(1):460
Wang J et al (2022) MicroRNA-124 expression in Kupffer cells modulates liver injury by targeting IL-6/STAT3 signaling. Antiviral Res 311:105510
Wu HH et al (2019) Mesenchymal stem cell-based drug delivery strategy: from cells to biomimetic. J Control Release 294:102–113
Wu K et al (2022) Recent advances in nanoplatforms for the treatment of osteosarcoma. Front Oncol 12:805978
Xu C et al (2021) Curcumin in osteosarcoma therapy: combining with immunotherapy, chemotherapeutics, bone tissue engineering materials and potential synergism with photodynamic therapy. Front Oncol 11:672490
Yang M et al (2022) TIPE1 inhibits osteosarcoma tumorigenesis and progression by regulating PRMT1 mediated STAT3 arginine methylation. Cell Death Dis 13(9):815
Yin J et al (2020) MXene-based hydrogels endow polyether ether ketone with effective osteogenicity and combined treatment of osteosarcoma and bacterial infection. ACS Appl Mater Interfaces 12(41):45891–45903
Zhang W, Huang X (2022) Stem cell membrane-camouflaged targeted delivery system in tumor. Mater Today Bio 16:100377
Zhang M et al (2020) Polydopamine nanoparticles camouflaged by stem cell membranes for synergistic chemo-photothermal therapy of malignant bone tumors. Int J Nanomed 15:10183–10197
Zou S et al (2020) Targeting STAT3 in cancer immunotherapy. Mol Cancer 19(1):145
Acknowledgements
We are grateful to the following staffs for the guidance and support of flow cytometry: Yucheng Zhang, Liu Yingnan, Lv Huiying, Gao Puyu and Yu Weijia. We are grateful to Xiaoyue Zhang from the Middle School of Northeast Normal University for the support in animal experiments.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 82172230), the Jilin Scientific and Technological Development Program (Grant Nos. 20210101334JC, 20190901007JC and 20190908002TC), the Capital Construction Funds Planned Projects in the Provincial Budget of 2019 (Grant No. 2019C016), the Changchun Science and Technology Development Plan Project (Grant No. 21ZGY29), the Health Special Project of Jilin Provincial Finance Department (Grant No. 2019SCZ509), and Health Research Talents Project of Jilin Province (Grant No. 2020SCZ44).
Author information
Authors and Affiliations
Contributions
KW collected, analyzed the data, and drafted the manuscript. ZZ, TL and CL participated in the data analysis and interpretation. JJ and XM revised and edited the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the China-Japan Union Hospital of Jilin University, Changchun, China based on the ethical guidelines for Animal Experimentation of the National Research Council of China.
Consent for publication
All the authors agree to the publication of this manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Preparation and morphology of PDA nanoparticles. A. Schematic representation of the PDA nanoparticle polymerization process for 24 h. B. Morphology of PDA nanoparticles observed under transmission electron microscope. Fig. S2. Characterization of hydrated particle size of PDA, CPDA/siSTAT3, and CPDA/siSTAT3@SCM nanoparticles. Fig. S3. Toxicity experiment of PDA@SCM nanoparticles was conducted on LO2 cells. Fig. S4. IC-50 curves were measured by the CCK8 methodto determine the concentration of CUR required to inhibit cell growth by 50% in MG63 cells, with concentrations ranging from 5 μg/mL to 65 μg/mL. Fig. S5. The calcein-AM/PI staining of MG63 cells in each group was visualized and analyzed. The histogram shows the quantification of calcein-AM/PI staining of MG63 cells in each group. Fig. S6 Hematological analysis of ALP, ALT, ALB, AST, CK, LDH, BUN, and CREA levels. ALP: alkaline phosphatase, ALT: alanine aminotransferase, ALB: albumin, AST: aspartate transaminase, CK: creatine kinase, LDH: lactate dehydrogenase, BUN: blood urea nitrogen, CREA: creatinine
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, K., Zhou, Z., Liu, T. et al. Co-delivery of curcumin and si-STAT3 with a bioinspired tumor homing for polydopamine nanoparticles for synergistic osteosarcoma therapy. Cancer Nano 14, 66 (2023). https://doi.org/10.1186/s12645-023-00215-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12645-023-00215-5